
Abstract
Rocaglates are a series of structurally complex secondary metabolites with considerable cytotoxicity that have been isolated from plants of the Aglaia genus (Meliaceae). A new rocaglate (aglapervirisin A, 1) and its eight new biosynthetic precursors of rocaglate (aglapervirisins B-J, 2–9) together with five known compounds, were isolated from the leaves of Aglaia perviridis. Their structures were elucidated based on a joint effort of spectroscopic methods [IR, UV, MS, ECD, 1D- and 2D-NMR, HRESIMS], chemical conversion and single-crystal X-ray diffraction. Among these isolates, three (1, 10–11) were silvestrols, a rare subtype rocaglates, exhibiting notable cytotoxicity against four human tumor cell lines, with IC50 values between 8.0 and 15.0 nM. Aglapervirisin A (1) induces cell cycle arrest at the G2/M-phase boundary at concentration 10 nM accompanied by reductions in the expression levels of Cdc2 and Cdc25C in HepG2 cells after 72h co-incubation and further induces the apoptosis of HepG2 cells at concentrations over 160 nM.
Similar content being viewed by others
Introduction
Rocaglate, a class of structurally complex secondary metabolites from plants of the Aglaia genus (Meliaceae), have attracted great attentions for their considerable cytotoxicity1,2,3,4,5. Rocaglates could block cell cycle progression6,7,8,9,10,11,12,13 from G2 to M and exhibited promising activity in human tumor cell lines and xenograft models. Their cytostatic effects are comparable to those of established anticancer drugs such as vinblastine sulphate, actinomycin D and hydroxycamptothecine14,15. Rocaglates possess cyclopenta[b]benzofuran skeleton, which originates from the [3 + 2] cyclization products of flavonol and diamide (cyclopenta[bc]benzofurans). Silvestrols, featuring an unprecedented dioxanyloxy unit attached to phenyl ring A of the cyclopenta[b]benzofuran skeleton, is a rare subtype of rocaglate, mostly of marked cytotoxicity8,11,13. The structure complexity and the potent activities of silvestrols have attracted significant interest in their biosynthesis and total synthesis16,17,18,19.
In the plant kingdom, silvestrol analogues are characteristically and exclusively present in Aglaia species and only four silvestrols have been reported4 as yet. Certain Aglaia species have been used as traditional medicines for treating fever, cough, diarrhoea and contused wounds4,5. In continuation of the discovery of novel and bioactive natural products from plants of the Meliaceae family20,21,22, the species A. perviridis, a wild shrub indigenous to Yunnan Province of China20,21,22, was investigated to find cytotoxic rocaglates, particularly silvestrols. As a result, three silvestrol analogues including a new one (aglapervirisin A, 1), eight new biosynthetic precursors of rocaglates (cyclopenta[bc]benzopyrans, 2–9) and three known precursors (12–14) were isolated and purified from the leaves of A. perviridis (Fig. 1). Their structures were mainly elucidated through comprehensive analysis using spectroscopic methods, including IR, UV, MS, HRESIMS, 1D-NMR and 2D-NMR. The absolute configuration of 1 was determined by ECD analysis and chemical conversion and that of 2 was established by single-crystal X-ray diffraction using Cu Kα radiation. These isolates (except for 6 and 12) were evaluated for their cytotoxicity against four human cancer cell lines: three silvestrol analogues (1, 10 and 11) showed potent activity with IC50 values between 8.0 and 15.0 nM. Of them, 1 induced cell cycle arrest by reducing the Cdc2 and Cdc25C expression levels in a dose-dependent manner and induced the apoptosis of these cells at concentrations over 160 nM. Herein, we report the separation and structural elucidation of these isolated rocaglate derivatives, as well as the bioassay results.

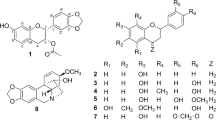
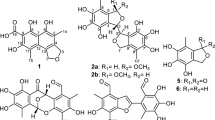
Chemical structures of compounds 1–9, 1a, 10a and 11a.
Results and Discussion
Aglapervirisin A (1),  −82.1 (c, 0.11, MeOH), was obtained as colourless powder with the molecular formula C28H27NO8, as deduced from the [M + Na]+ ion peak in HRESIMS data (C36H40O14Na, m/z 719.2313). The 1H NMR spectrum of 1 displayed resonances for the four aromatic protons of a 1,4-disubstituted benzene, five aromatic protons of a monosubstituted benzene, two aromatic protons of a 1,2,3,5-tetrasubstituted benzene and four methoxy groups. Its 1D-NMR (Table 1) data, particularly the three characteristic proton signals at δH 5.11(1H, d, J = 7.0 Hz), δH 3.91 (1H, dd, J = 14.5, 7.0 Hz) and δH 4.32(1H, d, J = 14 Hz), featured a cyclopenta[b]benzofuran derivative nature of 123. A series of proton signals at δH 3.5 to 5.5, carbon signals at δC 60.0 to 70.0 and two characteristic acetal carbons at δC 93.7 and 95.3, suggested the presence of a dioxanyloxy unit in the structure of 1. The above-described analysis indicated that compound 1 was a silvestrol (10) analogue. Compared with 10, the presence of an acetyl group in 1 was evidenced by characteristic NMR signals (δH 1.89, δC 51.9, 171.7) and 42 mass unit more than 10. The key HMBC correlation (Fig. 2) from H-6′′′ (δH 4.08 and 3.99) to the acetyl group (δC 20.5) indicated that the acetyl group was located at 6′′′-OH. The key ROESY correlations (Fig. 2) of H-1/H-2, H-2′, 6′ and H-2/H-2′, 6′, H-2″, 6″, also observed for 1023, indicate that H-1, H-2, 1,4-disubstituted benzene and monosubstituted benzene were co-facial. Thus, the planar structure and relative configuration of 1 was determined. The similar ECD spectra of 1 and silvestrol (10) indicated that the absolute stereochemistry of the basic skeleton of 1 was the same as that of silvestrol. However, the absolute configuration of C-5′′′ in 1 was difficult to determine based on the ECD comparison. To completely determine the absolute configuration of 1, the acetyl derivative of 1 (1a), silvestrol (10a) and episilvestrol (11a) (see Figure S16) were prepared by acylation. The identical HRESIMS (1a, m/z 756.2857, [M + NH4]+, 10a, m/z, 756.2859, [M + NH4]+), retention times in HPLC (1a, tR = 8.1 min, 10a, tR = 8.1 min, 70% MeOH- H2O) and the NMR data (see in SI), as well as the similar optical values obtained for 1a and 10a and the different retention times in HPLC (1a, tR = 8.1 min, 11a, tR = 9.3 min, 70% MeOH- H2O) and the NMR data (see in SI) obtained for 1a and 11a, indicate that the absolute configuration of 1 was consistent with that of silvestrol (10)23. The C-5′′′ in 1 was adopted as R based on the R configuration of C-5′′′ in compound 1023. Thus, the structure of 1 was determined and named as aglapervirisin A.
