
Abstract
FSD-C10, a Fasudil derivative, was shown to reduce severity of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS), through the modulation of the immune response and induction of neuroprotective molecules in the central nervous system (CNS). However, whether FSD-C10 can promote neuroregeneration remains unknown. In this study, we further analyzed the effect of FSD-C10 on neuroprotection and remyelination. FSD-C10-treated mice showed a longer, thicker and more intense MAP2 and synaptophysin positive signal in the CNS, with significantly fewer CD4+ T cells, macrophages and microglia. Importantly, the CNS of FSD-C10-treated mice showed a shift of activated macrophages/microglia from the type 1 to type 2 status, elevated numbers of oligodendrocyte precursor cells (OPCs) and oligodendrocytes, and increased levels of neurotrophic factors NT-3, GDNF and BDNF. FSD-C10-treated microglia significantly inhibited Th1/Th17 cell differentiation and increased the number of IL-10+ CD4+ T cells, and the conditioned medium from FSD-C10-treated microglia promoted OPC survival and oligodendrocyte maturation. Addition of FSD-C10 directly promoted remyelination in a chemical-induced demyelination model on organotypic slice culture, in a BDNF-dependent manner. Together, these findings demonstrate that FSD-C10 promotes neural repair through mechanisms that involved both immunomodulation and induction of neurotrophic factors.
Similar content being viewed by others
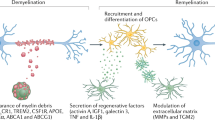


Introduction
Multiple sclerosis (MS) is a chronic inflammatory debilitating disease in the Central Nervous System (CNS) that affects over 2 million people worldwide. Oligodendrocyte death is believed to be essential in the pathogenesis of MS as CNS myelin is produced by oligodendrocytes, and the loss of these cells results in demyelination, axonal damage and severe impairment of neurological function1,2,3,4,5. Concurrently with inflammation and demyelinating processes, repair mechanisms are initiated in primary demyelinated lesions. Extensive remyelination can be observed during the early stage of MS by recruitment, proliferation and differentiation of oligodendrocyte precursor cells (OPC)5. However, the remyelination is reduced after successive relapses and failure of effective remyelination in progressive MS lesions is associated with a lack of oligodendrocyte maturation6,7 and increased axonal degeneration8. Therefore, stimulation of remyelination through an increase in oligodendrocyte maturation in the CNS lesions is critical to the functional recovery in MS6,9.
Fasudil, an inhibitor of Rho kinases (ROCK), has been shown to have beneficial effects on CNS-related disorders10,11. In EAE, Fasudil reduced the severity of disease through the stimulation of an anti-inflammatory response and a shift of M1 towards M2 macrophage/microglia12,13. M1 microglia secrete toxic molecules that destruct axon-supporting myelin and oligodendrocytes, whereas M2 cells release anti-inflammatory cytokines and growth factors that contribute to efficient remyelination and protect neurons from damage5,14,15. Manipulating the switch from M1- to M2-dominant polarization of microglia is a desirable strategy for efficient remyelination therapies. In addition, failure of spontaneous remyelination is also associated with a lack of sufficient amount of neurotrophic factors (BDNF, NT-3 and GDNF) in the CNS during inflammation16,17,18. In this context, our previous study showed that nasal administration of FSD-C10, a derivative of Fasudil with less toxic effect, effectively suppressed the clinical severity of experimental autoimmune encephalomyelitis (EAE), an animal model of MS. This effect was associated with a upregulated Tregs19. Still, whether FSD-C10 presents a neuroregenerative and neuroprotective effect has yet to be elucidated.
In the present study, we found that FSD-C10 significantly promoted neurological recovery, oligodendrogenesis, and remyelination. The mechanisms underlying these effects relayed on immunomodulation and direct neuroregeneration. Our data show that FSD-C10 has a beneficial effect on EAE acting through the modulation of the immune response and neuroregeneration.
Results
Intranasal FSD-C10 has a neuroprotective potential in EAE
Similar to our previous study19, nasal administration of FSD-C10 effectively suppressed clinical severity of EAE, with reduced CNS inflammation and demyelination (Figure S1). Extensive CD4+ T cells and CD68+ macrophages were found in brains from untreated EAE mice whereas the frequency of these cells were significantly reduced in mice treated with nasal FSD-C10 (Figure S2). In order to study the neural protection effect of FSD-C10, we treated MOG35–55-immunized mice with FSD-C10 (2.5 mg/kg/d). Treatment regimen started from day 3 p.i. until day 27 p.i. At the end of treatment, mice were euthanized and the CNS tissue was collected and analyzed for the expression of microtubule-associated protein 2 (MAP2), which is specifically expressed in dendrites and plays a key role in dendritic outgrowth, branching and synaptogenesis20,21,22. Our data show that FSD-C10 increased MAP2 expression and improved MAP2 positive dendritic morphology in prefrontal cortex and hippocampus compared to tissue from untreated EAE mice (Fig. 1a). Similar results were also observed in spinal cord tissues (data not shown). We further examined the synaptic structure in brain and the spinal cord by analyzing the expression of synaptophysin, a protein found in presynaptic vesicles. This parameter allows a general analysis of spinal cord circuit integrity23,24. The synaptophysin positive staining in the cortex of brain and the grey matter of spinal cord of FSD-C10-treated mice was significantly higher than in the control mice (Fig. 1b). These results indicate that FSD-C10 performed a neuropreservation in the CNS of EAE mice.

EAE was induced in C57BL/6 mice by immunization with MOG35–55/CFA. Mice received FSD-C10 or ddH2O by intranasal pathway beginning on day 3 p.i. until day 27 p.i. Brain and lumbar sections of spinal cord were harvested at day 28 p.i. and immunostained with MAP2 and synaptophysin. (a) MAP2 expression in the cortex (areas 1, 2 and 3) and hippocampal region (area 4) of brain. (b) Representative microphotographs and quantitative analysis for synaptophysin expression in the cortex of brain and the grey matter of spinal cord. Quantitative analyses represent mean ± SEM (n = 7 each group). ***P < 0.001. One representative of three experiments is shown.
Characterization of proliferative cells in the CNS of FSD-C10-treated mice
To study the mechanism underlying the neuroprotective effect of FSD-C10 in EAE, we first explored and characterized cell proliferation in the CNS after treatment. Mice received 3 i.p. injections of BrdU at days 25–27 post immunization (p.i.) and were euthanized at 28 days p.i. Analysis of coronal brain sections showed that, while the BrdU+ proliferative cells were mainly concentrated in perivascular space in tissue from untreated mice, these cells were extensively detected in the whole brain parenchyma of FSD-C10-treated EAE mice (Fig. 2a).

