
Abstract
Lysobacter species are Gram-negative bacteria that are emerging as new sources of antibiotics, including HSAF (Heat Stable Antifungal Factor), which was identified from L. enzymogenes with a new mode of action. LesR, a LuxR solo, was recently shown to regulate the HSAF biosynthesis via an unidentified mechanism in L. enzymogenes OH11. Here, we used a comparative proteomic approach to identify the LesR targets and found that LesR influenced the expression of 33 proteins belonging to 10 functional groups, with 9 proteins belonging to the TBDR (TonB-Dependent Receptor) family. The fundamental role of bacterial TBDR in nutrient uptake motivates us to explore their potential regulation on HSAF biosynthesis which is also modulated by nutrient condition. Six out of 9 TBDR coding genes were individually in-frame deleted. Phenotypic and gene-expression assays showed that TBDR7, whose level was lower in a strain overexpressing lesR, was involved in regulating HSAF yield. TBDR7 was not involved in the growth, but played a vital role in transcribing the key HSAF biosynthetic gene. Taken together, the current lesR-based proteomic study provides the first report that TBDR7 plays a key role in regulating antibiotic (HSAF) biosynthesis, a function which has never been found for TBDRs in bacteria.
Similar content being viewed by others
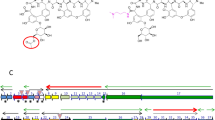


Introduction
TonB-dependent receptors (TBDRs) are a family of proteins that are located in the outer membrane of Gram-negative bacteria1. These receptors share a common structural feature of two domains; a C-terminal membrane-embedded β-barrel domain that is sealed by a conserved N-terminal globular domain (plug domain)2. TBDRs typically act as channels that open in response to outside ligands to allow import of extracellular nutrients, such as iron-siderophore complexes or non-Fe compounds (e.g. vitamin B12), into the periplasmic space4. The best-characterized examples include FecA, FhuA, FepA and BtuB, which are necessary for the active transport of the corresponding iron siderophores of ferric citrate, ferrichrome, or enterobactin, as well as vitamin B12, respectively1,2,3. The TBDR-dependent substrate transport is an active process that requires energy input from the proton motive force across the cytoplasmic membrane. Such a process requires that the ligand-loaded TBDRs interact with the TonB protein complex consisting of three inner membrane proteins (TonB/ExbB/ExbD)5. Although the basic role of TBDRs is believed mainly in nutrient transport, some TBDRs are also shown to trigger pathogenesis in several animal and plant bacterial pathogens8. Nevertheless, TBDR is never reported to play a role in regulating bacterial antibiotic biosynthesis to our knowledge.
LuxR solo is defined as a group of LuxR-family proteins possessing a classical AHL (N-acyl-homoserine lactones)-binding domain at the N terminus and a HTH DNA-binding domain at the C terminus, as other LuxR proteins in the canonical LuxI/R system10. However, LuxR solo lacks any cognate LuxI protein synthesizing the QS (Quorum sensing) signal AHL10,11. Bacterial LuxR solo thus potentially responds to signals produced by the bacteria itself, the neighboring bacteria, or the eukaryotes (e.g. plants) to exert the corresponding regulations, such as biofilm formation, virulence and biocontrol activity in several bacteria10,12–18. The genus Lysobacter, belonging to the Xanthomonadaceae family, is a group of Gram-negative bacteria with several conserved features, such as high genomic G+C content (approximately 70%), flagella-independent twitching motility, production of abundant lytic enzymes, as well as generation of bioactive natural products19,20,21. These distinct characteristics differentiate Lysobacter from its ecological/taxonomic related species, such as Xanthomonas19,22. A well-characterized species of this genus is L. enzymogenes, which is emerging as a biological control agent against fungal pathogens of crop plants, such as Bipolaris sorokiniana and Rhizoctonia solani23,24. L. enzymogenes is also currently recognized as a new sources of antibiotics20, including HSAF (Heat Stable Antifungal Factor) that belongs to the distinct PTM (polycyclic tetramate macrolactam) antifungal antibiotic with a new mode of action20,25,26. Furthermore, our previous reports show that the hsaf pks/nrps gene, encoding a hybrid polyketide synthase-nonribosomal peptide synthetase, is responsible for the HSAF biosynthesis in L. enzymogenes27,28. However, the yield of HSAF in L. enzymogenes is relatively low even in the HSAF inducing medium26,27. Therefore, elucidation of the regulatory mechanism(s) of HSAF biosynthesis in L. enzymogenes is necessary for improving the HSAF yield by genetic engineering or molecular biotechnology. Recent advancements have started to shed light into this issue21,22,29. Intriguingly, we previously found that overexpression, but not deletion of lesR, the only LuxR solo coding gene in L. enzymogenes, almost entirely impaired the HSAF production29. However, how the overexpressed lesR performed this critical control on the HSAF biosynthesis remains to be investigated.