−82.1 (c, 0.11, MeOH), was obtained as colourless powder with the molecular formula C28H27NO8, as deduced from the [M + Na]+ ion peak in HRESIMS data (C36H40O14Na, m/z 719.2313). The 1H NMR spectrum of 1 displayed resonances for the four aromatic protons of a 1,4-disubstituted benzene, five aromatic protons of a monosubstituted benzene, two aromatic protons of a 1,2,3,5-tetrasubstituted benzene and four methoxy groups. Its 1D-NMR (Table 1) data, particularly the three characteristic proton signals at δH 5.11(1H, d, J = 7.0 Hz), δH 3.91 (1H, dd, J = 14.5, 7.0 Hz) and δH 4.32(1H, d, J = 14 Hz), featured a cyclopenta[b]benzofuran derivative nature of 123. A series of proton signals at δH 3.5 to 5.5, carbon signals at δC 60.0 to 70.0 and two characteristic acetal carbons at δC 93.7 and 95.3, suggested the presence of a dioxanyloxy unit in the structure of 1. The above-described analysis indicated that compound 1 was a silvestrol (10) analogue. Compared with 10, the presence of an acetyl group in 1 was evidenced by characteristic NMR signals (δH 1.89, δC 51.9, 171.7) and 42 mass unit more than 10. The key HMBC correlation (Fig. 2) from H-6′′′ (δH 4.08 and 3.99) to the acetyl group (δC 20.5) indicated that the acetyl group was located at 6′′′-OH. The key ROESY correlations (Fig. 2) of H-1/H-2, H-2′, 6′ and H-2/H-2′, 6′, H-2″, 6″, also observed for 1023, indicate that H-1, H-2, 1,4-disubstituted benzene and monosubstituted benzene were co-facial. Thus, the planar structure and relative configuration of 1 was determined. The similar ECD spectra of 1 and silvestrol (10) indicated that the absolute stereochemistry of the basic skeleton of 1 was the same as that of silvestrol. However, the absolute configuration of C-5′′′ in 1 was difficult to determine based on the ECD comparison. To completely determine the absolute configuration of 1, the acetyl derivative of 1 (1a), silvestrol (10a) and episilvestrol (11a) (see Figure S16) were prepared by acylation. The identical HRESIMS (1a, m/z 756.2857, [M + NH4]+, 10a, m/z, 756.2859, [M + NH4]+), retention times in HPLC (1a, tR = 8.1 min, 10a, tR = 8.1 min, 70% MeOH- H2O) and the NMR data (see in SI), as well as the similar optical values obtained for 1a and 10a and the different retention times in HPLC (1a, tR = 8.1 min, 11a, tR = 9.3 min, 70% MeOH- H2O) and the NMR data (see in SI) obtained for 1a and 11a, indicate that the absolute configuration of 1 was consistent with that of silvestrol (10)23. The C-5′′′ in 1 was adopted as R based on the R configuration of C-5′′′ in compound 1023. Thus, the structure of 1 was determined and named as aglapervirisin A.

Selected key HMBC and ROESY correlations observed for 1.
Aglapervirisin B (2) was obtained as colourless crystal ( −28.3) and gave a sodiated molecular ion [M + Na]+ at m/z 675.2672 (calcd 675.2677) in the HRESIMS corresponding to a molecular formula of C38H40N2O8, which requires 20 indices of hydrogen deficiency. In the 1H NMR spectrum, the 16 aromatic hydrogen signals in the low-field region (δH 6.09–7.85) presented four benzene rings, including two monosubstituted benzene rings, a 1,4-disubstituted benzene ring and a 1,2,3,5-tetrasubstituted benzene ring. Two amide protons at δH 6.47 (NH-17) and 5.26 (NH-12) and the remaining methylenes suggested the presence of a 1,4-butanediamide chain. These characteristic proton signals revealed that 2 was a cyclopenta[bc]benzopyran derivative24,25,26,27.
−28.3) and gave a sodiated molecular ion [M + Na]+ at m/z 675.2672 (calcd 675.2677) in the HRESIMS corresponding to a molecular formula of C38H40N2O8, which requires 20 indices of hydrogen deficiency. In the 1H NMR spectrum, the 16 aromatic hydrogen signals in the low-field region (δH 6.09–7.85) presented four benzene rings, including two monosubstituted benzene rings, a 1,4-disubstituted benzene ring and a 1,2,3,5-tetrasubstituted benzene ring. Two amide protons at δH 6.47 (NH-17) and 5.26 (NH-12) and the remaining methylenes suggested the presence of a 1,4-butanediamide chain. These characteristic proton signals revealed that 2 was a cyclopenta[bc]benzopyran derivative24,25,26,27.
In the HMBC spectrum (Fig. 3), a cross peak between signals at δH 7.76 (H-20, 24) and δC 167.6 (C-18) established the connection of one of the monosubstituted benzene rings to the butanediamide chain. The correlated peaks from H-3 to C-11 and from H-4 to C-1″ and C-2″, 6″ allowed the placement of the 1, 4-butanediamide moiety and the second monosubstituted benzene ring at C-3 and C-4, respectively. Thus, the planar structure of 2 was depicted in Fig. 3. The relative configuration of 2 was determined according to the coupling constants between H-3 and H-4 and the cross peaks in the ROESY spectrum (Fig. 3). The vicinal coupling constant value between H-3 and H-4 was 6.5 Hz, allowed the assignment of H-3β and H-4α configuration24,25,26,27. The ROESY correlations between H-10 and H-2″, 6″ and the lack of any correlation between H-3 and H-10 in compound 2 further confirmed the H-3β and H-4α. A single-crystal X-ray diffraction experiment (Fig. 4) using Cu Kα radiation was performed and the absolute configuration of five asymmetric carbons in 2 was unambiguously established as 2R, 3S, 4R, 5R and 6S. The absolute configuration of such a flavonol and diamide [3 + 2] adduct containing a benzoyl-1,4-butanediamide moiety was determined for the first time via the single-crystal X-ray diffraction method.

Selected key HMBC and ROESY correlations observed for 2.

Single-crystal X-ray structure of 2.
Aglapervirisin C (3) was obtained as a white powder { −22.1 (c 0.10, MeOH)} and the molecular formula of C38H40N2O8, which is equal to that found for 2, was determined based on the HRESIMS data (m/z, 675.2675, [M + Na]+). The NMR data (Table 2) of 3 were similar to those of 2, which indicated that 3 was also a cyclopenta[bc]benzopyran derivative with a benzoyl-1,4-butanediamide moiety28. However, the methine signals of H-10 at δH 4.67 and C-10 (δC 76.1) in 3 showed notable differences from those of 2 (δH 4.19, δC 82.6) (Table 1), which combined with the observed obvious ROESY correlation between H-3 (δH 3.51) and H-10 (δH 4.68) in 3, absent in 2, revealed that 2 and 3 were epimers at C-10.
−22.1 (c 0.10, MeOH)} and the molecular formula of C38H40N2O8, which is equal to that found for 2, was determined based on the HRESIMS data (m/z, 675.2675, [M + Na]+). The NMR data (Table 2) of 3 were similar to those of 2, which indicated that 3 was also a cyclopenta[bc]benzopyran derivative with a benzoyl-1,4-butanediamide moiety28. However, the methine signals of H-10 at δH 4.67 and C-10 (δC 76.1) in 3 showed notable differences from those of 2 (δH 4.19, δC 82.6) (Table 1), which combined with the observed obvious ROESY correlation between H-3 (δH 3.51) and H-10 (δH 4.68) in 3, absent in 2, revealed that 2 and 3 were epimers at C-10.
Aglapervirisin D (4),  −0.3 (c 0.12, MeOH), was obtained as a white amorphous powder and its molecular formula was elucidated to be C38H38N2O8 (m/z 673.2516, [M + Na]+) based on its 13C NMR data and HRESIMS, with one more degree of unsaturation than 2. The similarity between the NMR data (Table 2) of 4 and 2 and the key HMBC correlations between H-3/ C-2″, 6″ and H-4/C-11 suggest that 4 was also a cyclopenta[bc]benzopyran4,27,28 derivative, similar to 2. Compared with 2, the presence of an additional carbonyl group (δC 209.5) and the absence of the characteristic signal of C-10 (δH 4.19, δC 82.6) indicated that 4 was an oxidation derivate at C-10 of 2. This deduction was confirmed by the HMBC correlations between H-4 (δH 4.66) and C-10. The coupling constant J(H-3, H-4) (11.5 Hz)3,27 together with the important ROESY correlation between H-4 to H-2′, 6′ (δH 7.30), made the assignments of H-3α and H-4β. Thus, the new compound 4 was assigned as an oxidation derivate at C-10 of 2.
