
Abstract
Mowat-Wilson syndrome (MOWS) is a congenital disease caused by de novo heterozygous loss of function mutations or deletions of the ZEB2 gene. MOWS patients show multiple anomalies including intellectual disability, a distinctive facial appearance, microcephaly, congenital heart defects and Hirschsprung disease. However, the skin manifestation(s) of patients with MOWS has not been documented in detail. Here, we recognized that MOWS patients exhibit many Ehlers-Danlos syndrome (EDS)-like symptoms, such as skin hyperextensibility, atrophic scars and joint hypermobility. MOWS patients showed a thinner dermal thickness and electron microscopy revealed miniaturized collagen fibrils. Notably, mice with a mesoderm-specific deletion of the Zeb2 gene (Zeb2-cKO) demonstrated redundant skin, dermal hypoplasia and miniaturized collagen fibrils similar to those of MOWS patients. Dermal fibroblasts derived from Zeb2-cKO mice showed a decreased expression of extracellular matrix (ECM) molecules, such as collagens, whereas molecules involved in degradation of the ECM, such as matrix metalloproteinases (MMPs), were up-regulated. Furthermore, bleomycin-induced skin fibrosis was attenuated in Zeb2-cKO mice. We conclude that MOWS patients exhibit an EDS-like skin phenotype through alterations of collagen fibrillogenesis due to ZEB2 mutations or deletions.
Similar content being viewed by others


Introduction
Mowat-Wilson syndrome (MOWS; OMIM 235730) is a congenital disease with the prevalence of 1 per 50,000–90,000 live births1,2,3. MOWS is characterized by a distinctive facial appearance, intellectual retardation, epilepsy and malformations including Hirschsprung disease, congenital heart defects and urogenital anomalies4,5. In 2001, the underlying cause of MOWS was identified as mutations or deletions in the Zinc finger E-box-binding homeobox 2 gene (ZEB2, previously called ZFHX1B or SIP1) located at chromosome 2q226,7. ZEB2 encodes Smad Interacting Protein 1 (SIP1), which is a multidomain protein characterized by a central homeodomain, a CtBP-binding domain and a Smad-binding domain, as well as two separated, highly conserved zinc finger clusters at the N- and C-terminals8. Each zinc finger cluster can independently bind to CACCT(G) sequences in the promoter regions of genes involved in development and differentiation, including the E-cadherin promoter9, by which E-cadherin is down-regulated, leading to initiation of the epithelial-to-mesenchymal transition10. ZEB2 was originally identified as a transcriptional co-repressor for the transforming growth factor β (TGF-β) signaling pathway through binding to Smad proteins8,11. Since ZEB2 is a highly evolutionarily conserved gene that is widely expressed during embryological development, mice that are homozygous deficient in the Zeb2 gene have defects in neural crest cell migration, compromised development of the peripheral nervous system, craniofacial tissues and the heart, which results in lethality between E9.5 and E10.512,13. Notably, neural crest-specific ablation of Zeb2 caused craniofacial and gastrointestinal malformations that resemble symptoms in human patients with MOWS14. Patients with typical MOWS harbor deletions or truncating mutations of ZEB2, suggesting that haploinsufficiency is the pathogenic basis of MOWS, although no genotype-phenotype correlations have been suggested2.
Among various anomalies in multiple organs, skin symptoms in MOWS patients have not been frequently reported, if any. Some patients with MOWS were reported with fair complexion15 or raindrop depigmentation16. Here we report for the first time that most MOWS patients exhibit skin manifestations similar to those of patients with Ehlers-Danlos syndrome (EDS). EDS is a diverse group of heritable connective tissue disorders, which include 6 major types and additional minor subtypes typically characterized by skin hyperextensibility and hypermobile joints2,17. Since the pathogenesis of EDS generally involves abnormalities in collagen fibrillogenesis18, we investigated whether MOWS patients also show abnormalities in the dermis. Further, we generated mesoderm-specific Zeb2 conditional knockout (Zeb2-cKO) mice, which reproduced the skin phenotype found in MOWS patients.