Mice from each group received three i.p. injections of BrdU (sigma) on days 25–27 p.i. and brains were harvested from mice described in Fig. 1. (a) Proliferating cells were identified by a mouse anti-BrdU antibody. (b) Immunostaining of BrdU (red) and cell-specific markers (green) including CD4, CD68, CD11b, NG2 and GalC were performed. Representative microphotographs of double positive cells for BrdU and different cell types were shown, and percentages of these double positive cells in each cell type were analyzed. Data represent mean ± SEM (n = 7 each group). ***P < 0.001. One representative of three experiments is shown.
We then analyzed the proliferative status of different cell types in the CNS after FSD-C10 treatment. In ddH2O-treated EAE mice, 30–40% of immune cells, e.g., CD4+ T cells and CD68+/CD11b+ microglia/macrophages, were BrdU+, while the percentages of BrdU+ cells were significantly decreased by FSD-C10 treatment. In contrast, only low numbers of BrdU+ were colocalized with GFAP+ (astrocytes), NG2+ (OPCs) and GalC+ (mature oligodendrocytes) in ddH2O-treated EAE mice, and FSD-C10 treatment largely increased the proportions of BrdU+/NG2+ and GalC+ cells (Fig. 2b). Thus, FSD-C10 treatment effectively stimulated the proliferation of OPC/oligodendrocyte lineage cells, but suppressed proliferation of immune cells.
Elevated numbers of OPCs and oligodendroglia in FSD-C10-treated mice
To evaluate the effects of FSD-C10 on OPC recruitment and oligodendrocyte maturation, immunohistofluorescence was carried out on brain section with antibodies to NG2 and GalC, as markers of OPCs and oligodendrocytes, respectively. A low number of NG2+ cells was observed in brain of EAE control mice, and a significantly increased number of these cells was observed in brain of FSD-C10-treated EAE mice (Fig. 3a and b). Similarly, about a 2-fold increase of GalC+ oligodendrocytes in the brain of FSD-C10-treated EAE mice was seen compared to those in the untreated mice (Fig. 3a and b). These results were further confirmed by analysis of total protein (Fig. 3c). In contrast, administration of FSD-C10 in naïve mice increased the number of OPCs, but not oligodendrocytes (Fig. d,e), indicating that FSD-C10 affects OPC lineage cells in the physiological condition, without disturbing the regulation of mature myelinating cells in vivo.

EAE (a,b,c) or naïve mice (d,e) received FSD-C10 or ddH2O by intranasal pathway. Brains were harvested from mice described in Fig. 1. Immunohistofluorescence of OPCs (NG2+) and oligodendrocytes (GalC+) within brains were examined. Representative microphotographs (a,d) and quantitative analyses for NG2+ and GalC+ cells from ddH2O-treated and FSD-C10-treated EAE mice (b,e), as well as representative Western blot photos and their quantitative analysis (c) were shown. Data represent mean ± SEM (n = 7 each group for brain section staining, and n = 4 each group for Western blot). *P < 0.05, ***P < 0.001. One representative of three experiments is shown.
Intranasal FSD-C10 stimulated the production of neurotrophic factors
It was shown that induction of neurotrophins, including NT-3, GDNF, and BDNF, plays an important role in neurogenesis, remyelination, and brain repair16,25,26. We therefore determined if FSD-C10 treatment induced these neurotrophic factors. Our data show that while low numbers of cells producing these neurotrophic factors appeared in the brain of ddH2O-treated EAE mice, significantly increased numbers were observed in the brain of FSD-C10-treated EAE mice (Fig. 4a and b). Similarly, total protein analysis confirmed that a strong up-regulation of these neurotrophic factors was found in brain of FSD-C10-treated mice compared with EAE mice (Fig. 4c). These results indicate a stimulatory effect of FSD-C10 in the production of neurotrophic factors in vivo.

Brains were harvested from mice described in Fig. 1, and sections were immunostained with NT-3, GDNF and BDNF antibodies. Representative microphotographs (a) and their quantitative analyses (b) were shown. (c) Expression of these neurotrophic factors in brain tissues was also determined by Western blot and quantitatively analyzed. Data represent mean ± SEM (n = 7 each group for brain section staining, and n = 4 each group for Western blot). *P < 0.05, **P < 0.01, ***P < 0.001. One representative of three experiments is shown.
Intranasal FSD-C10 shifted activated microglia from M1 to M2 status
Given the important roles of different microglia/macrophages in CNS myelination, we then determined the functional states of microglia after FSD-C10 treatment. In normal brain, few ramified microglia (resting microglia) can be detected, while fully ramified microglia (activated microglia) are numerous in middle-injured brain; elongated microglia (triangles, phagocytic microglia) became rod cells, and probably fuse to form cell clusters in severe-injured brain27. In our experiment, numerous hyper-ramified and elongated microglia with large cell body were detected in brain of ddH2O treated EAE, indicating activated microglia. In contrast, microglia in FSD-C10-treated mice showed in a thin branched form (Fig. 5a and b).

Brains were harvested from mice described in Fig. 1. (a) Brains were immunostained with CD11b antibody for analyzing the functional states and morphological diversity of microglia. FSD-C10 treatment reduced the numbers of fully ramified and triangle microglia (activated microglia). The single microglia area (polygon) was quantitatively analyzed by Image-pro plus 6.0 software in (b). (c) Expression of M1 markers (CD16/32 and iNOS) and M2 markers (CD206 and IL-10) in CD11b+ cells was determined by immunohistochemistry and (d) double positive cells were quantitatively analyzed. (e) Expression of iNOS and Arg-1 was also measured by Western blot. (f) Brains were homogenized on ice, and equal amounts of protein (10 mg) were measured by ELISA for cytokine production. Quantitative analyses represent mean ± SEM (n = 7 each group for brain section staining, and n = 4 each group for Western blot and ELISA). *P < 0.05, **P < 0.01, ***P < 0.001. One representative of three experiments is shown.
Functionally, microglia can have different states, including the inflammatory and neurotoxic M1 and anti-inflammatory and neuroprotective M2 phenotypes28. Our results showed that, compared with control mice, FSD-C10-treated mice exhibited substantially reduced numbers of CD11b+ microglia/macrophages expressing the M1 markers iNOS and CD16/32, while numbers of these cells expressing the M2 markers IL-10 and CD206 were significantly increased (Fig. 5c and d). FSD-C10 inhibited the protein expression of iNOS while enhanced the levels of Arg-1 (Fig. 5e). Similarly, levels of M1 cytokines IL-1β, IL-6 and TNF-α were significantly suppressed while levels of IL-10 were upregulated after FSD-C10 treatment (Fig. 5f). These results indicate that intranasal administration of FSD-C10 blocked the microglia activation and switched these cells from a M1 to M2 phenotype. While FSD-C10 treatment in naïve mice increased Arg-1 expression in microglia, it did not affect the expression of iNOS and CD16/32, IL-10 and CD206 (Figure S3).