To further understand the mechanism used by LesR in regulating HSAF biosynthesis, we have endeavored to identify the LesR targets that contribute to HSAF biosynthesis. By a combination of proteomics, bioinformatics and genetic approaches, we discovered that a certain TBDR protein, whose level was affected by the lesR overexpression, played an important role in regulating the antibiotic HSAF biosynthesis in L. enzymogenes. Our findings represent the first report about a novel functionality of TBDR proteins in bacteria.
Results
The levels of 9 TBDR proteins were affected by the lesR overexpression
Given that overexpression, but not deletion, of lesR in the wild-type OH11 was found to almost shut down the HSAF biosynthesis29, we therefore selected the lesR overexpression strain, as well as the wild-type OH11 possessing an empty vector as a control for 2-D gel proteome analysis to identify potential LesR targets in L. enzymogenes. To achieve this point, the growth ability of the lesR overexpression strain and control strain was first determined and compared in the HSAF-inducing medium (1/10 TSB broth). As shown in Fig. 1A, overexpression of lesR in the wild-type OH11 did not alter its growth ability in comparison to that of the control strain. On the basis of this result, a good time point at 12.5 h after initial inoculation that corresponds to the logarithmic phase of both strains was chosen for cell collection (Fig. 1A). We showed that regulation of lesR in the HSAF biosynthesis was functional at this designated point, because the control strain produced HSAF, whereas no HSAF was detected in the lesR overexpression strain at this time (Fig. 1B).

(A) Monitoring and comparison of the growth curve between the lesR overexpression and control strains in 1/10 TSB broth. The time point used for cell collection was set at OD6001.0 (indicated by the dotted line). Data are from three independent biological experiments. Each experiment involved three replicates for each strain. (B) Overexpression of lesR almost impaired the HSAF production in strain OH11. The HSAF production (indicated by the red arrow) from the collected cells, as noted in part (A), was extracted and determined by HPLC. (C) Functional classification of the identified 33 proteins affected by the lesR overexpression. The detailed information of each gene described in this figure was provided in the Table 1. OH11(lesR), the lesR overexpression strain; OH11(pBBR), the wild-type strain containing an empty expressing vector.
Next, total proteins were extracted from the collected cells of the lesR overexpression strain and control strain. After purification and quantification, these proteins were separated by 2-D gel electrophoresis. In this way, a total of 98 differentially expressed protein spots (with a threshold of larger than 1.5-fold change) were excised from silver-stained gels and subject to MALDI-TOF-TOF analysis; 33 of them were confidentially identified in the genome of strain OH11 (Table 1). In silico analysis further divided these 33 proteins into 10 groups (Fig. 1C and Table 1). The largest percentage of annotated proteins (27%) affected by lesR overexpression corresponds to the group of ‘inorganic ion transport and metabolism’ (Fig. 1C), which comprised 9 TBDR proteins (Table 1). These TBDR proteins were then further investigated for their potential roles in regulating HSAF biosynthesis, because the basic role of TBDR in nutrient uptake in L. enzymogenes is speculated to be correlated with the nutrient-dependent property of HSAF biosynthesis25,26. After a detailed sequence analysis of these 9 TBDR proteins as shown in Fig. 2A, we found that each TBDR possessed two conserved domains, a C-terminal membrane-embedded β-barrel domain (ligand_gated_channel) and an N-terminal plug domain (Plug domain; ~150–200 residues) that is similar to the well-characterized TBDR protein BtuB30,31. Furthermore, TBDR2, TBDR4 and TBDR9 also contained an additional domain, the TonB_dep_Rec domain (TonB dependent_Receptor). Notably, all 9 TBDR proteins had a TonB-box region at their corresponding N terminus as BtuB (Fig. 2B).

(A) The domain analyses of nine TBDR (TonB-Dependent Receptor) proteins. The well-characterized vitamin B12 receptor, BtuB from E. coli (gi|948468), served as a reference TBDR. (B) Multiple alignment of the TonB-box region of TBDR1 to TBDR9 with that of BtuB. The predicted TonB-box region was highlighted with a red box, similar to that of BtuB30. The conserved amino acid (Val74) that was marked with a red asterisk was selected for point mutation in further study.