−0.3 (c 0.12, MeOH), was obtained as a white amorphous powder and its molecular formula was elucidated to be C38H38N2O8 (m/z 673.2516, [M + Na]+) based on its 13C NMR data and HRESIMS, with one more degree of unsaturation than 2. The similarity between the NMR data (Table 2) of 4 and 2 and the key HMBC correlations between H-3/ C-2″, 6″ and H-4/C-11 suggest that 4 was also a cyclopenta[bc]benzopyran4,27,28 derivative, similar to 2. Compared with 2, the presence of an additional carbonyl group (δC 209.5) and the absence of the characteristic signal of C-10 (δH 4.19, δC 82.6) indicated that 4 was an oxidation derivate at C-10 of 2. This deduction was confirmed by the HMBC correlations between H-4 (δH 4.66) and C-10. The coupling constant J(H-3, H-4) (11.5 Hz)3,27 together with the important ROESY correlation between H-4 to H-2′, 6′ (δH 7.30), made the assignments of H-3α and H-4β. Thus, the new compound 4 was assigned as an oxidation derivate at C-10 of 2.
Aglapervirisin E (5) was obtained as a white powder { −4.4 (c 0.28, MeOH)} with a molecular formula of C39H42N2O9 according to the pseudomolecular ion at m/z 705.2784 [M + Na]+ (calcd C39H42N2O9Na, 705.2783). Four sets of signals for benzene-ring, including one monosubstituted, two p-disubstituted and one 1,2,3,5-tetrasubstituted benzene rings, three methoxyl groups in the 1H NMR spectrum and its similar HMBC correlations to those of 2 suggested that 5 was also a typical cyclopenta[bc]benzopyran4,27 derivative. The 1D-NMR data (Table 2) for 5 and 2 showed one more 4-hydroxybenzyl group [δH 7.06 (2H, d, J = 8.5 Hz, δC 129.4), δH 6.84 (2H, d, J = 8.5 Hz, δC 113.9)] and (δH 3.45, δC 43.1), in 5 than in 2, which suggested that a p-hydroxybenzyl group moiety presented in 5 other than a benzoyl group in 2. The obvious HMBC correlations from H-16 (δH 2.95) and H-19 (δH 3.45) to the amide carbonyl carbon (172.0, C-18) placed the p-hydroxybenzyl group at N-17. Thus, the planar structure of 5 was determined as depicted. In the 1H NMR spectrum of 5 (Table 3), an extremely upfield shifted methoxyl signal was observed at δH 3.09. The coupling constant value (8.5 Hz) of J(H-3,H-4) was also differ greatly from that of 2 (6.5 Hz). These data characterized H-3α and H-4β in 53,24,28, respectively, in opposite to those in 2. A key ROESY correlation between H-10 (δH 4.89) and H-3 (δH 3.86) confirmed that 5 had opposite relative configurations at position C-3 and C-4 compared with 2, which also established an endo relationship between H-3 and H-1028. Thus, the structure of 5 was established as shown.
−4.4 (c 0.28, MeOH)} with a molecular formula of C39H42N2O9 according to the pseudomolecular ion at m/z 705.2784 [M + Na]+ (calcd C39H42N2O9Na, 705.2783). Four sets of signals for benzene-ring, including one monosubstituted, two p-disubstituted and one 1,2,3,5-tetrasubstituted benzene rings, three methoxyl groups in the 1H NMR spectrum and its similar HMBC correlations to those of 2 suggested that 5 was also a typical cyclopenta[bc]benzopyran4,27 derivative. The 1D-NMR data (Table 2) for 5 and 2 showed one more 4-hydroxybenzyl group [δH 7.06 (2H, d, J = 8.5 Hz, δC 129.4), δH 6.84 (2H, d, J = 8.5 Hz, δC 113.9)] and (δH 3.45, δC 43.1), in 5 than in 2, which suggested that a p-hydroxybenzyl group moiety presented in 5 other than a benzoyl group in 2. The obvious HMBC correlations from H-16 (δH 2.95) and H-19 (δH 3.45) to the amide carbonyl carbon (172.0, C-18) placed the p-hydroxybenzyl group at N-17. Thus, the planar structure of 5 was determined as depicted. In the 1H NMR spectrum of 5 (Table 3), an extremely upfield shifted methoxyl signal was observed at δH 3.09. The coupling constant value (8.5 Hz) of J(H-3,H-4) was also differ greatly from that of 2 (6.5 Hz). These data characterized H-3α and H-4β in 53,24,28, respectively, in opposite to those in 2. A key ROESY correlation between H-10 (δH 4.89) and H-3 (δH 3.86) confirmed that 5 had opposite relative configurations at position C-3 and C-4 compared with 2, which also established an endo relationship between H-3 and H-1028. Thus, the structure of 5 was established as shown.
The molecular formula of aglapervirisin F (6) { −5.4 (c 0.28, MeOH)} was determined to be C38H40N2O9 by HRESIMS (m/z 691.2625, [M + Na]+, calcd 691.2626), with one CH2 unit less than 5. Its 1H and 13C NMR data, particularly the characteristic methoxyl signal at δH 3.10 and the vicinal coupling constant value (8.5 Hz) of H-4/H-3, resemble greatly with those of 5, which indicated that the basic skeleton of 6 was the same as that of 5. The absence of a characteristic methine (δH 3.45, δC 43.1, C-19) indicated that the p-hydroxyphenylacetyl group at N-17 in 5 was replaced by a 4-hydroxybenzoyl group moiety in 6, as supported also by the observed HMBC correlations from H-16 (δH 3.17) and H-20/24 (δH 7.67) to C-18 (δC 170.0). The ROESY correlations of H-10/H-3 indicated that the relative configuration of 6 was also the same as that of 5. Finally, the structure of 6 was assigned as shown.
−5.4 (c 0.28, MeOH)} was determined to be C38H40N2O9 by HRESIMS (m/z 691.2625, [M + Na]+, calcd 691.2626), with one CH2 unit less than 5. Its 1H and 13C NMR data, particularly the characteristic methoxyl signal at δH 3.10 and the vicinal coupling constant value (8.5 Hz) of H-4/H-3, resemble greatly with those of 5, which indicated that the basic skeleton of 6 was the same as that of 5. The absence of a characteristic methine (δH 3.45, δC 43.1, C-19) indicated that the p-hydroxyphenylacetyl group at N-17 in 5 was replaced by a 4-hydroxybenzoyl group moiety in 6, as supported also by the observed HMBC correlations from H-16 (δH 3.17) and H-20/24 (δH 7.67) to C-18 (δC 170.0). The ROESY correlations of H-10/H-3 indicated that the relative configuration of 6 was also the same as that of 5. Finally, the structure of 6 was assigned as shown.