FSD-C10 treatment inhibited Th1 and Th17 cells in EAE mice
We then explored whether intranasal administration of FSD-C10 could suppress the development of Th1 and Th17 cells. MOG35–55-immunized mice received ddH2O or FSD-C10 starting from day 3 p.i., their splenocytes obtained on 15 p.i., and treated with or without MOG35–55 (25 μg/ml) for 72 h. As shown in Fig. 6a, splenocytes of FSD-C10-treated EAE mice showed a lower production of IL-17, TNF-α, IFN-γ and a higher production of IL-10 than those of the PBS-treated group. In addition, FSD-C10 treatment also reduced MOG-induced T cell proliferation in the presence or absence of MOG (Fig. 6b).

Splenocytes from each group of EAE mice (n = 3) were harvested on day 12 p.i., (a) In vivo inhibition of pro-inflammatory cytokines. Splenocytes were cultured at 5 × 106 cells/ml and stimulated with MOG35–55 for 3 days. Cytokine production in supernatants was analyzed by ELISA. (b) Splenocytes were cultured at 2 × 106 cells/ml in 100 μl/well and stimulated with or without MOG35–55 for 3 days and labeled with BrdU for cell proliferation assay by an ELISA reader following the manufacturer’s instructions (Abcam) (n = 8 each group). **P < 0.01, comparisons between ddH2O-treated animals and FSD-C10-treated groups. One representative of three experiments is shown.
FSD-C10 shifted M1 to M2 phenotype in vitro
To test whether FSD-C10 treatment can directly induce a shift of M1 microglia towards a M2 phenotype, BV-2 microglia were firstly polarized to M1 by exposure to lipopolysaccharide (LPS), and then were treated with FSD-C10. The polarization was examined by immunofluorescence. Results showed that LPS stimulated high expression levels of M1 markers iNOS, CD16/32 and IL-12, but low levels of M2 markers CD206, IL-10 and Arg-1. FSD-C10 treatment significantly inhibited the expression of all M1 markers, but enhanced those M2 markers (Fig. 7). These results indicate a direct effect of FSD-C10 in the shift to the M2 phenotype in microglia.

BV-2 microglia were stimulated with PBS or LPS (0.5 μg/ml) in the absence or presence of FSD-C10 (15 μg/ml) for 24 h. Cells were stained with M1 microglia markers CD16/32, iNOS as well as M2 markers CD206, Arg-1. IL-12 and IL-10 were checked with ELISA. Representative photographs and quantitative analyses (mean ± SEM; n = 5 each group) from immunostaining and ELISA are shown. Results are expressed as number of immunostaining positive cells per view field and pg/ml. Groups designated by the same letter are not significantly different, while those with different letters (a,b,c) are significantly different (p < 0.05). One representative of three experiments is shown.
FSD-C10-treated microglia have a reduced ability to prime autoreactive T cells in vitro
We then determined the direct effect of FSD-C10 on Th1/Th17 cells in purified CD4+ T cells, and showed that adding FSD-C10 in vitro significantly inhibited Th1/Th17 cell differentiation (Fig. 8). However, whether FSD-C10 can inhibit the capacity of microglia in priming effector T cell response remains unclear. To answer this question, we first determined the effect of FSD-C10-treated microglia on MOG-reactive T cell proliferation. While LPS-stimulated microglia promoted T cell proliferative responses, this capacity was inhibited when microglia were treated with FSD-C10 (Fig. 9a, P < 0.01). In contrast, microglia without LPS stimulation did not affect T cell proliferation (Fig. 9a).

For differentiation of Th1 and Th17 cells, purified CD4+ T cells of normal female C57BL/6 mice (8 wks) were stimulated with anti-CD3 and anti-CD28 for 3 days under (a,b) Th1 polarization conditions (IL-12 and anti-IL-4) and (c,d) Th17 polarization conditions (TGF-β1, IL-6, IL-1β, anti-IL-4 and anti-IFN-γ) in the presence or absence of FSD-C10. Percentages of IFN-γ+ CD4+ Th1 cells and IL-17+ CD4+ Th17 cells were determined by flow cytometry. Data represent mean ± SEM (n = 5 each group). ***P < 0.001. One representative of three experiments is shown.

CD4+ T cells from EAE mice were labeled with CFSE and co-cultured with BV-2 microglia that were stimulated with or without LPS and treated with PBS- or FSD-C10 at an estimated ratio of 10:1, in the presence of MOG35–55 (25 μg/ml) for 72 h. (a) Percentage of proliferating cells was determined through the measurement of CFSE dilution using flow cytometry. Data represent mean ± SEM (n = 5 each group). (b) IFN-γ, IL-10, and IL-17 secretion was analyzed using flow cytometry. Data represent mean ± SEM (n = 5 each group). (c) The levels of IFN-γ, IL-10, IL-17, and TNF-α in culture supernatants were detected using ELISA kits. Data represent mean ± SEM (n = 8 each group). *P < 0.05, **P < 0.01, ***P < 0.001. One representative of three experiments is shown.
Next, we examined whether polarization of the BV-2 microglia mediated by FSD-C10 could affect the population and function of the encephalomyelitic T cells. FSD-C10-treated BV-2 microglia were co-cultured with MOG-reactive T cells. As shown in Fig. 9b, FSD-C10-treated microglia following LPS maturation induced CD4+ IL-10+ T cells, whereas the percentages of MOG-reactive Th1 (CD4+ IFN-γ+) and Th17 (CD4+ IL-17+) cells were reduced. Consistent with the FACS data, ELISA assays confirmed that FSD-C10-treated BV-2 microglia inhibited IFN-γ, IL-17 and TNF-α production by T cells and induced them to produce IL-10, compared with the PBS-treated BV-2 microglia (Fig. 9c, P < 0.05–0.01). These results indicate that FSD-C10-treated microglia can convert the function of MOG-reactive T cells from pro-inflammatory to an anti-inflammatory one.
LPS/FSD-C10-stimulated microglia cells produce factors that promote oligodendrocyte survival and maturation
We further explored the function of FSD-C10-treated microglia on OPC/oligodendrocytes. OPCs were incubated with conditioned media (CM) obtained from cultures where microglia cells were treated with PBS (M0), LPS (M1), or LPS/FSD-C10 (M2) as described above. After 24 h of incubation with CM, OPCs were stained with the Live/dead cell imaging kit to assess cell viability. The percentages of leaky OPCs were significantly lower in cells cultured with the M2-CM than those with M1-CM. The M1-CM also markedly induced OPC death, however, the CM from PBS- and LPS/FSD-C10-stimulated microglia exhibited a significantly lower death rate than the LPS treatment group (Fig. 10a). These results indicate that, in contrast to M1-CM, conditioned media from FSD-C10-treated microglia cells has no toxic effect over OPC.