The analysis also showed that all detected TBDR proteins did not contain a long N-terminal signaling domain, a distinct structural feature of the TBDT (TonB-dependent transducers) family that differentiates them from the conventional TBDRs32. Furthermore, none of all detected TBDR proteins was adjacent to a ECF (extracytoplasmic function) sigma factor and anti-sigma factor in their respective genetic organization (Supplementary Fig. S2), which is another typical characteristic of the TBDT-based CSS (cell-surface signaling) system in bacteria33. All these results strongly suggest that the 9 detected TBDR proteins belong to the conventional TBDR family, but not TBDT. Collectively, this 2-D proteomic study indicates that the levels of all 9 TBDR proteins were influenced by the lesR overexpression in L. enzymogenes.
Systematic mutation revealed that only TBDR7 played a key positive role in controlling HSAF biosynthesis
To test whether all identified TBDR proteins contribute to HSAF biosynthesis, each TBDR gene in L. enzymogenes was mutated by an in-frame deletion. In this way, 6 gene-deletion mutants, including the ΔTBDR1, 2, 4, 7, 8 and 9, were generated and further validated (Supplementary Table S2). The TBDR3, 5 and 6 coding genes appear to be essential for bacterial survival under the test condition, because these gene knockout bacteria failed to grow under a similar condition. Next, the HSAF yield was quantified in each TBDR mutant. As shown in Fig. 3, only 2 out of the 6 TBDR mutants were found to change the HSAF level, and knockout of the gene TBDR2, 4, 8 or 9 had no effect on the HSAF yield (Fig. 3). In particular, inactivation of TBDR7 almost abolished the HSAF production, whereas missing of TBDR1 significantly enhanced the HSAF level (Fig. 3). We also generated a ΔTBDR1&7 double mutant (Table 2 and Supplementary Table S2), and found that it produced approximately 55% HSAF to that of the wild type (Fig. 3), suggesting that TBDR1 and TBDR7 played opposing roles in regulating HSAF biosynthesis in L. enzymogenes. Collectively, the above results suggest that both TBDR1 and TBDR7 potentially controlled the HSAF biosynthesis in L. enzymogenes.

Peak area indicated the area of HSAF determined by HPLC method, while the OD600 represents the growth status of tested strains at the time points used for the extraction of HSAF. ΔTBDR (number) indicated the deletion mutant of target TBDR gene; ΔTBDR1&7, the double mutant of TBDR1 and TBDR7. Three replicates for each treatment were used, and the experiment was repeated three times. Vertical bars represent standard errors. The asterisk above the bars indicate a significant difference between the wild-type strain OH11 and the tested strains (*p < 0.05).
In the following study, we focused our efforts on TBDR7, because it seems to play a greater role in HSAF production than TBDR1. To verify the role of TBDR7, its complemented strain of ΔTBDR7 was constructed and verified by RT-PCR (Fig. 4A). As shown in Fig. 4B, the in trans TBDR7 complementation restored the HSAF production of the ΔTBDR7 mutant to the wild-type level, whereas the ΔTBDR7 mutant was deficient when complemented with an empty vector (Fig. 4B). In addition, we also created a point mutation (V74A) at the predicted TonB-box region of TBDR7 (Fig. 2B), because this amino acid (Val74) was previously shown to be important in transporting vitamin B12 in BtuB34. As shown in Fig. 4B, the TBDR7 containing the V74A indeed failed to complement the HSAF deficiency of the ΔTBDR7 mutant, revealing the importance of the TBDR7 TonB-box region in controlling HSAF biosynthesis. Collectively, these results suggest that TBDR7 played a vital role in regulating the HSAF production in L. enzymogenes.

(A) RT-PCR assay velidated the transcription of TBDR7 in the corresponding transformed strain. The size of expected DNA fragment was 388 bp. The gene, 16S rRNA (abbreviation for 16s) was used as an internal control, as described previously22. (B) Quantification of the HSAF level from the complemented strain of the TBDR7 deletion mutant. (C) Growth curves of the wild type and TBDR7 mutant in 1/10 TSB medium. (D) Mutation of TBDR7 almost entirely shut down the transcription of the key HSAF biosynthetic gene, hsaf pks/nrps. OH11, the wild-type strain of L. enzymogenes; ΔTBDR7, the TBDR7 mutant; ΔTBDR7 (pBBR), ΔTBDR7 containing an empty vector; ΔTBDR7 (TBDR7), the complemented strain of ΔTBDR7; ΔTBDR7 (TBDR7-V74A), ΔTBDR7 containing the plasmid-born TBDR7, where the amino acid Val74 within the TonB-box region was substituted by Ala74. Three replicates for each treatment were used, and the experiment was repeated three times. Vertical bars represent standard errors. The asterisk above the bars indicate a significant difference between the wild-type strain OH11 and the tested strains (*p < 0.05).