Results
Patients with MOWS exhibit an EDS-like phenotype
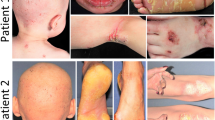
We enrolled 12 patients with MOWS (2–16 years of age), 8 of whom had been previously reported3,19 along with diagnoses based on genotype findings in their ZEB2 gene and/or clinical features. Those features include a characteristic facial appearance, microcephaly, congenital heart disease such as ventricular septal defect, neurological symptoms such as intellectual disability and seizure, Hirschsprung disease, and agenesis or hypoplasia of the corpus callosum (Supplementary Table 1). We recognized that all 12 of those MOWS patients had soft and velvety skin. Furthermore, they exhibited some of the characteristic features of EDS, including skin hyperextensibility (Fig. 1b,c,h,j), joint hypermobility (Fig. 1a,d,f,k), atrophic cutaneous scars and a molluscoid pseudotumor of the elbow (Fig. 1e), which are characterized as classical symptoms of EDS20. Joint laxity was evident in the fingers, wrists and toes (Fig. 1a,d,f,k), although their symptoms did not reach a Beighton score of 5 or more, which is the standard assessment of joint disease in EDS21. Some patients had thin, almost translucent skin and therefore their veins were easily visible (Fig. 1g,i). However, they did not develop easy bruising as do patients with vascular EDS. It should be noted that the skin phenotypes found in MOWS patients did not correlate with ZEB2 mutations or the severity of their clinical features (Supplementary Table 1). Ultrasonic examination of two representative patients (patients #11 and #12) revealed that the dermal thickness in various parts of their body was generally thinner than in age-matched healthy controls (Supplementary Fig. 1).

(a,d,f,k) Joint hypermobility. (b,c,h,j) Skin hyperextensibility. (e) Atrophic cutaneous scars (arrow), molluscoid pseudotumor of the elbow (arrowhead). (g,i) Skin thinning. Patient numbers are shown in each panel (see Supplementary Table 1 for listing).
Ultrastructural characteristics of the dermis of a MOWS patient
Electron microscopic examination of a skin biopsy from a representative MOWS patient (patient #11) revealed marked miniaturization in the diameter of collagen fibrils compared with a healthy control (Fig. 2; mean nm ± SD; 92.96 ± 11.94 in the healthy control, 50.6 ± 9.32 in MOWS; P < 0.001 by Student’s t-test). Similarly, the dermis of EDS patients shows abnormalities in collagen fibrils by electron microscopy, including heterogeneity in size with a flower-like shape22. Although the features were not identical, the ultrastructural abnormality in collagen fibrils strongly suggests that MOWS patients exhibit the EDS-like skin phenotype possibly due to abnormal fibrillogenesis of collagen in the dermis.

(a) Electron micrographs of the dermis of a healthy control (top) and a MOWS patient (#11, bottom). Scale bars, 500 nm, 100 nm (insets). (b) Diameters of collagen fibrils in the healthy control (top) and in the MOWS patient (bottom). Diameters of 500 fibrils were counted and are shown in 10 nm increments. ***P < 0.001 by Student’s t-test.
Decreased ZEB2 expression in dermal fibroblasts from a MOWS patient
MOWS is caused by de novo heterozygous mutations or deletions in the ZEB2 gene2, which leads to haploinsufficiency7. RT-PCR revealed that ZEB2 gene expression in dermal fibroblasts derived from MOWS patient #11, who had a frameshift mutation (p.T761Kfs_26)3, was decreased by approximately 40% compared to healthy controls, whereas the level of ZEB1 mRNA was not affected (Supplementary Fig. 2a). Correspondingly, the ZEB2 protein level of fibroblasts derived from MOWS patient #11 was reduced close to half of the levels in control fibroblasts (Supplementary Fig. 2b,c). Collectively, these results confirmed the ZEB2 haploinsufficiency in MOWS patients.
Generation of Zeb2-cKO mice using the Cre-loxP system
To study the role of Zeb2 in vivo, we generated Cre-mediated Zeb2-cKO mice (Supplementary Fig. 3a), since a germline Zeb2 deficiency leads to embryonic lethality13. To this end, we crossed Zeb2 floxed mice with mice harboring the Cre transgene under the promoter of the Prx1 gene, which is specific for the mesoderm/mesenchyme23,24. Prx1-Cre:Zeb2flox/flox (Zeb2-cKO) mice were born in accordance with the Mendelian law. Genomic PCR analysis of newborn Zeb2-cKO mice revealed that the gene targeting occurred in dermal fibroblasts and in subcutaneous fat tissues, but not in epidermal cells or the liver, indicating that it was mesenchymal-specific (Supplementary Fig. 3b).