OPCs were generated from brain of neonatal mice and cultured with conditioned media (CM) from microglia treated with PBS (M0), LPS (M1), or LPS+ FSD-C10 (M2) for 24 hrs. (a) OPCs were stained using Live/Dead staining kit as given in the material and methods section, and representative images of viable cells, dead cells, and leaky cells were shown. These cells were then counted and quantified under fluorescent microscopy by Image-Pro Plus software in a blinded fashion. (b) We added M0-CM, M1-CM and M2-CM to the OPC differentiation media in a 1:1 ratio and the OPCs were cultured for 7 days. Cells were then stained by anti-MBP antibody. Representative images and quantification of length of cell branches were shown, and measurements were done with Image-Pro Plus software. Data in this figure represent mean ± SEM (n = 5 each group). **P < 0.01; **P < 0.001. One representative of three experiments is shown.
We also studied the effect of different CMs on OPC maturation. After culturing OPCs with CM for 7 days, these cells were stained with MBP, a marker of oligodendrocytes. Although all 3 groups of OPCs expressed MBP, M2-CM-treated group had elevated multipolar branches, with a significant increase in arborization (Fig. 10b). Taken together, the addition of M2-CM not only promoted the OPC survival but also increased the arborization/maturation of oligodendrocytes.
FSD-C10 directly promotes remyelination in vitro in a BDNF-dependent manner
To examine whether FSD-C10 can directly promote remyelination, a LPC-induced demyelination model in organotypic slice culture was conducted. In this experimental approach, demyelination is achieved after incubation of cultures with LPC for 18 h. Treatment with LPC leads to a decreased expression of MBP in organotypic cultures. When these demyelinated slices were treated with FSD-C10 for 14 days, MBP expression was significantly enhanced compared with the PBS-treated slices, suggesting a significant promotion in remyelination by FSD-C10 treatment (Fig. 11).

Postnatal day 5 (P5) mouse pups were sacrificed and consecutive slices (350 μm) were cut from the cerebellums. Three slices in each 6-well insert plate were cultured in 1 ml medium. LPC was added into cerebellum slice cultures for 18 hrs to induce demyelination and then slices were cultured with 15 μg/ml FSD-C10 or PBS for 14 days, with or without antibodies to CNTF, NT-3 and BDNF. Slices were then stained with anti-MBP and anti-NF-H. Results were visualized by confocal microscopy and MBP and NF-H expression was analyzed by ImageJ software. Quantitative analyses represent mean ± SEM (n = 5 each group). Groups designated by the same letter are not significantly different, while those with different letters (a, b) are significantly different (p < 0.05). One representative of three experiments is shown.
Given that FSD-C10 stimulated the production of the neurotrophic factors NT-3, GDNF and BDNF in vivo (Fig. 4), we aimed to evaluate whether these proteins played a role in FSD-C10-induced remyelination. For that matter, demyelinated organotypic cultures were treated with FSD-C10 in the presence of neutralizing antibodies against these factors. Our results showed that FSD-C10-induced remyelination was dramatically blocked by anti-BDNF antibodies (Fig. 11). Blockage of GDNF or NT-3 did not revert the effect of FSD-C10 (data not shown). Similarly, NF-H expression (neurons) was also largely reduced when anti-BDNF antibodies were added (Fig. 11). These results indicate that FSD-C10 can directly induce remyelination through a BDNF-dependent pathway.
Discussion
In this paper we defined some of the mechanisms of FSD-C10-induced EAE amelioration. We found that FSD-C10 stimulates neurogrowth and regeneration, shifts macrophage/microglia activation profile and promotes remyelination. Inhibition of Rho associated kinase (ROCK) has been shown to promote OPC differentiation and myelin formation29,30,31,32. We have recently found that nasal administration of FSD-C10, a novel and efficient ROCK inhibitor, suppressed the activity and expression of ROCK, alleviated disease severity and CNS inflammation of EAE mice, and preserved myelin sheath19,33. Our data showed a higher proliferation rate of OPCs and mature oligodendrocytes in the CNS of FSD-C10-treated mice, which confirms the protective effect of FSD-C10 on CNS myelination in vivo. In a series of in vitro experiments, FSD-C10 treatment induced type 2 macrophages/microglia and the condition media of these cells protected OPCs from inflammation-induced death and promoted the maturation of these cells into oligodendrocytes. Further, addition of FSD-C10 into LPC-induced demyelination cultures effectively promoted remyelination in a BDNF-dependent manner.
MS/EAE is initiated through the activation of encephalitogenic myelin-responsive CD4+ T cells, which invade the CNS1,2. Together with lymphocyte infiltration, resident microglia are activated and present self-antigens to lymphocytes. A large proportion of circulating monocytes infiltrate the CNS as well. Monocytes/Macrophages and microglia are the ultimate effector cells in neuroinflammation, leading to the destruction of the myelin sheath and oligodendrocyte death1,2. Microglia cells play a pivotal role in the pathogeny of MS and EAE34. In EAE, activated microglia cells release inflammatory mediators, which lead to neurotoxic consequences. The existence of activated microglia is the most prominent feature of chronic neuroinflammation or neurodegenerative disease35. Our data show that FSD-C10 treatment significantly reduced the numbers of these immune cells in the CNS. Their proliferation in the disease foci was also significantly inhibited following treatment. These results indicate that FSD-C10 can alleviate EAE through a reduction in either the recruitment of peripheral immune cells as well as their proliferation in the CNS. We also found that FSD-C10 kept microglia in a thin branched form or possibly reverted the reactive microglia back to the resting state. In addition, intranasal FSD-C10 treatment significantly decreased microglia proliferation and their IL-1β and TNF-α expression. As a result, the cytotoxic outcome of these pathologic mediators on neural cells was significantly diminished.
In CNS, microglia undergo M1 or M2 polarization in response to different environmental stimuli36. For instance, while M1 microglia are typically found in of MS lesions and their frequency directly correlates with the extent of axonal damage, M2-polarized microglia contributes to regenerative response in the CNS37. Importantly, M2 microglia are lacking in chronic lesion of EAE5. Blockage of M2 profile in microglia has been shown to contribute to impaired remyelination38. Therefore, it is reasonable to speculate that stimulating the M2-polarized phenotype of microglia would confer protection from inflammation-induced neurodegeneration and demyelination. Our results show a clear M2 phenotype of macrophage/microglia after nasal FSD-C10 treatment. Interestingly, FSD-C10-treated microglia effectively induced IL-10-producing, but inhibited IFN-γ/IL-17 producing T cells, and conditioned media from M2 cultures increased the survival rate of OPCs and arborization/maturation of oligodendrocytes. Our observations suggest that FSD-C10 treatment induces M2 microglia, which have the capacity to enhance OPC survival and their maturation into oligodendrocytes.