TBDR7 positively regulated the hsaf pks/nprs transcription
To investigate whether the deficiency of HSAF production of the ΔTBDR7 mutant in 1/10 TSB is due to the different growth rates, the growth capacity of the ΔTBDR7 mutant and the wild-type OH11 in this medium was examined. As shown in Fig. 4C, deletion of TBDR7 did not seem to alter the bacterial growth rate under the similar test condition, suggesting that TBDR7 was not involved in the growth, but controlled the HSAF production in L. enzymogenes. To further address this point, we determined the transcriptional level of hsaf pks/nrps, the key gene for HSAF biosynthesis in L. enzymogenes27. As shown in Fig. 4D, transcription of hsaf pks/nrps was shut down almost entirely in the ΔTBDR7 mutant compared to that of the wild-type OH11. This finding was consistent with the decreasing HSAF level in the ΔTBDR7 mutant (Fig. 3), and further suggests that the contribution of TBDR7 on the HSAF biosynthesis was, at least partially, due to decreasing transcription of the key HSAF biosynthetic gene in L. enzymogenes.
Discussion
In the present study, an omics-based strategy was utilized to investigate how LesR, the LuxR solo from a biological control agent L. enzymogenes, is able to regulate the antibiotic HSAF biosynthesis. A comparative proteomic analysis led to the finding that LesR affects the expression of 98 protein spots when the threshold was set at 1.5-fold change. For these proteins, we have paid attention most to a series of TBDR proteins, because they are closely associated with the nutrient-dependent trait of HSAF biosynthesis. By using a combination of systematic mutation, phenotypic analysis and quantitative gene expression methods, we have further found that TBDR7 was not involved in the growth, and acted as a key protein in controlling the HSAF production. Although bacterial TBDRs have been reported to play a key fundamental role in various nutrient transport, and some of them also have an important role in pathogen-host interaction in several pathogenic bacteria8, to our knowledge, no TBDR has been reported to control antibiotic biosynthesis in bacteria35. In the present study, we have used L. enzymogenes as a model bacterium and provide the first result that TBDR7 was involved in generating HSAF, a unique antifungal antibiotic. The present results therefore reveal a novel function of TBDR in bacteria.
Although the contribution of TBDR7 to the HSAF biosynthesis is well revealed in the present manuscript, the mechanism(s) is still unclear at this moment. It is well accepted that the fundamental function of TBDR is to uptake nutrient in nearly all bacterial species in an energy-dependent way, which requires direct interaction between the periplasmic domain of TonB and the TonB-box region of the ligand-loaded TBDRs33. In the present study, we did not yet know the nature of the TBDR7-loading ligand, and also lack the data for the direct interaction between TBDR7 and TonB. However, we did also find that TonB can regulate the HSAF biosynthesis, as mutation of tonB (Supplementary Table S2) almost completely impaired the HSAF yield (Supplementary Fig. S3). This result was correlated with the phenotype change of the ΔTBDR7 mutant on HSAF production. It will be intriguing to identify the potential TBDR7-loading ligand(s) and to explore the possible TBDR7-TonB interaction for a better understanding on how TBDR7 regulates the HSAF biosynthesis.
TBDR7 is one of the 9 TBDR proteins that were identified from a lesR-based proteomic study presented in this work. These 9 TBDR proteins account for approximately 16% of all TBDR proteins (55) distributed in the genome of strain OH11 which belong to the group of ‘inorganic ion transport and metabolism’. The functionality of TBDR has been shown to be directly related to different nutrient uptake, such as iron34. Competition for iron has long been known to be an important trait for beneficial rhizosphere colonization and for antagonism of plant deleterious microorganisms36. This finding has also been reported by PsoR, a LuxR solo of Pseudomonas fluorescens that responds to plant compounds17. As reported previously, LesR is a LuxR solo, but does not belong to the novel subgroup of plant-responding LuxR solo regulators (e.g. PsoR, OryR and XccR)29. However, we found that overexpression of either LesR or PsoR in the background of the relevant wild-type strain affected the gene/proteins involved in iron acquisition17 (Fig. 1C). This suggests that different types of LuxR solos from different bacterial biological control agents might share a similar role in controlling expression of the certain genes/proteins, such as those relating to ‘inorganic ion transport and metabolism’.