Zeb2-cKO mice show a skin phenotype similar to MOWS
Although the gross appearance of newborn Zeb2-cKO mice was normal, from two weeks of age onward they showed some growth retardation and hair loss in the scalp and extremities (Fig. 3a,b). Strikingly, they exhibited redundant, hyperextensive skin, which resembled that found in EDS patients (Fig. 3c,d). Furthermore, histopathology revealed that the dermis of Zeb2-cKO mice had a reduced thickness compared with wild-type control mice (Fig. 4a,b). In addition, subcutaneous fat tissues in the chest and limbs, but not in the back, were atrophic compared with controls (Fig. 4a). Electron microscopic analysis of the dermis revealed uniform miniaturization of collagen fibrils in Zeb2-cKO mice both at 7 (Fig. 5) and at 14 weeks of age (Supplementary Fig. 4) compared with wild-type mice. Therefore, the ultrastructural characteristics of Zeb2-cKO mice were identical to those found in MOWS patients. Collectively, the skin abnormalities of MOWS patients were faithfully reproduced in Zeb2-cKO mice, suggesting that ZEB2 gene disruption might lead to impairment in the homeostasis of the dermal structure such as collagen synthesis.

(a) Wild-type (WT) and Zeb2-cKO (cKO) mice at 2 weeks of age. Growth retardation and local alopecia in the scalp were found in Zeb2-cKO mice. (b) Alopecia developed in the scalp, limbs, chest and abdomen of Zeb2-cKO mice at 4 weeks of age. (c) Redundant skin in the upper limb of Zeb2-cKO mice (right) at 8 weeks of age. (d) Zeb2-cKO mice (right) at 16 weeks of age showed redundant skin and hyperextensibility.

(a) Histological features of the back, chest and limb skins of wild-type mice (WT, top panels) and Zeb2-cKO mice (bottom panels) at 7 weeks of age. Scale bar, 50 μm. (b) Dermal thickness (μm) of WT (white bars, n = 3) and Zeb2-cKO mice (black bars, n = 3). **P < 0.01 by Student’s t-test.

(a) Electron micrographs of the dermis of wild-type (WT, top) and Zeb2-cKO (bottom) mice at 7 weeks of age. Scale bars, 500 nm, 100 nm (insets). (b) Diameters of collagen fibrils in WT (top) and Zeb2-cKO (bottom) mice. Diameters of 500 fibrils were counted and are shown in 10 nm increments. ***P < 0.001 by Student’s t-test.
Abnormal teeth and craniofacial development in Zeb2-cKO mice
In addition to changes in skin phenotypes, Zeb2-cKO mice showed abnormalities in the development of their craniofacial bones and teeth (Supplementary Fig. 5a,b). Histological examination of the teeth of Zeb2-cKO mice revealed a reduction of alveolar bone volume, cellular cementum hyperplasia and a morphological abnormality in the root apex (Supplementary Fig. 5c). The abnormalities in craniofacial bone and teeth observed in Zeb2-cKO mice might be relevant to the characteristic microcephaly and frequently found malalignment of teeth in MOWS patients.
Abnormalities in gene expression related to collagenogenesis in dermal fibroblasts of Zeb2-cKO mice
To examine the changes in gene expression in dermal fibroblasts derived from Zeb2-cKO mice, we performed DNA microarray analysis. Transcripts with at least a 4-fold difference in expression level between Zeb2-cKO fibroblasts and wild-type fibroblasts were selected and sorted with ECM-related genes (Supplementary Fig. 6). Many genes encoding collagen, Timps and other proteins known to be involved in connective tissue diseases such as EDS and Marfan syndrome, including Adamts2, Fbn1 and Fbn2, had more than a 4-fold decreased expression in Zeb2-cKO fibroblasts compared with wild-type fibroblasts. In contrast, genes encoding Mmps were on the list of genes with increased expression in Zeb2-cKO fibroblasts. Real time RT-PCR analysis reproduced, in part, the results of the microarray analysis, such as the down-regulation of Col1a1, Tmp2 and Adamts2 and the up-regulation of Mmp13 (Fig. 6a). Furthermore, Western blot analysis revealed a reduced expression level of Type 1 collagen and a markedly increased expression level of Mmp13 protein (Fig. 6b). Taken collectively, dermal fibroblasts derived from Zeb2-cKO mice exhibited changes in ECM-related molecules, which are involved in the down-regulation of collagenogenesis and in the up-regulation of collagenolysis.

(a) mRNA levels of dermal fibroblasts from Zeb2-cKO mice (cKO) relative to wild-type (WT), which are set to 100. *, **, ***P < 0.05, 0.01, 0.001, respectively, by Student’s t-test. n = 3–5. (b) Western blot analysis. Numbers indicate relative folds of proteins from Zeb2-cKO fibroblasts compared with WT fibroblasts after normalization with β-actin proteins. Bands are displayed as cropped images from original blots on the gel (see Supplementary Information).