It has been shown that loss of trophic support for oligodendrocytes and neurons leads to the failure of spontaneous remyelination and neural recovery in MS/EAE39. A treatment that has capacity to induce the production of neurotrophic factors in the disease foci would be greatly beneficial. Indeed, overexpression of NT-3 in neural stem cells reduces CNS inflammation and neurological deficits in ongoing EAE through the stimulation of proliferation and differentiation of neural stem cells into oligodendrocytes and neurons culminating in remyelination and neuronal repopulation16. Local release of NT-3 also reduces astrogliosis, a main cause of MS plaque formation40,41. GDNF, a member of the TGF-β superfamily42, promotes survival and differentiation of dopaminergic and motor neurons43,44, selectively enhances regrowth of damaged axons45, and induces myelin formation18,46. BDNF is well-known to promote neuronal survival and axonal growth47,48. BDNF also stimulates oligodendrocyte differentiation and maturation49,50, and protect the myelinated axons51,52. Although FSD-C10 treatment upregulated the production of NT-3, GDNF and BDNF, we found that blockage of BDNF abolished the beneficial effect of FSD-C10. Thus, BDNF plays an important role in FSD-C10-induced remyelination.
Our study showed that FSD-C10 is an efficient therapeutic drug for EAE. By inhibiting the migration and expansion of peripheral immune cells in the CNS, shifting macrophages/microglia from M1 to an M2 phenotype, and enhancing the production of neurotrophic factors, FSD-C10 demonstrated to exert several immune and non-immune beneficial effects in the course of neuroinflammation. Ultimately, FSD-C10 promoted remyelination and neuroprotection. Together, these findings demonstrate that FSD-C10 promotes neural repair in CNS autoimmunity through mechanisms involved both immunomodulation and induction of neurotrophic factors.
Materials and Methods
Mice
Female C57BL/6 mice (10–12 weeks and 18–20 g) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). All animal experiments were conducted in adherence with the International Council for Laboratory Animal Science guidelines. The study was approved by the Council for Laboratory and Ethics Committee of Shanxi Datong University, Datong, China. All mice were housed under pathogen-free conditions and kept in a reversed 12:12-h light/dark cycle in a temperature-controlled room (25 ± 2 °C) for one week prior to experimental manipulation.
Induction of chronic EAE
EAE were induced as described previously19. Mice were subcutaneously immunized with 300 μg mouse myelin oligodendrocyte glycoprotein 35–55 peptide (MOG35–55, MEVGWYRSPFSRVVHLYRNGK) in Freund’s complete adjuvant (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 3 mg/ml of Tuberculosis H37Ra (BD Difco, NJ, USA). In addition, 750 ng Pertussis toxin (Enzo Life Sciences, NY, USA) was injected intraperitoneally on days 0 and 2 p.i. MOG35–55 was produced in an automatic synthesizer (CL. Bio-Scientific., Xi’an, China). Purity of the peptide was > 95% as determined by HPLC.
Administration of Fasudil derivative FSD-C10
MOG35–55 immunized mice were divided into 2 groups (n = 12 each group), and normal mice were also divided into 2 groups (n = 6 each group); all were treated with either FSD-C10 (FSD-C10-treated) or ddH2O (ddH2O-treated control). FSD-C10 is a Fasudil derivative that is easily soluble in ethanol, and suitably water-soluble. FSD-C10 (from Tianjin Chase Sun Pharmaceutical Co., Ltd, Tian’jin, China) was dissolved in sterile ddH2O. For intranasal administration, mice were slightly anesthetized with diethylether, and then received 10 μl/each nostril (2.5 mg/kg/day) of FSD-C10 at the opening of the nostrils on day 3 until day 27 p.i., allowing the animal to sniff the solution into the upper nasal cavity. Mice that received the same volume of ddH2O nasally served as untreated control.
Cell proliferation
To study in vivo cell proliferation, mice received daily i.p. injections of BrdU (50 mg/kg) (Sigma) on days 25 until 27 p.i, and brain tissues were harvested at day 28 p.i. Proliferating cells were identified by using a mouse anti-BrdU antibody (1:500, Thermo Fisher Scientific, Waltham MA, USA) following experimental procedure recommended by the manufacturer. Briefly, brain sections were pretreated 10 min in paraformaldehyde (PFA) 4%, rinsed in PBS, and incubated 30 min in a solution of 2 N HCL. Slides were rinsed twice in PBS, and sections were incubated 10 min at 37 °C with the 0.1% trypsin. Slides were rinsed three times in PBS and then stained with anti- BrdU antibody.
Tissue processing
Mice underwent intracardiac perfusion under deep anesthesia with saline and 4% PFA solution in PBS. Brains were removed, and incubated overnight in sucrose solution 10%, 20%, and 30% respectively at 4 °C. 10 μm coronal brain sections were collected on poly-lys-coated slides by leica cryotome (Leica, Rueil-Malmaison, France). Brain slices were cut (−0.82 to −2.54 mm posterior to bregma based on the paxinos atlas) for 170 sequential sections.
Immunohistochemistry
Brain slides were air-dried, permeabilized, and blocked for 1 h and primary antibody applied overnight at 4 °C in a humid chamber. Fluorescently-conjugated secondary antibodies were applied for 2 h at room temperature in a humid chamber. Antibodies used to detect M1 microglia are as follows: mouse anti-iNOS (Enzo Life Sciences), rat anti-CD16/32 (BD Pharmingen, NJ, USA), anti-TNFα (Peprotech, NJ, USA), and IL-12 (Peprotech). Antibodies used to detect M2 markers are as follows: goat anti-Arginase-1 (anti-Arg-1, eBioscience, San Diego, CA, USA), IL-10 (Abcam, Camb, UK), CD206 (eBioscience). Antibodies against microglia/macrophage markers include rat anti-CD68 (Serotec, Kidlington, UK), CD4 (BD Pharmingen), CD11b (eBioscience). Oligodendrocyte, OPC neuron were detected by anti-NG2 (Promega, Madison, WI, USA), and GalC (Promega), anti-microtubule associated protein-2 (anti-MAP2, Millipore, Billericay, MA, UK) and anti-synaptophysin (Abcam). Other antibodies include rabbit anti-GFAP (Millipore), anti-NT-3 (Abcam), anti-GDNF (Novus Biologicals, CO, USA, 1:1000), anti-BDNF (Abcam). As a negative control, additional sections were treated similarly, but the primary antibodies were omitted. Results were visualized under fluorescent microscopy by Image-Pro Plus software or confocal laser scanning microscope (CLSM, Olympus, Tokyo, Japan) by FV-1200 software in a blinded fashion. Quantification was performed on three coronal sections per mouse, and six mice per group were analyzed.