Since LesR is a transcription factor, one possible mechanism of LesR, therefore, is to regulate transcription of TBDR7 via a direct or indirect manner. In partial support of this hypothesis, we found that overexpression of lesR significantly increased the transcript of TBDR7 compared to that of the control strain (Supplementary Fig. S4), although the protein level of TBDR7 was decreased in the lesR overexpression strain. These results suggest that a post-transcriptional modification may occur in influencing the protein level of TBDR7 in the overexpressed lesR strain. We have also attempted to use gel shifting assay to test the direct interaction between LesR and the promoter of TBDR7 in vitro, but failed, due to the great difficulty in getting purified recombinant LesR protein. This situation is consistent with the previous report on the preparation of representative LuxR solo OryR from X. oryzae pv. Oryzae14,37. In a future study, we will try to fuse the LesR with the MBP tag to obtain soluble fused LesR to check its binding with the TBDR7 promoter.
Materials and Methods
Strains, plasmids and culture conditions
The bacterial strains and plasmids used in this study are listed in Table 2. Escherichia coli strain DH5α was used for plasmid constructions, and was grown in LB medium at 37 °C. Lysobacter enzymogenes OH11 (CGMCC No. 1978) and its derivative strains were grown in LB or 1/10 TSB (Trypic Soy Broth, Sigma) at 28 °C, shaking at 200 rpm. When required, antibiotics were added into the medium at final concentrations: kanamycin (Km) 100 μg/ml and gentamicin (Gm) 150 μg/ml for L. enzymogenes strains; Gm 25 μg/mL for E. coli strains.
Protein extraction and purification, and 2-D gel analyses
Total protein was extracted from bacterial cells according to the previous description38. In brief, cells were harvested from the culture (10% TSB broth) with an OD600 (Optical Density at 600nm) of 1.0 by centrifugation (10,000 × g at 4 °C for 10 min). The cell pellets were re-suspended in 20-mL washing buffer (50 mM Tris-HCI, pH 7.2), and this step (centrifugation and washing) was repeated twice. The final washed cells were resuspended in 400-μl alklysis buffer (addition of 0.1 mM PMSF), and fragmentized by an ultrasonic machine. Then the lysate was discarded by centrifugation at 10,000 × g at 4 C for 20 min. The supernatant was transferred into a fresh 2-mL reaction tube. Total protein of each strain was purified by CKEAN UP kit (GE, USA). Protein samples were subsequently used for 2-D gel analyses, which was described in details in a previous study from our laboratory39.
Trypsin digestion, MALDI-TOF MS analysis and sequence analysis
Protein spots were excised from the 2-D gel. Removal of silver ions in gels, trypsin digestion and peptide extraction were performed as described previously39,40. In brief, after the peptides were cocrystallized with CHCA (alpha cyano-4-hydroxy cinnamic acid) by evaporating organic solvents, tryptic-digested peptide masses were measured using a MALDI-TOF-TOF mass spectrometer (ABI4700 System, USA). All mass spectra were recorded in positive reflector mode and generated by accumulating data from 1000 laser shots. The following threshold criteria and settings were used: detected mass range of 700–3200 Da (optimal resolution for the quality of 1500 Da), using a standard peptide mixture [des-Argl-Bradykinin Mr904.468, Angiotensin I Mr1296.685, Glu-l-Fihrinopeptide B Mr1570.677, ACTH (1–17) Mr2093.087, ACTH(18–39) Mr2465.199; ACTH (7–38) Mr3657.929] as an external standard calibration, with laser frequency of 50 Hz, repetition rate of 200 Hz, UV wavelength of 355 nm, and accelerated voltage of 20,000 V. Peptide mass fingerprint data were matched to the comprehensive non-redundant sequence database NCBInr using Profound program under 50 ppm mass tolerance.
Data was processed via the Data Explorer software and proteins were unambiguously identified by searching against the database NCBInr using the MASCOT software search engine (http://www.matrixscience.com/cgi/search form.pl?FORMVER=2&SEARCH=MIS). The search parameters were as follows: (1) peptide quality of 800–4000 Da, mass tolerance for the fragment ion of 0.25 Da; (2) a minimum of seven matching peptides; (3) one missed cleavage; (4) taxonomy: L. enzymogenes (bacterium); and (5) allowed modifications, alkylation of cysteine by carbamidomethylation of Cys (complete) acetylation of the N-terminus and oxidation of methionine (partial). Moreover, in order to evaluate protein identification, we considered the percentage of sequence coverage, the observation of distribution of matching peptides (authentic hit is often characterized by peptides that are adjacent to one another in the sequence and that overlap), the distribution of error (distributed around zero), the gap in probability and score distribution from the first to other candidate; only matches with over 90% sequence identity and a maximum e-value of 10−10 were considered. Domain and functional analyzing of proteins identified was carried out using NCBI (http://www.ncbi.nlm.nih.gov/) and Pfam (http://pfam.sanger.ac.uk/).