Impairment of bleomycin-induced collagen biosynthesis in Zeb2-cKO mice
Finally, we investigated whether the Zeb2 deletion affected inducible skin fibrosis. Bleomycin was subcutaneously injected every other day in the backs of 6 to 8-week-old mice for 4 weeks. Strikingly, the fibrogenic response of the dermis to bleomycin was greatly attenuated in Zeb2-cKO mice compared with wild-type mice (Fig. 7). This result indicated that Zeb2 is required not only for constitutive fibrogenesis but also for the inducible fibrogenesis of the skin.

(a) Histological views of the dermis in 12-week-old wild-type (WT) and Zeb2-cKO mice after treatment with PBS or bleomycin (BLM) (H&E). Scale bar, 100 μm. (b) Mean dermal thickness (μm) ± SD in WT mice (n = 4) and in Zeb2-cKO mice (n = 3). **P < 0.01; NS, not significant, by Student’s t-test.
Discussion
ZEB2 has been recognized to be a multifunctional regulator of nervous system development25, since mutations in the ZEB2 gene lead to severe neurological consequences in animal models12,13,14,26, which recapitulates a number of symptoms of MOWS patients. Heterozygous Zeb2 knockout mice display decreased thermal pain responses, suggesting that Zeb2 contributes to thermal pain sensitivity via coordinated changes in DRG-neuron voltage-gated ion channels27. To circumvent embryonic lethality and/or underscore the role for ZEB2 in detail, homozygous deletion of Zeb2 has been conducted through the Cre-loxP system under organ-specific promoters. Neural crest-specific Zeb2 knockout mice display craniofacial and gastrointestinal malformations14. Zeb2 deletion specifically in the cerebral cortex affects development of the hippocampus and dentate gyrus28. Further, conditional deletion of Zeb2 in postmitotic neurons results in premature generation of layer 2–5 neurons and defects in ipsilateral neocortical axonal growth29,30. A recent study demonstrated that de novo heterozygous Zeb2 KO mice, which were established by inducing the Zeb2 mutation in germ cells, develop multiple defects relevant to MOWS, including craniofacial abnormalities and defective corpus callosum formation26. However, no description was made as to whether they exhibit a skin phenotype.
Indeed, there has been a paucity of literature on the skin phenotypes in patients with MOWS, but only brief descriptions of a fair complexion15 or raindrop depigmentation have been made16. In this study, we report for the first time that most patients with MOWS have skin symptoms similar to EDS patients, which are characterized by skin hyperextensibility, thin, translucent skin with visible veins, atrophic scarring and joint hypermobility. While the typical electron microscopy findings of the dermis in EDS are called ‘collagen flowers’ or ‘cauliflowers’20, those in patients with MOWS are miniaturized fibrils, indicating a distinct difference from EDS but a common mechanism of abnormal fibrillogenesis in the dermis. A ZEB2 deficiency should be the underlying cause of that abnormality, since the same change was found in Zeb2-cKO mice.
To explain this hypothesis, we generated Zeb2-cKO mice under control of the Prx1 promoter, which is expressed throughout the early limb bud mesenchyme and in a subset of the craniofacial mesenchyme24. Indeed, Cre-mediated recombination occurred in dermal fibroblasts and in subcutaneous fat tissues, but not in epidermal keratinocytes or liver tissues. Abnormal craniofacial bone and teeth development in Zeb2-cKO mice might be attributed to the Zeb2 deletion in the craniofacial/teeth mesenchyme and mimic, in part, the characteristic facial and dental features of MOWS patients, including hypertelorism, medially flared and broad eyebrows, a prominent columella, pointed chin, uplifted earlobes and malpositioned teeth5,15.
Although patients with MOWS have heterozygous mutations in the ZEB2 gene, mice having a heterozygous Zeb2 gene deletion in the present study (Prx1-Cre:Zeb2flox/wild) showed no symptoms in the skin, craniofacial or teeth development. The discordance with this mouse model might be attributed to the nature of the Prx1 promoter for Cre recombinase. The reduction of the dermal thickness and the occurrence of alopecia somewhat depended on the sites of Zeb2-cKO mice; that is, it was more pronounced on the limbs, scalp and interlimb flanks, but less on the back. This could be due to site-specific differences in expression of the Prx1 enhancer24.