Aglapervirisin G (7) was obtained as an optically active, white amorphous powder ( −22.1). The molecular formula of 7 was determined to be C38H40N2O8 (m/z 675.2675, [M + Na]+, calcd 675.2677), the same as 2. The 1H NMR resonances of 7 resembled those of 2, including five benzene ring signals, a characteristic singlet for H-10 and two apparent doublets (H-3, H-4), indicated that 7 was an isomer of 2. The key HMBC correlations of H-3 (δH 4.25) /C-2″6″ (δC 129.4), H-2″6″ (δH 6.90) /C-3 (δC 53.8) and H-4 (δH 3.09) /C-11 (δC 169.3) in 7, were not consistent with those of 2, which indicated that the substituents at C-3 and C-4 were mutually exchanged in 73,24,27. Thus, the planar structure of 7 was determined. The relative configurations of H-3α and H-4β were determined based on the vicinal coupling constant (J = 9.0 Hz)28 and the ROESY correlations between H-3/H-2′, 6′, H-3/H-2″, 6″, H-4/H-2″, 6″ and H-4/NH-1224,28. The key cross peak between H-10 and H-4 indicated an endo relationship between H-10 and H-428. Thus, the structure of 7 was proposed as depicted.
−22.1). The molecular formula of 7 was determined to be C38H40N2O8 (m/z 675.2675, [M + Na]+, calcd 675.2677), the same as 2. The 1H NMR resonances of 7 resembled those of 2, including five benzene ring signals, a characteristic singlet for H-10 and two apparent doublets (H-3, H-4), indicated that 7 was an isomer of 2. The key HMBC correlations of H-3 (δH 4.25) /C-2″6″ (δC 129.4), H-2″6″ (δH 6.90) /C-3 (δC 53.8) and H-4 (δH 3.09) /C-11 (δC 169.3) in 7, were not consistent with those of 2, which indicated that the substituents at C-3 and C-4 were mutually exchanged in 73,24,27. Thus, the planar structure of 7 was determined. The relative configurations of H-3α and H-4β were determined based on the vicinal coupling constant (J = 9.0 Hz)28 and the ROESY correlations between H-3/H-2′, 6′, H-3/H-2″, 6″, H-4/H-2″, 6″ and H-4/NH-1224,28. The key cross peak between H-10 and H-4 indicated an endo relationship between H-10 and H-428. Thus, the structure of 7 was proposed as depicted.
Aglapervirisin H (8) was obtained as a colourless powder,  + 96.9 (c 0.10, MeOH), exhibited a sodicated molecular ion at m/z 600.2202 [M + Na]+ (calcd for C32H35NO9Na, 600.2204) in the HRESIMS. The eleven characteristic aromatic protons at δH (7.71−5.78) and 18 carbon signals observed in the 1H and 13C NMR data of 8, indicated that this compound had four aromatic protons and six aromatic carbons less than 5. The presence of a characteristic methoxyl at δH 3.62 and obvious HMBC correlations from H-15 (δH 1.94) and the methoxyl signal (δH 3.62) to a carbonyl carbon at δC 173.4 indicated that the benzoyl-1,4-butanediamide moiety in 2 was replaced by a 4-aminobutanoate methyl ester moiety in 83. The key HMBC correlations (Fig. 5) of the resonances of H-3 (δH 3.88) with C-11 (δC 173.8) and H-4 (δH 4.10) with C-2″, 6″ (δC 128.7) indicated that the basic connection of the core planar structure in 8 was the same as that of 5. The characteristic deshielded shift of the 6-OMe (δH 3.08) signal and the coupling constant J(H-3,H-4) (8.5 Hz) indicated that the relative configurations of 8 at C-3 and C-4 were the same as those of 53,24,28. The absence of a cross peak between H-10 (δH 4.89) and H-3 (δH 3.88) indicated an exo relationship between H-10 and H-328. The absolute configuration of 8 was assigned as 2R, 3R, 4S, 5R and 6S based on the calculated ECD (Fig. 6). Thus, 8 was a new cyclopenta[bc]benzopyran-type4,27 derivative, depicted in Fig. 5.
+ 96.9 (c 0.10, MeOH), exhibited a sodicated molecular ion at m/z 600.2202 [M + Na]+ (calcd for C32H35NO9Na, 600.2204) in the HRESIMS. The eleven characteristic aromatic protons at δH (7.71−5.78) and 18 carbon signals observed in the 1H and 13C NMR data of 8, indicated that this compound had four aromatic protons and six aromatic carbons less than 5. The presence of a characteristic methoxyl at δH 3.62 and obvious HMBC correlations from H-15 (δH 1.94) and the methoxyl signal (δH 3.62) to a carbonyl carbon at δC 173.4 indicated that the benzoyl-1,4-butanediamide moiety in 2 was replaced by a 4-aminobutanoate methyl ester moiety in 83. The key HMBC correlations (Fig. 5) of the resonances of H-3 (δH 3.88) with C-11 (δC 173.8) and H-4 (δH 4.10) with C-2″, 6″ (δC 128.7) indicated that the basic connection of the core planar structure in 8 was the same as that of 5. The characteristic deshielded shift of the 6-OMe (δH 3.08) signal and the coupling constant J(H-3,H-4) (8.5 Hz) indicated that the relative configurations of 8 at C-3 and C-4 were the same as those of 53,24,28. The absence of a cross peak between H-10 (δH 4.89) and H-3 (δH 3.88) indicated an exo relationship between H-10 and H-328. The absolute configuration of 8 was assigned as 2R, 3R, 4S, 5R and 6S based on the calculated ECD (Fig. 6). Thus, 8 was a new cyclopenta[bc]benzopyran-type4,27 derivative, depicted in Fig. 5.

Selected key HMBC and ROESY correlations observed for 8.

Theoretically calculated and experimentally determined ECD of 8.
Aglapervirisin I (9) was obtained as a colourless powder { + 85.4 (c, 0.15, MeOH)} with a molecular formula of C31H35NO8, as deduced from its HRESIMS (m/z 572.2253 [M + Na]+, calcd 572.2255). The 1H and 13C NMR data of 9 were similar to those of 8. The obvious differences between them were the presence of two multiplet protons at δH 3.40 and the absence of a carbonyl carbon and a methoxy group in 9. The above information indicated that the methyl side chain was reduced to a hydroxymethyl group, as confirmed by the key HMBC correlations between H-15 (δH 1.10) and C-16 (δC 62.5). The coupling constant of J(H-3,H-4) (8.5 Hz) and characteristic deshielded shift of the 6-OMe (δH 3.07) signal indicated that the planar structure and relative configuration of 9 were consistent with those of 8. Furthermore, the absolute configuration of 9 was elucidated as 2R, 3R, 4S, 5R and 6S based on its ECD data, which was similar to that of 8. Thus, 9 was elucidated to be a new cyclopenta[bc]benzopyran derivative and was named as aglapervirisin I.
+ 85.4 (c, 0.15, MeOH)} with a molecular formula of C31H35NO8, as deduced from its HRESIMS (m/z 572.2253 [M + Na]+, calcd 572.2255). The 1H and 13C NMR data of 9 were similar to those of 8. The obvious differences between them were the presence of two multiplet protons at δH 3.40 and the absence of a carbonyl carbon and a methoxy group in 9. The above information indicated that the methyl side chain was reduced to a hydroxymethyl group, as confirmed by the key HMBC correlations between H-15 (δH 1.10) and C-16 (δC 62.5). The coupling constant of J(H-3,H-4) (8.5 Hz) and characteristic deshielded shift of the 6-OMe (δH 3.07) signal indicated that the planar structure and relative configuration of 9 were consistent with those of 8. Furthermore, the absolute configuration of 9 was elucidated as 2R, 3R, 4S, 5R and 6S based on its ECD data, which was similar to that of 8. Thus, 9 was elucidated to be a new cyclopenta[bc]benzopyran derivative and was named as aglapervirisin I.