In-frame deletion, complementation and site-directed mutagenesis
The protocol of gene in-frame deletion in L. enzymogenes was utilized as described previously41. In brief, two flanking regions of target gene were generated by PCR using a set of primer pairs (Supplementary Table S1), and cloned into the corresponding sites of the suicide vector pEX18Gm (Table 2). The final constructs were transformed into the wild-type OH11 or its derivatives by electroporation. Then Lysobacter transformants were selected on LB plates without sucrose and with Km (100 μg/ml) and Gm (150 μg/ml). The positive colonies were plated on LB plates containing 1/10 (w/v) sucrose and Km (100 μg/ml) to select for resolution of the construct by a second cross-over event. The resultant mutants were confirmed by a PCR method (Supplementary Table S2). For generation of complemented strains, the target gene, TBDR7 together with its native promoter region (Supplementary Table S1) was amplified from strain OH11 and cloned into the broad-host vector pBBR1-MCS5 (Table 2). The final construct was transformed into the TBDR7-deletion mutant for complementation. The expression of TBDR7 in the transformed strains was determined by reverse transcription PCR (RT-PCR), which was described as below. Site-directed mutagenesis of the TonB-box region of TBDR7 was carried out by using the Fast Mutagenesis System kit (FM111-01, Transgen Biotech, China) according to the product manufacturer. The point-mutated DNA was validated by sequencing.
HSAF extraction and quantification
Extraction and HPLC (High-Performance Liquid Chromatography) based quantification of HSAF from various Lysobacter strains was performed as described previously22,25,27. Three replicates for each treatment were used, and the experiment was performed three times.
Growth determination
Various Lysobacter strains were cultured in LB liquid at 28 °C overnight. Then 500 μl of the overnight culture for each strain was transferred into the fresh 1/10 TSB broth (50 ml) to grow until the OD600 reached to 1.0 (the logarithmic phase of growth). Next, 1 ml of each culture was transferred again into the fresh 1/10 TSB broth (50 ml) to start the detection of growth curve. All inoculation broths were grown at 28 °C with shaking at 200 rpm, and the OD600 value was determined every 2 h until bacterial growth reached the stationary phase. Each sample involves three technical replicates and the experiment was performed three times.
RNA extraction, qRT-PCR and RT-PCR
The wild-type OH11 and its derivatives, including the TBDR7 deletion mutant, OH11 containing an empty vector and the lesR overexpression strain, were grown on 1/10 TSB. Cells of the wild-type OH11 and the TBDR7 mutant were collected at the time point 10 h and 11 h, respectively, corresponding to the same cell density (OD600 = 1.0). Similarly, OH11 containing the empty vector and the lesR overexpression strain were collected at the time point 12.5 h (OD600 = 1.0). Then the total RNA was extracted from the collected cells of each strain using a kit from OMIGA Company (China). Next, qRT-PCR (quantitative RT-PCR) and RT-PCR assays, including cDNA synthesis and PCR amplification were performed as described previously22,29. Primer sequences used in this assay were listed in Supplementary Table S1.
Data submission
The sequence data of the present study has been submitted to the NCBI Genbank. The details were provided in Table 1.
Data analysis
All analyses were conducted using SPSS 14.0 (SPSS Inc., Chicago, IL, USA). The hypothesis test of percentages (t-test, *p < 0.05) was used to determine significant differences in production of antibiotic metabolite and gene expression between the control and treatment of the present study.
Additional Information
How to cite this article: Wang, R. et al. A TonB-dependent receptor regulates antifungal HSAF biosynthesis in Lysobacter. Sci. Rep. 6, 26881; doi: 10.1038/srep26881 (2016).
References
Ferguson, A. D. et al. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science 282, 2215–2220 (1998).
Buchanan, S. K. et al. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nat. Struct. Biol. 6, 56–63 (1999).
Ferguson, A. D. et al. Structural basis of gating by the outer membrane transporter FecA. Science 295, 1715–1719 (2002).
Moeck, G. S. & Coulton, J. W. TonB-dependent iron acquisition: mechanisms of siderophore-mediated active transport. Mol. Microbiol. 28, 675–681 (1998).
Postle, K. & Kadner, R. J. Touch and go: tying TonB to transport. Mol. Microbiol. 49, 869–882 (2003).
Cadieux, N. & Kadner, R. J. Site-directed disulfide bonding reveals an interaction site between energy-coupling protein TonB and BtuB, the outer membrane cobalamin transporter. Proc. Nat. Acad. Sci. 96, 10673–10678 (1999).