Western blot analysis
The brains were homogenized on ice with a Ultrasonic crusher (HD3000, Wiggens, Beijing, China) in ice-cold lysis buffer (1 × PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, and 0.1% SDS, RIPA) supplemented with protease inhibitors PMSF. Lysates were centrifuged at 10,000 × g for 20 min at 4 °C for two times, and the supernatants were collected. Protein concentrations were determined by a Bradford protein assay. Equal amounts of protein (30 μg) were separated by SDS-PAGE, and transferred onto a polyvinylidene fluoride filter (PVDF) membrane (Millipore). Membranes were blocked with 5% non-fat milk, and incubated at 4 °C overnight with anti-NG2, anti-GalC, anti-GDNF, anti-NT-3, anti-BDNF, anti-iNOS, anti-Arginase-1 and anti-β-actin (Cell signaling, Davers, MA, USA). Bands were visualized by HRP-conjugated secondary antibodies (Thermo Scientific, MA, USA) and chemiluminescence (ECL) kit under ECL system (Millipore).
Cytokine measurement by ELISA
Levels of IL-1β, IL-6, TNF-α, IL-12, and IL-10 were measured by sandwich ELISA kits (Peprotech or R&D Systems, Minneapolis, MN) in accordance to manufacturer’s instructions. Determinations were performed in 3 independent experiments.
Splenocyte proliferation and cytokine measurement ex vivo
Splenocytes were isolated from ddH2O-treated and FSD-C10-treated EAE mice on day 12 p.i., and cultured with or without 25 μg/ml MOG35–55 peptide for 3 days. Cell proliferation was determined by BrdU-incorporation test using BrdU Cell Proliferation ELISA Kit (Abcam). Supernatants from splenocytes were collected at 72 h to measure concentration of IFN-γ, IL-17, TNF-α, and IL-10 using ELISA kits (R&D System, Minneapolis, MN).
Microglia polarization
The BV2 immortalized microglial cell line was obtained from ShenKe Biological Technolology CO., Ltd., Shanghai, PR China, and cultured in Dulbecco′s modified Eagle medium (DMEM; Gibco, Waltham MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco) at 37 °C in a humidified cell incubator with a 95%/5% (v/v) mixture of air and CO2. BV2 cells were allowed to attach to the culture dish for 2 h. Later, cells were either left untreated or treated overnight with lipopolysaccharide (LPS, Sigma-Aldrich, St. Louis, MO, USA, 500 ng/ml), FSD-C10 (15 μg/ml) or LPS plus FSD-C10. Conditioned media was collected and stored at −20 °C. Cells were acquired for immunostaining analyses.
Th1/Th17 cell polarization of CD4+ T cells in vitro
Differentiation of Th1 and Th17 cells was induced in vitro following previously described protocols53. Briefly, single-cell suspensions derived from spleen of normal female C57BL/6 mice (6–8 week) were purified by negative selection with a mouse CD4+ T Cell Isolation Kit II (Miltenyi Biotec). Purified naïve CD4+ T cells were cultured with soluble anti-CD3e (5 μg/ml) and anti-CD28 (2 μg/ml) under their respective polarizing conditions. IL-12 (5 ng/ml) was added to induce differentiation into Th1 cells. TGF-β1 (2 ng/ml), IL-6 (20 ng/ml), IL-1β (10 ng/ml), anti-IL-4 (10 μg/ml), and anti-IFN-γ (10 μg/ml) were added in Th17 polarizing conditions. FSD-C10 at 15 μg/ml was also added to define its direct effect on Th1/Th17 cell differentiation, and cultures without FSD-C10 served as control. Cells were collected 3 days later for flow cytometric analysis.
Co-culture of microglia and MOG-reactive T cells
Splenocytes were harvested from the spleen of EAE mice on day 8 p.i., and CD4+ T cells were purified using anti-mouse CD4 magnetic beads (Miltenyi Biotech, Auburn, CA, USA). These cells were then labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) for 20 min and washed for co-culture assay. The number of proliferating cells was determined through CFSE dilution with flow cytometry.
BV-2 microglia were cultured in the presence or absence of LPS (500 ng/ml) and/or FSD-C10 (15 μg/ml) for 24 h, washed, and then co-cultured with MOG-reactive CD4+ T cells prepared as described above at an estimated ratio of 1:10 (microglia: T cells) in the presence of MOG35–55 (25 μg/ml) for 72 h. Culture supernatants were collected to analyze cytokine production, and the cells obtained for flow cytometric analysis.
Flow cytometry
Splenocytes were stimulated with 50 ng/ml PMA and 500 ng/ml ionomycin in the presence of GolgiPlug for 4 h. Cells were surface-stained with mAb against CD4. Cells were then washed, fixed, and permeabilized with Fix & Perm Medium (Invitrogen), and intracellular cytokines were stained with Abs against IL17, and IFN-γ, or IL10 (BD Biosciences). Flow cytometric analysis was performed on FACSAria (BD Biosciences, San Jose, CA) and data were analyzed with FlowJo software (Treestar, Ashland, OR).
Immunocytochemistry staining
BV-2 cells were plated in 24 wells plates with coverslips. Cells were fixed with 4% PFA solution, washed in PBS for three times. Antibodies used to detect M1 microglia are as follows: anti-iNOS, anti-CD16/32. Antibodies used to detect M2 markers are as follows: anti-Arg-1, CD206. Results were visualized by confocal microscopy (FV1200; Olympus, Japan).
OPC culture and conditioned medium treatment
OPCs were isolated from CNS tissue of mice using magnetic isolation beads according to manufacturer’s instruction (CD140a (PDGFRa), Miltenyi Biotec, Cologne, Germany) and cultivated as follows. 2 × 104/ml OPCs were plated in 24-well plates with 1 ml DMEM/F12 containing 10 ng/ml PDGF AA, 20 ng/ml b-FGF, 2% N2, 2% B27, 1% p/o29,54. OPCs were treated with microglia media as conditioned media (CM) added in a 1:1 ratio to OPC culture media. After culturing for 24 h, viability of OPCs was assessed by using the Live/Dead® Cell Imaging kit (Invitrogen, Cal, USA) in accordance with the protocol provided by the manufacturer. Twenty to 60 images were made from each sample and in each image the cells were counted. Cells with a green fluorescent cytoplasm and a dark nucleus were counted as “viable cells”, while cells with only a red nucleus were counted as “dead cells”. Cells that had a red nucleus with green cytoplasm, and were consequently in an intermediate state between viable and dead, were denoted as “leaky cells”. The sum number of viable cells, dead cells and leaky cells were defined as “total cells”55,56,57. Results were expressed as number of different type of cell per view field. Data are presented as mean ± SEM. To study the effect of conditioned media (CM) on OPC differentiation, we cultured OPCs in DMEM/DF12 medium containing 2% N2, 2% B27, 40 ng/ml triiodothyronine and 10 ng/ml ciliary neurotrophic factor (CNTF). We added CM to the differentiation medium in a 1:1 ratio and the OPCs were cultured for 7 days. Cells were then stained by anti-myelin basic protein (MBP, Abcam) antibody.