Five known compounds were identified as silvestrol (10) (m/z 655.2387, [M + H]+), episilvestrol (11) (m/z 655.2380, [M + H]+), foveoglin A (12) (m/z 653.2854, [M + H]+), foveoglin B (13) (m/z 653.2856, [M + H]+) and cyclofoveoglin (14) (m/z 651.2701, [M + H]+) based on comparisons of their 1H NMR, 13C NMR and ESIMS data with reported values in the published literatures23,28.
Aglapervirisin A (1) inhibits proliferation of HepG2 cells by causing G2/M-phase arrest in HepG2 cells
The cytotoxicity of all of the isolates (except for 6 and 12) against four human tumour cell lines was tested. Silvestrol analogues (1, 10 and 11) exhibited significant cytotoxicity towards the tested tumor cell lines and that the cyclopenta[bc]benzopyrans (2–5, 7–9, 13, 14) were inactive or only of moderate cytotoxicity (Table 4).
Cell cycle arrest is one of the important factors that influence the proliferation of tumour cells29. Thus, the regulation of the cell cycle of HepG2 cells by aglapervirisin A (1), a new silvestrol analogue possessing significant cytotoxicity, was investigated through flow cytometry analysis. Treatment of HepG2 cells with 1 at concentrations of 40 or 160 nM for 48 h increased percentage of HepG2 cells arrested at the G2/M boundary from 10% to 25% or 35%, respectively. These results indicated that 1 can induce the arrest of HepG2 cells at the G2/M boundary in a dose-dependent manner, similarly to hydroxycamptothecin14,15, as shown in Fig. 7. The western blotting results indicated that 1 down-regulates the expression of Cdc2 and Cdc25C, which may be responsible for the induction of G2/M arrest.

Compound 1 induces cell cycle arrest and apoptotic cell death in HepG2 cells.
(A) Compound 1 induces cell cycle arrest at the G2/M boundary. The cells were treated with vehicle or with 1 at 0 μM, 6.25 μM, 12.5 μM, or 25 μM for 48 h and the cell cycle distribution was assessed by flow cytometry; (B) Percentages of cells in different phases of the cell cycle; (C) The levels of Cdc2 and Cdc25C were measured by western blotting using GAPDH as the loading control. Data are presented as the means ± SD of three experiments. *P < 0.05, **P < 0.01 compared to the control group.
Aglapervirisin A (1) causes HepG2 cell death by apoptosis
The induction of apoptosis is also a mechanism through which antitumour agents exert their therapeutic effects30,31. Thus, the induction of apoptosis by 1 was also examined in this study (Fig. 7). The assay showed that incubation with 1 for 72 h induced the apoptosis of HepG2 cells in a dose-dependent manner. The percentages of apoptotic cells were 6.2% and 14.5% after treatment with 1 at concentrations of 160 and 2560 nM, respectively, as compared with 0.6% in the negative control group. Thus, these results demonstrated that 1 induced HepG2 cell death by apoptosis (Fig. 8).

Analysis of apoptosis in 1-treated HepG2 cells.
(A) The cells were treated with 1 at 160 nM, 640 nM or 2560 nM for 72 h. (B) The numbers of apoptotic cell were calculated by flow cytometry. Data are presented as the means ± SD of three experiments. *P < 0.05, **P < 0.01 compared to the control group.
Conclusion
In summary, this paper describes the isolation and structural elucidation of a rare silvestrol analogue (aglapervirisin A, 1), eight new biosynthetic precursors of rocaglate (cyclopenta[bc]benzopyrans, aglapervirisins A-H, 2–9) and five known analogues isolated from the leaves of A. perviridis. The absolute configurations of 1, 2 and 8 were confirmed through chemical conversion, single-crystal X-ray diffraction and quantum chemical calculations of ECD, respectively. The new silvestrol analogue 1 showed significant cytotoxicity at the nanomolar level against several cancer cell lines and a further mechanistic study indicated that this cytotoxicity was associated with the induction of G2/M phase arrest through reductions in the expression levels of Cdc2 and Cdc25C and the induction of apoptosis. Taken together, these findings indicate that the isolated compounds are potential natural prodrugs with anticancer activity.
Methods
General Experimental Procedures
The optical rotations were measured on a JASCO P-1020 polarimeter at room temperature. The melting points were measured using an X-4 digital display micromelting apparatus and are uncorrected. The IR spectra were recorded on a Bruker Tensor 27 spectrometer using KBr pellets. The 1D- and 2D-NMR spectra were measured on a Bruker AVIII-500 NMR instrument (1H: 500 MHz, 13C: 125 MHz) using TMS as the internal standards. HRESIMS was performed on an Agilent 6529B Q-TOF mass instrument using electrospray ionization. All of the solvents used were of analytical grade (Jiangsu Hanbang Science and Technology. Co., Ltd.). Silica gel (200–300 mesh, Qingdao Haiyang Chemical Co., Ltd, China), Sephadex LH-20 (Pharmacia, Sweden), MCI (Mitsubishi, Japan) and RP-C18 silica (40–63 μm, Fuji, Japan) were used for the column chromatography. Preparative HPLC was conducted using an Agilent 1260 Series instrument with a Shim-Pak RP-C18 column (20 × 200 mm), at a flow rate of 10.0 mL/min and detection by a binary channel UV detector. The fractions obtained from CC were monitored by TLC with precoated silica gel GF254 (Qingdao Haiyang Chemical Co., Ltd, China) plates.
Plant Material
Air-dried leaves of A. perviridis Hiern were collected from Xishuangbanna, Yunnan Province, People’s Republic of China, in June 2013 and were identified by Professor Shun-Cheng Zhang, Xishuangbanna Tropical Botanical Garden, Chinese Academy of Sciences, People’s Republic of China. A voucher specimen (No. AA201308) was deposited in the Department of Natural Medicinal Chemistry, China Pharmaceutical University.
Extraction and Isolation
The air-dried leaves (15.0 kg) were percolated with 95% aqueous ethanol (4 × 80 L) at room temperature. After removal of the solvent under reduced pressure, the crude extract (1510.5 g) was suspended in H2O (1.5 L) and partitioned with petroleum ether (3 × 1 L). The petroleum ether (PE) extract (502.7 g) was suspended in PE and was reverse partitioned with 50% aqueous methanol. The objective extract (100 g) was subjected to a macroporous resin (D101) column and was eluted with water and EtOH (90:10, 50:10, 30:70, 5:95, v/v) to obtain four fractions (A-D). Fraction C (20.5 g) was separated by chromatography on a silica gel column and eluted with a gradient of PE-acetone (20:1 to 5:1, v/v) to yield five fractions (C1-C5). Fraction C1-C5 were then separated by chromatography on a Sephadex LH-20 column and purified by semi-preparative-HPLC with MeOH-H2O, to obtain 1 (10 mg), 2 (50 mg), 3 (30 mg), 4 (10 mg), 5 (5 mg), 6 (6 mg), 7 (3 mg), 8 (2 mg), 9 (5 mg), 10 (18 mg), 11 (17 mg), 12 (300 mg), 13 (12 mg) and 14 (9 mg).
X-ray crystallographic data for 2
C38H40N2O8 (M = 652.72): orthorhombic, space group P212121 (no. 19), a = 7.5679 (3) Å, b = 9.6137 (3) Å, c = 46.6023 (14) Å, V = 3390.6 (2) Å3, Z = 4, T = 291 (2) K, μ (Cu Kα) = 0.734 mm−1, Dcalc = 1.279 g/mm3, 17740 reflections measured (7.588 ≤ 2θ ≤ 139.168), 6251 unique (Rint = 0.0376, Rsigma = 0.0396) which are used in all of the calculations. The final R1 was 0.0477 [I > 2σ (I)] and wR2 was 0.1591 (all data). Flack parameter: −0.07(14).