The most striking feature of Zeb2-cKO mice was their skin phenotype, including a redundant, hyperextensible thin skin, which resembled, in part, the skin phenotype found in MOWS patients that display EDS-like symptoms. EDS is a heterogeneous connective tissue disorder through various pathomechanisms consisting of dominant-negative effects of mutant or haploinsufficiency of procollagen α-chains, and deficiency of collagen-processing enzymes18. Zeb2-cKO mice display a redundant, thin dermis with histological and ultrastructural changes and an attenuated dermal fibrogenesis by bleomycin treatment, which represents an EDS-like abnormality in the homeostasis of the dermis. However, it remains undefined so far whether Zeb2-cKO mice have joint hypermobility or vascular changes, as found in classical EDS. Together with the results from experiments using Zeb2-cKO mice, we speculate that the EDS-like skin phenotype in MOWS patients might result from abnormal collagen biosynthesis by dermal fibroblasts due to ZEB2 mutations or deletions. However, Zeb2-cKO mice display hair loss (alopecia), which MOWS and EDS patients do not develop.
ZEB2, previously termed Smad interacting protein 1 (SIP1), acts as a transcriptional co-repressor in the TGF-β signaling pathway8,11, but it can also act as a transcriptional activator31. ZEB2 also interacts with nucleosome remodeling and the histone deacetylation (NuRD) complex32. Through these molecular interactions, ZEB2 plays a critical role in neural crest cell migration, and therefore, embryos harboring homozygous deletions of the Zeb2 gene exhibit compromised development of the peripheral nervous system, craniofacial tissue and heart12,13. We did not find any impairment in in vitro cell proliferation, invasive or migratory activity of fibroblasts of Zeb2-cKO mice (unpublished observations). In contrast, fibroblasts derived from Zeb2-deficient mice show altered expression profiles of genes that would increase collagenolysis but would reduce collagenogenesis. Some of the down-regulated genes identified in the DNA array analysis involve those responsible for hereditary connective tissue disorders, including collagens and ADAMTS2 for EDS and fibrillin-1 for Marfan syndrome. Further studies will be necessary to investigate how ZEB2, as a transcriptional regulator, mechanistically contributes to ECM homeostasis such as collagen fibrillogenesis in the skin. In conclusion, we demonstrate for the first time that mesoderm-specific deletion of Zeb2 leads to the abnormality in the dermis of an EDS-like skin phenotype, which was also observed in patients with MOWS. In addition, ZEB2 would be a novel therapeutic target for pathogenic diseases involving fibrosis, including liver cirrhosis, cardiac fibrosis, pulmonary fibrosis and scleroderma.
Methods
Patients
Twelve patients, who had been diagnosed with MOWS based on clinical symptoms and mutation or deletion analysis of the ZEB2 gene, were inspected in detail for their skin appearance by 3 dermatologists (M.T., S.S., N.K.). Informed consent was obtained from all patients whose information and images are presented in this study. Skin biopsies were taken from patients #11 and 12 after obtaining written informed consent. The experiments were conducted at the Department of Dermatology, Kochi Medical School Hospital after approval by the Institutional Review Board at Kochi University in accordance with the Declaration of Helsinki Principles.
Ultrasound measurement of skin thickness
Patients and age-matched healthy controls were measured with an ACUCON P300 ultrasound system using a LA435 convex transducer (Siemens Healthineers, Tokyo, Japan), to measure skin thickness at different sites of the body, including the arm, breast, abdomen, back, lumbar, femoral and popliteal areas. Dermal thickness was measured on the B-mode image.
Electron microscopic examination
Skin specimens were fixed in 2.5% glutaraldehyde for 2 h, post-fixed in 1% osmium tetroxide for 2 h, dehydrated in a graded ethanol series, and embedded in epoxy resin. Semithin sections (1 μm) were stained with toluidine blue. Ultrathin sections (100 nm) were stained with uranyl acetate and lead citrate and were examined with a JEM-1400Plus transmission electron microscope (JEOL Ltd. Tokyo, Japan).
Primary fibroblast culture
The dermis of biopsied skin specimens after removal of the epidermis using dispase (BD Biosciences, Tokyo, Japan) was cut into pieces with a scalpel, then soaked in Dulbecco’s modified Eagle’s medium with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, 100 U/mL streptomycin and 2 mM L-glutamine (Invitrogen, Carlsbad, CA). Five to 7 days later, propagated fibroblasts were collected and split. Cells were used for experiments at the 2nd to 4th passages.