Ferguson, A. D. & Deisenhofer, J. Metal import through microbial membranes. Cell 116, 15–24. (2004).
Tauseef, I. et al. Influence of the combination and phase variation status of the haemoglobin receptors HmbR and HpuAB on meningococcal virulence. Microbiology 157, 1446–1456 (2011).
Blanvillain, S. et al. Plant carbohydrate scavenging through TonB-Dependent receptors: A feature shared by phytopathogenic and aAquatic bacteria. PLoS ONE 2, e224 (2007).
Gonzalez, J. F., Myers, M. P. & Venturi, V. The inter-kingdom solo OryR regulator of Xanthomonas oryzae is important for motility. Mol. Plant. Pathol. 14, 211–221 (2013).
Fuqua, C., Parsek, M. R. & Greenberg, E. P. Regulation of gene expression by cell-to-cell communication: acyl-homoserine lactone quorum sensing. Annu. Rev. Genet. 35, 439–468 (2001).
Volf, J. et al. Role of SdiA in Salmonella enterica serovar Typhimurium physiology and virulence. Arch. Microbiol 178, 94–101 (2002).
Ahmer, B. M. Cell-to-cell signalling in Escherichia coli and Salmonella enterica . Mol Microbiol 52, 933–945 (2004).
Ferluga, S. et al. A LuxR homologue of Xanthomonas oryzae pv. oryzae is required for optimal rice virulence. Mol. Plant. Pathol. 8, 529–538 (2007).
Zhang, L. et al. A proline iminopeptidase gene upregulated in planta by a LuxR homologue is essential for pathogenicity of Xanthomonas campestris pv. campestris . Mol. Microbiol. 65, 121–136 (2007).
Lee, J. et al. Reconfiguring the quorum-sensing regulator SdiA of Escherichia coli to control biofilm formation via indole and N-acyl homoserine lactones. Appl. Environ. Microbiol. 75, 1703–1716 (2009).
Subramoni, S. et al. Bacterial subfamily of LuxR regulators that respond to plant compounds. Appl. Environ. Microbiol. 77, 4579–4588 (2011).
Chatnaparat, T. et al. XagR, a LuxR homolog, contributes to the virulence of Xanthomonas axonopodis pv. glycines to soybean. Mol. Plant. Microbe. Interact. 25, 1104–1117 (2012).
Christensen, P. & Cook, F. D. Lysobacter, a new genus of nonfruiting, gliding bacteria with high base ratio. Int. J. Syst. Bacteriol. 28, 367–393 (1978).
Xie, Y. X. et al. Bioactive natural products from Lysobacter . Nat. Prod. Rep. 29, 1277–1287 (2012).
Zhou, X. et al. PilG is involved in the regulation of twitching motility and antifungal antibiotic biosynthesis in the biological control agent Lysobacter enzymogenes . Phytopathology 105, 1318–1324 (2015).
Qian, G. L. et al. Lysobacter enzymogenes uses two distinct cell-cell signaling systems for differential regulation of secondary-metabolite biosynthesis and colony morphology. Appl. Environ. Microbiol. 79, 6604–6616 (2013a).
Zhang, Z. & Yuen, G. Y. Biological control of Bipolaris sorokiniana on tall fescue by Stenotrophomonas maltophilia strain C3. Phytopathology 89, 817–822 (1999).
Yuen, G. Y. & Zhang, Z. Control of brown patch using the bacterium Stenotrophomonas maltophilia C3 and culture fluid. Int. Turfgrass . Soc. Res. 9, 742–747 (2001).
Yu, F. G. et al. Structure and biosynthesis of heat-stable antifungal factor (HSAF), a broad-spectrum antimycotic with a novel mode of action. Antimicrob. Agents. Ch. 51, 64–72 (2007).
Li, S. J. et al. Distinct ceramide synthases regulate polarized growth in the filamentous fungus Aspergillus nidulans . Mol. Biol. Cell. 17, 1218–1227 (2006).
Lou, L. L. et al. Biosynthesis of HSAF, a tetramic acid-containing macrolactam from Lysobacter enzymogenes . J. Am. Chem. Soc. 133, 643–645 (2011).
Li, Y. Y. et al. Iterative assembly of two separate polyketide chains by the same single-module bacterial polyketide synthase in the biosynthesis of HSAF. Angew. Chem. Int. Ed. Engl. 53, 7524–7530 (2014).
Qian, G. L. et al. Roles of a solo LuxR in the biological control agent Lysobacter enzymogenes strain OH11. Phytopathology 104, 224–231 (2014).