FSD-C10 treatment of LPC-induced demyelination in cerebellum slice cultures
Postnatal day 5 (P5) mice pups were killed, and the cerebellums were removed and dissected in the dissection medium containing 50% HBSS, 50% Opti-MEM, following previously described protocol58,59. Consecutive slices (350 μm) were cut from the cerebellums using a tissue chopper. We cultured 3 slices in each 6-well insert plate (Millipore) with 1 ml medium composed of 25% heat-inactivated horse serum, 25% HBSS, 50% Opti-MEM. Demyelination was performed by adding 10 mg/ml lysophosphatidylcholine (LPC, Sigma-Aldrich) in the media on day 10. After culturing 12 h of adding LPC, slices were washed and transplanted to new inserts, and 15 μg/ml FSD-C10 or PBS was added to the culture media. To study the effect of neurotrophins on remyelination, neutralizing antibodies to GDNF, NT-3 and BDNF (Abcam, USA) were also added to the media. Slices were stained with anti-MBP and anti-NF-H on day 24 after culturing. Results were visualized by confocal microscopy (LSM 510; Zeiss, Jena, Germany).
Statistical analysis
GraphPad Prism software was used for statistical analysis. Data are presented as mean ± SEM. Experiments were analyzed by Student’s t-tests, and P-values were considered statistically significant when P < 0.05.
Additional Information
How to cite this article: Li, Y.-H. et al. FSD-C10, a Fasudil derivative, promotes neuroregeneration through indirect and direct mechanisms. Sci. Rep. 7, 41227; doi: 10.1038/srep41227 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Jana, A. & Pahan, K. Sphingolipids in multiple sclerosis. Neuromolecular Med. 12, 351–361 (2010).
Segal, B. M. Experimental autoimmune encephalomyelitis: cytokines, effector T cells, and antigen-presenting cells in a prototypical Th1-mediated autoimmune disease. Curr Allergy Asthma Rep. 3, 86–93 (2003).
Way, S. W. et al. Pharmaceutical integrated stress response enhancement protects oligodendrocytes and provides a potential multiple sclerosis therapeutic. Nat Commun. 6, 6532 (2015).
Traka, M., Podojil, J. R., McCarthy, D. P., Miller, S. D. & Popko, B. Oligodendrocyte death results in immune-mediated CNS demyelination. Nat neurosci. 19, 65–74 (2016).
Miron, V. E. et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 16, 1211–1218 (2013).
Chang, A., Tourtellotte, W. W., Rudick, R. & Trapp, B. D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med. 346, 165–173 (2002).
Kuhlmann, T. et al. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 131, 1749–1758 (2008).
Zamvil, S. S. & Steinman, L. Diverse targets for intervention during inflammatory and neurodegenerative phases of multiple sclerosis. Neuron. 38, 685–688 (2003).
Takahashi, C., Muramatsu, R., Fujimura, H., Mochizuki, H. & Yamashita, T. Prostacyclin promotes oligodendrocyte precursor recruitment and remyelination after spinal cord demyelination. Cell Death Dis. 4, e795 (2013).
Watanabe, K. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol. 25, 681–686 (2007).
Liu, C. et al. Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS One. 8, e54841 (2013).
Hou, S. W. et al. Fasudil ameliorates disease progression in experimental autoimmune encephalomyelitis, acting possibly through antiinflammatory effect. 18, 909–917 (2012).
Liu, C. Y. et al. Fasudil mediates cell therapy of EAE by immunomodulating encephalomyelitic T cells and macrophages. Eur J Immunol. 45, 142–152 (2015).
Tam, W. Y. & Ma, C. H. Bipolar/rod-shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Sci Rep. 4, 7279 (2014).
Yu, Z. et al. MSX3 Switches Microglia Polarization and Protects from Inflammation-Induced Demyelination. J Neurosci. 35, 6350–6365 (2015).
Yang, J. et al. Neurotrophin 3 transduction augments remyelinating and immunomodulatory capacity of neural stem cells. Mol Ther. 22, 440–450 (2014).
Vondran, M. W., Clinton-Luke, P., Honeywell, J. Z. & Dreyfus, C. F. BDNF+/− mice exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia 58, 848–856 (2010).
Zhang, L. et al. GDNF-Enhanced Axonal Regeneration and Myelination Following Spinal Cord Injury is Mediated by Primary Effects on Neurons. Glia. 57, 1178–1191 (2009).
Li, Y. H. et al. Intranasal delivery of FSD-C10, a novel Rho kinase inhibitor, exhibits therapeutic potential in experimental autoimmune encephalomyelitis. Immunology. 143, 219–229 (2014).
Mundy, W. R., Robinette, B., Radio, N. M. & Freudenrich, T. M. Protein biomarkers associated with growth and synaptogenesis in a cell culture model of neuronal development. Toxicology 249, 220–229 (2008).
Tucker, R. P. The roles of microtubule-associated proteins in brain morphogenesis: a review. Brain Res Brain Res Rev. 15, 101–120 (1990).
Shoji, M. et al. Neurotrophic activity of jiadifenolide on neuronal precursor cells derived from human induced pluripotent stem cells. Biochem Biophys Res Commun. pii: S0006-291X(16)30092-4 (2016).
Thiel, G. Synapsin I, synapsin II, and synaptophysin: marker proteins of synaptic vesicles. Brain Pathol. 3, 87–95 (1993).
Zhu, B., Luo, L., Moore, G. R. W., Paty, D. W. & Cynade, r M. S. Dendritic and Synaptic Pathology in Experimental Autoimmune Encephalomyelitis. The American Journal of Pathology. 162, 1639–1650 (2003).
Shirazi, H. A., Rasouli, J., Ciric, B., Rostami, A. & Zhang, G. X. 1, 25-Dihydroxyvitamin D3 enhances neural stem cell proliferation and oligodendrocyte differentiation. Exp Mol Pathol. 98, 240–245 (2015).
KhorshidAhmad, T. et al. Transcriptional Regulation of Brain-Derived Neurotrophic Factor (BDNF) by Methyl CpG Binding Protein 2 (MeCP2): a Novel Mechanism for Re-Myelination and/or Myelin Repair Involved in the Treatment of Multiple Sclerosis (MS). Mol Neurobiol. 53, 1092–1107 (2016).
Streit, W. J., Walter, S. A. & Pennell, N. A. Reactive microgliosis. Prog Neurobiol. 57, 563–581 (1999).
Hu, X. et al. Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. 11, 56–64 (2015).
Pedraza, C. E. et al. Induction of oligodendrocyte differentiation and in vitro myelination by inhibition of rho-associated kinase. ASN Neuro. 6 (2014).
Miron, V. E. et al. Simvastatin regulates oligodendroglial process dynamics and survival. Glia. 55, 130–143 (2007).