A colorless crystal of 2 was obtained from a mixture of MeOH and H2O. The crystal data were obtained using a Bruker Smart 1000 CCD with a graphite monochromator, using Cu Kα radiation at 291 (2) K. The crystal was tested with a diffractometer using Olex232 and the structure was solved through direct methods with the ShelXS33 structure solution program and refined with the ShelXL33 refinement package using least squares minimization. The crystallographic data for 2 were deposited in the Cambridge Crystallographic Data Centre (deposition number CCDC 1042328). Copies of these data can be obtained free of charge via the Internet at www.ccdc.cam.ac.uk/conts/retrieving.html or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK [fax (+44) 1223 336 003; e-mail: deposit@ccdc.cam.ac.uk].
Theoretical calculated and experimental observed ECD of 8
The phenomena of ECD have been extensive applied in the determination of the absolute configurations of natural chiral molecules34. The conformations were generated and optimized by Gaussian 09 package35. For the TD calculations Gaussian 09 was used. TDDFT calculations employed the B3LYP functional and the 6-311+g (d, 2p) basis set (Nstates = 40, root = 3). An overall ECD spectrum was generated on the basis of Boltzmann weighting of 5 individual conformers (SI-Table 1) applying a shift based upon the difference between observed and calculated UV spectra. Comparisons of the experimental and calculated spectra were done using SpecDis with UV shift (36 nm) and a half-bandwidth of 0.21 eV. Finally, the calculated ECD spectrum of conformer 4 was adjacent to experimental ECD data. Through comparison with the experimental ECD of 8, the absolute configurations of 8 were assigned as 2R, 3R, 4S, 5R and 6S, respectively.
Determination of Cytotoxic Activities
As reported recently in the literatures, rocaglates exert significant cytotoxic activities24,25,26,27,28. In this investigation, the IC50 values in human leukemic (HL-60), colon cancer (HT-29), human breast cancer (MCF-7) and human liver hepatocellular carcinoma (HepG2) cell lines were determined to evaluate the cytotoxicity of the isolated compounds. The isolated compounds were evaluated based on their cytotoxic activities against the above-mentioned tumor cell lines. The silvestrol analogues exhibited more potent cytotoxic activity than the cyclopenta[bc]benzopyrans, as shown in Table 4. The cytotoxicity assay used in this study is based on the MTT method and was performed in 96-well microplates36. The cells were cultured in DMEM medium (Hyclone, Logan, UT, USA) with 10% foetal bovine serum in an atmosphere with 5% CO2 at 37 °C prior to the assay. Then, 150 μL of the cell suspension was seeded into each well of 96-well the cell culture plates and the cells were allowed to adhere for 12 h before testing. The initial density of the cells was 105/mL. Each tumor cell line was exposed to each test compound at concentrations of 0.001, 0.01, 0.1, 1 and 10 μM in triplicate for 48 h and paclitaxel and cis-platinum were used as positive controls. Using the Reed-Muench method, the IC50 values were calculated based on the obatained cell viability37.
Statistical analysis
Statistical analysis of the data was processed with GraphPad Prism 4.0 software. Statistical analysis of the data was expressed as mean ± SD. Values were analyzed by one-way analysis of variance (ANOVA) using SPSS version 12.0 software. p < 0.05 were considered statistically significant.
Preparation of the acetyl derivatives of 1, 10 and 11
Compounds 1 (1.0 mg), 10 (2.0 mg) and 11 (1.3 mg) were acetylated with acetic anhydride (0.5 mL), CH2Cl2 (1 mL) and DMAP (0.5 mg), respectively, at room temperature for 2 h. The reaction products were purified by semi-preparative-HPLC (MeOH-H2O, 70%) to give compounds 1a (0.5 mg, tR = 8.1 min, 50.0% yield), 10a (1.2 mg, tR = 8.1 min, 60.0% yield) and 11a (0.8 mg, tR = 9.3 min, 61.5% yield).
Aglapervirisin A (1)
colourless powder;  −82.1 (c, 0.11, MeOH); UV (MeOH) λmax (log ε) 209 (4.73), 273 (3.86) nm; IR (KBr) νmax 3424, 2944, 2840, 1620, 1516, 1454, 1385, 1252, 1220, 1201, 1148, 1114, 1049, 1007, 814, 696 cm−1; ECD (0.22 mg/mL, MeOH) λmax (ε) 200 (24.30), 216 (−17.72); 1H and 13C NMR, see Table 1; negative ESIMS m/z 539.8 [M + Cl]−; positive ESIMS m/z 506.0 [M + H]+; HRESIMS m/z 719.2313 [M + Na]+ (calcd for C36H40O14Na, 719.2313).
−82.1 (c, 0.11, MeOH); UV (MeOH) λmax (log ε) 209 (4.73), 273 (3.86) nm; IR (KBr) νmax 3424, 2944, 2840, 1620, 1516, 1454, 1385, 1252, 1220, 1201, 1148, 1114, 1049, 1007, 814, 696 cm−1; ECD (0.22 mg/mL, MeOH) λmax (ε) 200 (24.30), 216 (−17.72); 1H and 13C NMR, see Table 1; negative ESIMS m/z 539.8 [M + Cl]−; positive ESIMS m/z 506.0 [M + H]+; HRESIMS m/z 719.2313 [M + Na]+ (calcd for C36H40O14Na, 719.2313).
Aglapervirisin B (2)
colourless crystals (MeOH); mp: 168–169 °C;  −28.3 (c, 0.10, MeOH); UV (MeOH) λmax (log ε) 204 (4.85), 270 (3.49) nm; IR (KBr) νmax 3485, 2933, 1633, 1532, 1455, 1200, 1150, 1100, 1050, 814, 702 cm−1; ECD (0.2 mg/mL, MeOH) λmax (ε) 208 (−9.15), 226 (4.07); 1H and 13C NMR, see Table 2; negative ESIMS m/z 687.7 [M + Cl]−; positive ESIMS m/z 653.4 [M + H]+; HRESIMS m/z 653.2853 [M + H]+(calcd for C38H41N2O8, 653.2857); m/z 675.2672 [M + Na]+(calcd for C38H40N2O8Na, 675.2677).
−28.3 (c, 0.10, MeOH); UV (MeOH) λmax (log ε) 204 (4.85), 270 (3.49) nm; IR (KBr) νmax 3485, 2933, 1633, 1532, 1455, 1200, 1150, 1100, 1050, 814, 702 cm−1; ECD (0.2 mg/mL, MeOH) λmax (ε) 208 (−9.15), 226 (4.07); 1H and 13C NMR, see Table 2; negative ESIMS m/z 687.7 [M + Cl]−; positive ESIMS m/z 653.4 [M + H]+; HRESIMS m/z 653.2853 [M + H]+(calcd for C38H41N2O8, 653.2857); m/z 675.2672 [M + Na]+(calcd for C38H40N2O8Na, 675.2677).
Aglapervirisin C (3)
colourless powder;  −22.1 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 203 (4.73), 269 (3.43) nm; IR (KBr) νmax 3449, 2348, 1629, 1513, 1463, 1385, 1027, 915, 672 cm−1; ECD (0.2 mg/mL, MeOH) λmax (ε) 212 (−0.83), 227 (0.24); 1H and 13C NMR, see Table 2; negative ESIMS m/z 687.5 [M + Cl]−; positive ESIMS m/z 653.5[M + H]+; HRESIMS m/z 653.2861 [M + H]+(calcd for C38H41N2O8, 653.2857); m/z 675.2675 [M + Na]+(calcd for C38H40N2O8Na, 675.2677).