Generation of Prx1-Cre:Zeb2flox/flox mice
Mice that carried the Cre recombinase gene under control of the mesoderm-specific regulatory element Prx124 were crossed with mice that were homozygous for the Zeb2 floxed allele12 to generate Prx1-Cre/Zeb2flox/0 mice. The second cross with Zeb2flox/floxmice resulted in the generation of Prx1-Cre:Zeb2flox/flox mice. Allele-specific PCR was carried out as illustrated in Supplementary Fig. 3a. The primers were designed to detect the Zeb2 floxed allele using primers P1 and P2 (339 bp), and the truncated allele using P1 and P3 primers (288 bp). The DNA sequences of primers 1, 2 and 3 are as follows. P1,
5′-GAACTAGTTGAATTGGTAGAATCAATGGGG-3′, a sense sequence of intron 6; P2, 5′-GTAAAGGCTCTCTACGCCTTTTTCAGTTAG-3′; an antisense sequence of intron 6; P3, 5′-AAGCATGTCGGTAAGCTGACCAACTACTAG-3′, an antisense sequence of intron 7. Using a mixture of these primers, PCR was performed with 35 cycles of a reaction consisting of 30 sec of denaturation at 94 °C, 30 sec of annealing at 50 °C and 1 min of elongation at 72 °C.
DNA microarray analysis
Total RNAs were extracted from cells using the TRIzol reagent according to the manufacturer’s instructions (Life Technologies, Carlsbad, CA). The DNA microarray analysis was performed using Affymetrix GeneChip Mouse 430 2.0 Arrays according to the manufacturers’ instructions. Image files were scanned and processed by AGCC (Affymetrix GeneChip Command Console Software, Santa Clara, CA) and the microarray data were normalized with GCRMA. Transcripts with at least a 4-fold change difference in expression level between Zeb2-cKO and control samples were selected and used for further analysis. A gene ontology (GO) enrichment analysis33 was performed via a tool implemented in the RefDIC web server34 (http:// refdic.rcai.riken.jp/). A GO term with a corrected p-value < 0.05 was considered as enriched in the selected transcripts. The raw data for the microarray data are available from the RefDIC web server under the accession numbers RSM01584 and RSM01585.
Quantitative RT-PCR
Total RNAs were extracted using an RNA isolation kit (Promega, Madison, WI) according to the manufacturer’s protocol, and were reverse-transcribed using M-MLV reverse transcriptase (Invitrogen) with random oligonucleotide hexamers (Invitrogen). PCR reactions were performed using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA), and amplification conditions were as follows: 50 °C for 2 min, 90 °C for 10 min for 1 cycle, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The primers used were as follows (sense and antisense, respectively); mouse Zeb1, 5′-GCCAGCAGTCATGATGAAAA-3′ and 5′-TATCACAATACGGGCAGGTG-3′; mouse Zeb2, 5′-ACCAAATGCTAACCCAAGGA-3′ and 5′-GGCATTCGTAAGGTTTTTCA-3′; mouse Timp2, 5′-AGTGATTTCCCCGCCAACTC-3′ and 5′-AAGGGGGCCATCATGGTATC-3′; mouse Adamts2, 5′-CGTGGAGTGGCAGGGTGAG-3′ and 5′-CCTCCATCCGGATCAGACCA-3′; Mmp13, 5′-GCCAGAACTTCCCAACCAT-3′ and 5′-TCAGAGCCCAGAATTTTCTCC-3′; mouse HPRT, 5′-TGACCTTGATTTATTTTGCATACC-3′ and 5′-CGAGCAAGACGTTCAGTCCT-3′. The quantity of each transcript was analyzed using the 7300 Fast System Software (Applied Biosystems) and was normalized to hypoxanthine phosphoribosyltransferase (HPRT) according to the ∆∆Ct method.
Western blot analysis
Cell lysates were prepared using RIPA buffer (Sigma-Aldrich, Tokyo, Japan), separated on 4–15% gradient gels (Bio-Rad, Tokyo, Japan) and blotted on polyvinyl difluoride (PVDF) membranes (Bio-Rad). The membranes were incubated with antibodies including; anti-ZEB2 Ab (Santa Cruz Biotechnology, Dallas, TX), anti-HPRT Ab (GeneTex, Irvine, CA), anti-mouse type I collagen (CedarLane Laboratories, Burlington, Canada), anti-MMP13 Ab (Abcam, Cambridge, United Kingdom) and anti-beta actin Ab (Sigma-Aldrich), followed by HRP-conjugated secondary antibodies of relevant species (Cell Signaling, Tokyo, Japan). An ECL Prime kit (GE Healthcare, Tokyo, Japan) was used for signal detection. ImageJ (NIH) was used for quantification of the signals.