Paintlia, A. S. et al. HMG-CoA reductase inhibitor augments survival and differentiation of oligodendrocyte progenitors in animal model of multiple sclerosis. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 19, 1407–1421 (2005).
Paintlia, A. S., Paintlia, M. K., Singh, A. K. & Singh, I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Molecular Pharmacology. 73, 1381–1393 (2008).
Xin, Y. L. et al. FSD-C10: A more promising novel ROCK inhibitor than Fasudil for treatment of CNS autoimmunity. Biosci Rep (2015).
Napoli, I. & Neumann, H. Protective effects of microglia in multiple sclerosis. Exp Neurol. 225, 24–28 (2010).
Zindler, E. & Zipp, F. Neuronal injury in chronic CNS inflammation. Best Pract Res Clin Anaesthesiol. 24, 551–562 (2010).
Tang, Y. & Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol Neurobiol. 53, 1181–1194 (2016).
Ruckh, J. M. et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 10, 96–103 (2012).
Ponomarev, E. D., Veremeyko, T., Barteneva, N., Krichevsky, A. M. & Weiner, H. L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat Med. 17, 64–70 (2011).
Franklin, R. J. & Ffrench-Constant, C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 9, 839–855 (2008).
Sekimoto, M. et al. Functional expression of the TrkC gene, encoding a high affinity receptor for NT-3, in antigen-specific T helper type 2 (Th2) cells. Immunol Lett. 88, 221–226 (2003).
Girard, C. et al. Grafts of brain-derived neurotrophic factor and neurotrophin 3-transduced primate Schwann cells lead to functional recovery of the demyelinated mouse spinal cord. J Neurosci. 25, 7924–7933 (2005).
Razavi, S. et al. Neurotrophic factors and their effects in the treatment of multiple sclerosis. Adv Biomed Res. 4, 53 (2015).
Lu-Nguyen, N. B. et al. Transgenic expression of human glial cell line-derived neurotrophic factor from integration-deficient lentiviral vectors is neuroprotective in a rodent model of Parkinson’s disease. Hum Gene Ther, 25, 631–641 (2014).
Lin, L. F., Doherty, D. H., Lile, J. D., Bektesh, S. & Collins, F. GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurones. Science. 260, 1130–1132 (1993).
Ramer, M. S., Priestley, J. V. & McMahon, S. B. Functional regeneration of sensory axons into the adult spinal cord. Nature. 403, 312–316 (2000).
Deng, L. X. et al. A novel growth-promoting pathway formed by GDNF-overexpressing Schwann cells promotes propriospinal axonal regeneration, synapse formation, and partial recovery of function after spinal cord injury. J Neurosci. 33, 5655–5667 (2013).
Mariga, A., Mitre, M. & Chao, M. V. Consequences of Brain-Derived Neurotrophic Factor withdrawal in CNS neurons and implications in disease. Neurobiol Dis (2016).
Vilar, M. & Mira, H. Regulation of Neurogenesis by Neurotrophins during Adulthood: Expected and Unexpected Roles. Front Neurosci. (2016).
Van’t, V. A. et al. Brain-derived neurotrophic factor effects on oligodendrocyte progenitors of the basal forebrain are mediated through trkB and the MAP kinase pathway. J Neurosci Res. 87, 69–78 (2009).
VonDran, M. W., Singh, H., Honeywell, J. Z. & Dreyfus, C. F. Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J Neurosci. 31, 14182–14190 (2011).
Djalali, S. et al. Effects of brain-derived neurotrophic factor (BDNF) on glial cells and serotonergic neurons during development. J Neurochem. 92, 616–627 (2005).
Xiao, J. et al. Brain-derived neurotrophic factor promotes central nervous system myelination via a direct effect upon oligodendrocytes. Neurosignals. 18, 186–202 (2010).
Zhang, Y. et al. Therapeutic effect of baicalin on experimental autoimmune encephalomyelitis is mediated by SOCS3 regulatory pathway. Sci Rep. 5, 17407 (2015).
Yang, Z., Watanabe, M. & Nishiyama, A. Optimization of oligodendrocyte progenitor cell culture method for enhanced survival. J Neurosci Methods. 149, 50–56 (2005).
Verwer, R. W. H. et al. Cells in human postmortem brain tissue slices remain alive for several weeks in culture. FASEB J. 16, 54–60 (2002).
Wu, L. et al. Neural stem cells improve neuronal survival in cultured postmortem brain tissue from aged and Alzheimer patients. J Cell Mol Med. 12, 1611–1621 (2008).
Wu, H. M. et al. Microglial activation mediates host neuronal survival induced by neural stem cells. J Cell Mol Med. 18, 1300–1312 (2014).
Miron, V. E. et al. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am J. Pathol. 176, 2682–2694 (2010).
Morin-Brureau, M., De, B. F. & Lerner-Natoli, M. Organotypic brain slices: a model to study the neurovascular unit micro-environment in epilepsies. Fluids Barriers CNS. 10, 11 (2013).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Nos 81272163, 81501198), the Research Project Supported by Shanxi Scholarship Council of China (2014–7), the Department of Science and Technology, Shanxi Province of China (No. 2013081058). We thank Katherine Regan of the Department of Neurology, Thomas Jefferson University, Philadelphia, USA, for editorial assistance.
Author information
Authors and Affiliations
Contributions
Yan-hua Li and Cong Xie, Yuan Zhang and Xing Li participated in design of the study, conducted most of the experiments, analyzed the results, and wrote most of the paper. Bao-guo Xiao and Cun-gen Ma contributed to designing the study. Hai-fei Zhang, Qing Wang and Zhi Cai helped with data analysis. Rodolfo Thome and Guang-Xian Zhang participated in designing the study, analyzing data and rewriting the paper. We thank Katherine Regan for editorial assistance.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, YH., Xie, C., Zhang, Y. et al. FSD-C10, a Fasudil derivative, promotes neuroregeneration through indirect and direct mechanisms. Sci Rep 7, 41227 (2017). https://doi.org/10.1038/srep41227
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep41227
This article is cited by
-
Grape Seed Extract Attenuates Demyelination in Experimental Autoimmune Encephalomyelitis Mice by Inhibiting Inflammatory Response of Immune Cells
Chinese Journal of Integrative Medicine (2023)
-
Combined Treatment of Bone Marrow Mesenchymal Stem Cells and Fasudil Promotes Neurovascular Remodeling and Neurological Function Recovery in Ischemic Stroke
Applied Biochemistry and Biotechnology (2022)
-
Mdivi-1, a mitochondrial fission inhibitor, modulates T helper cells and suppresses the development of experimental autoimmune encephalomyelitis
Journal of Neuroinflammation (2019)
-
Therapeutic potentials of the Rho kinase inhibitor Fasudil in experimental autoimmune encephalomyelitis and the related mechanisms
Metabolic Brain Disease (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