−22.1 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 203 (4.73), 269 (3.43) nm; IR (KBr) νmax 3449, 2348, 1629, 1513, 1463, 1385, 1027, 915, 672 cm−1; ECD (0.2 mg/mL, MeOH) λmax (ε) 212 (−0.83), 227 (0.24); 1H and 13C NMR, see Table 2; negative ESIMS m/z 687.5 [M + Cl]−; positive ESIMS m/z 653.5[M + H]+; HRESIMS m/z 653.2861 [M + H]+(calcd for C38H41N2O8, 653.2857); m/z 675.2675 [M + Na]+(calcd for C38H40N2O8Na, 675.2677).
Aglapervirisin D (4)
colourless powder;  −3.4 (c 0.42, MeOH); UV (MeOH) λmax (log ε) 204 (4.85) nm; IR (KBr) νmax 3462, 2930, 1732, 1621, 1517, 1455, 1256, 1149, 1087, 1023, 833, 701 cm−1; 1H and 13C NMR, see Table 2; negative ESIMS m/z 685.5 [M + Cl]−; positive ESIMS m/z 651.5 [M + H]+; HRESIMS m/z 651.2694 [M + H]+ (calcd for C38H39N2O8, 651.2701); m/z 673.2516 [M + Na]+ (calcd for C38H38N2O8Na, 673.2520).
−3.4 (c 0.42, MeOH); UV (MeOH) λmax (log ε) 204 (4.85) nm; IR (KBr) νmax 3462, 2930, 1732, 1621, 1517, 1455, 1256, 1149, 1087, 1023, 833, 701 cm−1; 1H and 13C NMR, see Table 2; negative ESIMS m/z 685.5 [M + Cl]−; positive ESIMS m/z 651.5 [M + H]+; HRESIMS m/z 651.2694 [M + H]+ (calcd for C38H39N2O8, 651.2701); m/z 673.2516 [M + Na]+ (calcd for C38H38N2O8Na, 673.2520).
Aglapervirisin E (5)
colourless powder;  −4.4 (c 0.28, MeOH); UV (MeOH) λmax (log ε) 203 (4.82), 273 (3.65) nm; IR (KBr) νmax 3462, 2936, 1623, 1518, 1457, 1386, 1256, 1203, 1151, 1088, 835, 702 cm−1; 1H and 13C NMR, see Table 3; negative ESIMS m/z 717.5 [M + Cl]−; positive ESIMS m/z 683.5 [M + H]+; HRESIMS m/z 683.2964 [M + H]+ (calcd for C39H43N2O9, 683.2963); m/z 705.2784 [M + Na]+ (calcd for C38H40N2O8Na, 705.2783).
−4.4 (c 0.28, MeOH); UV (MeOH) λmax (log ε) 203 (4.82), 273 (3.65) nm; IR (KBr) νmax 3462, 2936, 1623, 1518, 1457, 1386, 1256, 1203, 1151, 1088, 835, 702 cm−1; 1H and 13C NMR, see Table 3; negative ESIMS m/z 717.5 [M + Cl]−; positive ESIMS m/z 683.5 [M + H]+; HRESIMS m/z 683.2964 [M + H]+ (calcd for C39H43N2O9, 683.2963); m/z 705.2784 [M + Na]+ (calcd for C38H40N2O8Na, 705.2783).
Aglapervirisin F (6)
colourless powder;  −5.4 (c 0.28, MeOH); UV (MeOH) λmax (log ε) 210 (4.58) nm; IR (KBr) νmax 3445, 2925, 2853, 2351, 1623, 1507, 1456, 1385, 1149, 823, 697, 663 cm−1; 1H and 13C NMR, see Table 3; negative ESIMS m/z 703.2 [M + Cl]−; positive ESIMS m/z 669.2 [M + H]+; HRESIMS m/z 669.2801 [M + H]+ (calcd for C38H41N2O9, 669.2807); m/z 691.2625 [M + Na]+ (calcd for C38H40N2O9Na, 691.2626).
−5.4 (c 0.28, MeOH); UV (MeOH) λmax (log ε) 210 (4.58) nm; IR (KBr) νmax 3445, 2925, 2853, 2351, 1623, 1507, 1456, 1385, 1149, 823, 697, 663 cm−1; 1H and 13C NMR, see Table 3; negative ESIMS m/z 703.2 [M + Cl]−; positive ESIMS m/z 669.2 [M + H]+; HRESIMS m/z 669.2801 [M + H]+ (calcd for C38H41N2O9, 669.2807); m/z 691.2625 [M + Na]+ (calcd for C38H40N2O9Na, 691.2626).
Aglapervirisin G (7)
colourless powder;  −35.8 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 203 (4.82), 273 (3.65) nm; IR (KBr) νmax 3446, 2932, 1620, 1457, 1385, 1147, 1099, 1029, 830, 699 cm−1; ECD (0.22 mg/mL, MeOH) λmax (ε) 214 (0.69), 236 (−1.12), 243 (−1.41); 1H and 13C NMR, see Table 2; negative ESIMS m/z 687.3 [M + Cl]−; positive ESIMS m/z 653.3 [M + H]+; HRESIMS m/z 653.2859 [M + H]+ (calcd for C38H41N2O8, 653.2857); m/z 675.2671 [M + Na]+ (calcd for C38H40N2O8Na, 675.2677).
−35.8 (c 0.11, MeOH); UV (MeOH) λmax (log ε) 203 (4.82), 273 (3.65) nm; IR (KBr) νmax 3446, 2932, 1620, 1457, 1385, 1147, 1099, 1029, 830, 699 cm−1; ECD (0.22 mg/mL, MeOH) λmax (ε) 214 (0.69), 236 (−1.12), 243 (−1.41); 1H and 13C NMR, see Table 2; negative ESIMS m/z 687.3 [M + Cl]−; positive ESIMS m/z 653.3 [M + H]+; HRESIMS m/z 653.2859 [M + H]+ (calcd for C38H41N2O8, 653.2857); m/z 675.2671 [M + Na]+ (calcd for C38H40N2O8Na, 675.2677).
Aglapervirisin H (8)
colourless powder;  +96.9 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 203 (4.73), 273 (3.54), 279 (3.59) nm; IR (KBr) νmax 3443, 2923, 1751, 1644, 1546, 1384, 1235, 1151, 1029, 703 cm−1; ECD (0.2 mg/mL, MeOH) λmax (ε) 200 (10.50), 218 (1.83), 242 (8.15); 1H and 13C NMR, see Table 3; negative ESIMS m/z 612.4 [M + Cl]−; positive ESIMS m/z 578.4 [M + H]+; HRESIMS m/z 578.2381 [M + H]+ (calcd for C32H36NO9, 578.2385); m/z 600.2202 [M + Na]+ (calcd for C32H35NO9Na, 600.2204).
+96.9 (c 0.10, MeOH); UV (MeOH) λmax (log ε) 203 (4.73), 273 (3.54), 279 (3.59) nm; IR (KBr) νmax 3443, 2923, 1751, 1644, 1546, 1384, 1235, 1151, 1029, 703 cm−1; ECD (0.2 mg/mL, MeOH) λmax (ε) 200 (10.50), 218 (1.83), 242 (8.15); 1H and 13C NMR, see Table 3; negative ESIMS m/z 612.4 [M + Cl]−; positive ESIMS m/z 578.4 [M + H]+; HRESIMS m/z 578.2381 [M + H]+ (calcd for C32H36NO9, 578.2385); m/z 600.2202 [M + Na]+ (calcd for C32H35NO9Na, 600.2204).