Bleomycin-induced fibrogenesis assay
Bleomycin hydrochloride (Wako Pure Chemical Industries, Ltd., Osaka, Japan) was dissolved in phosphate-buffered saline (PBS) at 500 μg/mL and sterilized with filtration. One hundred μl BLM (50 μg) or PBS were injected subcutaneously into the shaved backs of Zeb2-cKO mice or wild-type mice at 8 weeks of age every other day for 4 weeks. The injected skin was removed and processed for histological analysis.
Statistical analysis
Samples were compared with two-tailed, unpaired Student’s t-test. P-values less than 0.05 are considered significant.
Additional Information
How to cite this article: Teraishi, M. et al. Critical involvement of ZEB2 in collagen fibrillogenesis: the molecular similarity between Mowat-Wilson syndrome and Ehlers-Danlos syndrome. Sci. Rep. 7, 46565; doi: 10.1038/srep46565 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Mowat, D. R., Wilson, M. J. & Goossens, M. Mowat-Wilson syndrome. J. Med. Genet. 40, 305–310 (2003).
Mowat, D. Mowat-Wilson syndrome. Management of genetic syndromes (ed. Cassidy, S. B. & Allanson, J. E. ) 517–527 (Wiley-Blackwell, 2010).
Yamada, Y. et al. The spectrum of ZEB2 mutations causing the Mowat-Wilson syndrome in Japanese populations. Am. J. Med. Genet. 164A, 1899–1908 (2014).
Adam, M. P. et al. Mowat-Wilson Syndrome in GeneReviews[Internet] https://www.ncbi.nlm.nih.gov/books/NBK1412/ (2013).
Mowat, D. R. et al. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J. Med. Genet. 35, 617–623 (1998).
Wakamatsu, N. et al. Mutations in SIP1, encoding Smad interacting protein-1, cause a form of Hirschsprung disease. Nat. Genet. 27, 369–370 (2001).
Cacheux, V. et al. Loss-of-function mutations in SIP1 Smad interacting protein 1 result in a syndromic Hirschsprung disease. Hum. Mol. Genet. 10, 1503–1510 (2001).
Verschueren, K. et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5′-CACCT sequences in candidate target genes. J. Bio. Chem. 274, 20489–20498 (1999).
Remacle, J. E. et al. New mode of DNA binding of multi-zinc finger transcription factors: deltaEF1 family members bind with two hands to two target sites. EMBO J. 18, 5073–5084 (1999).
Comijn, J. et al. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 7, 1267–1278 (2001).
Postigo, A. A., Depp, J. L., Taylor, J. J. & Kroll, K. L. Regulation of Smad signaling through a differential recruitment of coactivators and corepressors by ZEB proteins. EMBO J. 22, 2453–2462 (2003).
Higashi, Y. et al. Generation of the floxed allele of the SIP1 (Smad-interacting protein 1) gene for Cre-mediated conditional knockout in the mouse. Genesis 32, 82–84 (2002).
Van de Putte, T. et al. Mice lacking ZFHX1B, the gene that codes for Smad-interacting protein-1, reveal a role for multiple neural crest cell defects in the etiology of Hirschsprung disease-mental retardation syndrome. Am. J. Hum. Genet. 72, 465–470 (2003).
Van de Putte, T., Francis, A., Nelles, L., van Grunsven, L. A. & Huylebroeck, D. Neural crest-specific removal of Zfhx1b in mouse leads to a wide range of neurocristopathies reminiscent of Mowat-Wilson syndrome. Hum. Mol. Genet. 16, 1423–1436 (2007).
Adam, M. P. et al. Clinical features and management issues in Mowat-Wilson syndrome. Am. J. Med. Genet. Part A 140, 2730–2741 (2006).
Wilson, M. et al. Further delineation of the phenotype associated with heterozygous mutations in ZFHX1B. Am. J. Med. Genet. Part A 119A, 257–265 (2003).
Beighton, P., De Paepe, A., Steinmann, B., Tsipouras, P. & Wenstrup, R. J. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am. J. Med. Genet. 77, 31–37 (1998).
Mao, J. R. & Bristow, J. The Ehlers-Danlos syndrome: on beyond collagens. J. Clin. Invest. 107, 1063–1069 (2001).
Ishihara, N. et al. Clinical and molecular analysis of Mowat-Wilson syndrome associated with ZFHX1B mutations and deletions at 2q22-q24.1. J. Med. Genet. 41, 387–393 (2004).