Schöffler, H. & Braun, V. Transport across the outer membrane of Escherichia coli K12 via the FhuA receptor is regulated by the TonB protein of the cytoplasmic membrane. Mol. Gen. Genet. 217, 378–383 (1989).
Peacock, R. S. et al. The solution structure of the C-terminal domain of TonB and interaction studies with TonB Box peptides. J. Mol. Biol. 345, 1185–1197 (2005).
Braun, V. & Endriss, F. Energy-coupled outer membrane transport proteins and regulatory proteins. Biometals 20, 219–231 (2007).
Ferguson, A. D. et al. Signal transduction pathway of TonB-dependent transporters. Proc. Nat. Acad. Sci. 104, 513–518 (2007).
Gudmundsdottir, A. et al. Point mutations in a conserved region (TonB box) of Escherichia coli outer membrane protein BtuB affect vitamin B12 transport. J. Bacteriol. 171, 6526–6533 (1989).
Hu, Y. H. et al. A TonB-dependent outer membrane receptor of Pseudomonas fluorescens: virulence and vaccine potential. Arch microbiol. 194, 795–802 (2012).
O’Sullivan, D. J. & O’Gara, F. Traits of fluorescent Pseudomonas spp. involved in suppression of plant root pathogens. Microbiol. Rev. 56, 662–676 (1992).
Ferluga, S. & Venturi, V. OryR is a LuxR-family protein involved in interkingdom signaling between pathogenic Xanthomonas oryzae pv. oryzae and rice. J. Bacteriol. 191, 890–897 (2009).
Watt, S. A. et al. Comprehensive analysis of the extracellular proteins from Xanthomonas campestris pv. campestris B100. Proteomics 5, 153–167 (2005).
Qian, G. L. et al. Proteomic analysis reveals novel extracellular virulence-associated proteins and functions regulated by the diffusible signal factor (DSF) in Xanthomonas oryzae pv. oryzicola . J. Proteome. Res. 12, 3327–3341 (2013b).
Joo, W. A. et al. Monitoring protein expression by proteomics: Human plama exposed to benzene. Proteomics 3, 2402–2411 (2003).
Qian, G. L. et al. Selection of available suicide vectors for gene mutagenesis using chiA (a chitinase encoding gene) as a new reporter and primary functional analysis of chiA in Lysobacter enzymogenes strain OH11. World. J. Microbiol. Biotechnol. 28, 549–557 (2012).
Hoang, T. T. et al. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–78 (1998).
Kovach, M. E. et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166, 175–176 (1995).
Acknowledgements
Dr. Guoliang Qian is a visiting scholar in Dr. Mark Gomelsky’s laboratory of University of Wyoming, USA, supported by China Scholarship Council. This study was supported by Jiangsu Agriculture Science and Technology Innovation Fund [CX(15)1023], National Basic Research (973) program of China (2015CB150600), National Natural Science Foundation of China (31371981; 31572046; 31329005), the grants from Jiangsu Academy of Agricultural Sciences and Jiangsu Province (ZX(15)1006; BE2014386; BE2015354), Program for New Century Excellent Talents in University of Ministry of Education of China (NCET-13-0863), Fundamental Research Funds for the Central Universities (KYTZ201403) and Special Fund for Agro-Scientific Research in the Public Interest (No. 201303015). Research in the Du lab is supported in part by NIH R01AI097260 and a UNL Redox Biology Center pilot grant. Dr. Chou is supported by the Ministry of Education, Taiwan, under the ATU plan, and by the National Science Council, Taiwan (grants 102-2113-M-005-006-MY3).
Author information
Authors and Affiliations
Contributions
R.W., H.X. and G.Q. conceived and designed experiments. R.W., H.X. and Y.L. carried out experiments. R.W., H.X., G.Q., F.L. and S.C analyzed data and prepared figures. R.W., H.X., Y.S., F.L. and G.Q. wrote the manuscript. L.D., Y.S. and S.C. reviewed and revised the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, R., Xu, H., Du, L. et al. A TonB-dependent receptor regulates antifungal HSAF biosynthesis in Lysobacter. Sci Rep 6, 26881 (2016). https://doi.org/10.1038/srep26881
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26881
This article is cited by
-
The recombination regulator RecX negatively regulates heat-stable antifungal factor (HSAF) biosynthesis in Lysobacter enzymogenes
Phytopathology Research (2023)
-
Efficient production of heat-stable antifungal factor through integrating statistical optimization with a two-stage temperature control strategy in Lysobacter enzymogenes OH11
BMC Biotechnology (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