Sobey, G. Ehlers-Danlos syndrome: how to diagnose and when to perform genetic tests. Arch. Dis. Child. 100, 57–61 (2015).
Beighton, P. & Horan, F. Orthopaedic aspects of the Ehlers-Danlos syndrome. J. Bone Joint Surg. Br. 51, 444–453 (1969).
Carlesimo, M. et al. Ehlers-Danlos syndrome: case report and an electron microscopy study. Rheum. Int. 32, 1507–1510 (2012).
Bergwerff, M. et al. Patterns of paired-related homeobox genes PRX1 and PRX2 suggest involvement in matrix modulation in the developing chick vascular system. Dev. Dyn. 213, 59–70 (1998).
Logan, M. et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 (2002).
Hegarty, S. V., Sullivan, A. M. & O’Keeffe, G. W. Zeb2: A multifunctional regulator of nervous system development. Prog. Neurobiol. 132, 81–95 (2015).
Takagi, T., Nishizaki, Y., Matsui, F., Wakamatsu, N. & Higashi, Y. De novo inbred heterozygous Zeb2/Sip1 mutant mice uniquely generated by germ-line conditional knockout exhibit craniofacial, callosal and behavioral defects associated with Mowat-Wilson syndrome. Hum. Mol. Genet 24, 6390–6402 (2015).
Jeub, M. et al. The transcription factor Smad-interacting protein 1 controls pain sensitivity via modulation of DRG neuron excitability. Pain 152, 2384–2398 (2011).
Miquelajauregui, A. et al. Smad-interacting protein-1 (Zfhx1b) acts upstream of Wnt signaling in the mouse hippocampus and controls its formation. Proc. Nat. Acad. Sci. USA 104, 12919–12924 (2007).
Seuntjens, E. et al. Sip1 regulates sequential fate decisions by feedback signaling from postmitotic neurons to progenitors. Nat. Neurosci. 12, 1373–1380 (2009).
Srivatsa, S., Parthasarathy, S., Molnar, Z. & Tarabykin, V. Sip1 downstream Effector ninein controls neocortical axonal growth, ipsilateral branching, and microtubule growth and stability. Neuron 85, 998–1012 (2015).
Long, J., Zuo, D. & Park, M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J. Biol. Chem. 280, 35477–35489 (2005).
Verstappen, G. et al. Atypical Mowat-Wilson patient confirms the importance of the novel association between ZFHX1B/SIP1 and NuRD corepressor complex. Hum. Mol. Genet. 17, 1175–1183 (2008).
Draghici, S., Khatri, P., Martins, R. P., Ostermeier, G. C. & Krawetz, S. A. Global functional profiling of gene expression. Genomics 81, 98–104 (2003).
Hijikata, A. et al. Construction of an open-access database that integrates cross-reference information from the transcriptome and proteome of immune cells. Bioinformatics 23, 2934–2941 (2007).
Acknowledgements
We thank Dr. Malcolm Logan for providing Prx1-Cre transgenic mice, Dr. Kenjiro Kosaki for providing patients information, Ms. Reiko Kamijima and Mr. Ken-ichi Yagyu for technical assistance. We also thank Dr. Atsushi Utani, who passed away Nov. 2016, for kind suggestion. This work was partly supported by Grants-in-Aid for Scientific Research form the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Contributions
S.S. designed the research; M.T. (M. Teraish), M.T. (M. Takaishi), K.N., M.I. performed the research; Y.H. (Y. Higashi). and T.F. contributed new reagents/analytic tools; S.S., Y.A., A.H., O.O., Y.H. (Y. Hiraki), S.M., T.F., N.W. analyzed data; and S.S., N.W. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Teraishi, M., Takaishi, M., Nakajima, K. et al. Critical involvement of ZEB2 in collagen fibrillogenesis: the molecular similarity between Mowat-Wilson syndrome and Ehlers-Danlos syndrome. Sci Rep 7, 46565 (2017). https://doi.org/10.1038/srep46565
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46565
This article is cited by
-
Mowat-Wilson syndrome: unraveling the complexities of diagnosis, treatment, and symptom management
Egyptian Journal of Medical Human Genetics (2024)
-
A de novo frameshift mutation in ZEB2 causes polledness, abnormal skull shape, small body stature and subfertility in Fleckvieh cattle
Scientific Reports (2020)
-
Craniofacial abnormality with skeletal dysplasia in mice lacking chondroitin sulfate N-acetylgalactosaminyltransferase-1
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