Aglapervirisin I (9)
colourless powder;  +85.4 (c, 0.15, MeOH); UV (MeOH) λmax (log ε) 205 (4.69), 210 (4.69), 273 (3.27), 279 (3.17) nm; IR (KBr) νmax 3446, 2931, 1623, 1519, 1457, 1149, 1007, 841, 701 cm−1; ECD (0.3 mg/mL, MeOH) λmax (ε) 200 (14.90), 217 (2.05), 242 (9.90); 1H and 13C NMR, see Table 3; negative ESIMS m/z 584.4 [M + Cl]−; positive ESIMS m/z 550.4 [M + H]+; HRESIMS m/z 550.2431 [M + H]+ (calcd for C31H36NO8, 550.2435); m/z 572.2253 [M + Na]+ (calcd for C31H35NO8Na, 572.2255).
+85.4 (c, 0.15, MeOH); UV (MeOH) λmax (log ε) 205 (4.69), 210 (4.69), 273 (3.27), 279 (3.17) nm; IR (KBr) νmax 3446, 2931, 1623, 1519, 1457, 1149, 1007, 841, 701 cm−1; ECD (0.3 mg/mL, MeOH) λmax (ε) 200 (14.90), 217 (2.05), 242 (9.90); 1H and 13C NMR, see Table 3; negative ESIMS m/z 584.4 [M + Cl]−; positive ESIMS m/z 550.4 [M + H]+; HRESIMS m/z 550.2431 [M + H]+ (calcd for C31H36NO8, 550.2435); m/z 572.2253 [M + Na]+ (calcd for C31H35NO8Na, 572.2255).
Compound 1a: white amorphous powder.  −44.4 (c, 0.05, MeOH); 1H NMR (CDCl3, 500 MHz), see Figure S10-1 in Supporting Information. HRESIMS m/z [M + NH4]+ 756.2857 (calcd for C38H46O15N, 756.2862).
−44.4 (c, 0.05, MeOH); 1H NMR (CDCl3, 500 MHz), see Figure S10-1 in Supporting Information. HRESIMS m/z [M + NH4]+ 756.2857 (calcd for C38H46O15N, 756.2862).
Compound 10: white amorphous powder. 1H NMR (CDCl3, 500 MHz), see Figure S11-1 in Supporting Information. HRESIMS m/z [M + H]+ 655.2387 (calcd for C34H39O13, 655.2385).
Compound 10a: white amorphous powder.  −52.9 (c, 0.12, MeOH); 1H NMR (CDCl3, 500 MHz), see Figure S11-3 in Supporting Information. HRESIMS m/z [M + NH4]+ 756.2864 (calcd for C38H46O15N, 756.2862).
−52.9 (c, 0.12, MeOH); 1H NMR (CDCl3, 500 MHz), see Figure S11-3 in Supporting Information. HRESIMS m/z [M + NH4]+ 756.2864 (calcd for C38H46O15N, 756.2862).
Compound 11: white amorphous powder. 1H NMR (CDCl3, 500 MHz), 13C NMR (CDCl3, 125 MHz), see Figure S12-1 in Supporting Information. HRESIMS m/z [M + H]+ 655.2380 (calcd for C34H39O13, 655.2385).
Compound 11a: white amorphous powder.  −61.5 (c, 0.08, MeOH); 1H NMR (CDCl3, 500 MHz), see Figure S12-3 in Supporting Information. HRESIMS m/z [M + NH4]+ 756.2859 (calcd for C38H46O15N, 756.2862).
−61.5 (c, 0.08, MeOH); 1H NMR (CDCl3, 500 MHz), see Figure S12-3 in Supporting Information. HRESIMS m/z [M + NH4]+ 756.2859 (calcd for C38H46O15N, 756.2862).
Compound 12: white amorphous powder. 1H NMR (CDCl3, 500 MHz), 13C NMR (CDCl3, 125 MHz), see Figure S13 in Supporting Information. HRESIMS m/z [M + H]+ 653.2854 (calcd for C38H41N2O8, 653.2857).
Compound 13: white amorphous powder. 1H NMR (CDCl3, 500 MHz), 13C NMR (CDCl3, 125 MHz), see Figure S14 in Supporting Information. HRESIMS m/z [M + H]+ 653.2856 (calcd for C38H41N2O8, 653.2857).
Compound 14: white amorphous powder. 1H NMR (CDCl3, 500 MHz), 13C NMR (CDCl3, 125 MHz), see Figure S15 in Supporting Information. HRESIMS m/z [M + H]+ 651.2703 (calcd for C38H39N2O8, 651.2701).
Additional Information
How to cite this article: An, F.-L. et al. Cytotoxic Rocaglate Derivatives from Leaves of Aglaia perviridis. Sci. Rep. 6, 20045; doi: 10.1038/srep20045 (2016).
References
Proksch, P., Edrada, R. A., Ebel, R., Bohnenstengel, F. I. & Nugroho, B. W. Chemistry and biological activity of rocaglamide derivatives and related compounds in Aglaia species (Meliaceae). Curr. Org. Chem. 5, 923–938 (2001).
Kim, S., Salim, A. A., Swanson, S. M. & Kinghorn, A. D. Potential of cyclopenta[b]benzofurans from Aglaia species in cancer chemotherapy. Anti-Cancer Agents Med. Chem. 6, 319–345 (2006).
Ribeiro, N., Thuaud, F., Nebigil, C. & Désaubry, L. Recent advances in the biology and chemistry of the flavaglines. Bioorg. Med. Chem. 20, 1857–1864 (2012).
Pan, L. et al. Rocaglamide, silvestrol and structurally related bioactive compounds from Aglaia species. Nat. Prod. Rep. 31, 924–939 (2014).
An, F. L. et al. Cytotoxic flavonol-diamide [3 + 2] adducts from the leaves of Aglaia odorata. Tetrahedron 71, 2450–2457 (2015).
Ohse, T. et al. Cyclopentabenzofuran lignan protein synthesis inhibitors from Aglaia odorata. J. Nat. Prod. 59, 650–652 (1996).
Lee, S. K. et al. Cytostatic mechanism and antitumor potential of novel 1h-cyclopenta[b]benzofuran lignans isolated from Aglaia elliptica. Chem.-Biol. Interact 115, 215–228 (1998).
Gerard, B., Cencic, R., Pelletier, J. & Porco, Jr. J. A. Enantioselective synthesis of the complex rocaglate (−)-silvestrol. Angew. Chem. Int. Ed. 46, 7831–7834 (2007).
Bordeleau, M. E. et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J. Clin. Invest. 118, 2651–2660 (2008).
Cencic, R. et al. Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS One 4, e5223 (2009)
Lucas, D. M. et al. The novel plant-derived agent silvestrol has b-cell selective activity in chronic lymphocytic leukemia and acute lymphoblastic leukemia in vitro and in vivo. Blood 113, 4656–4666 (2009)
Santagata, S. et al. Tight coordination of protein translation and hsf1 activation supports the anabolic malignant state. Science 341, 242–243 (2013).
Kogure, T. et al. Therapeutic potential of the translation inhibitor silvestrol in hepatocellular cancer. PLoS One 9, e76136 (2013).
Bohnenstengel, F. I. et al. 1H-cyclopenta[b]benzofuran lignans from Aglaia species inhibit cell proliferation and alter cell cycle distribution in human monocytic leukemia cell lines Z. Naturforsch. [C] 54, 1075–1083 (1999).
Bohnenstengel, F. I. et al. Structure activity relationships of antiproliferative rocaglamide derivatives from Aglaia species (Meliaceae). Z. Naturforsch. [C] 54, 55–60 (1999).
Adams, T. E. et al. Nazarov Cyclization Initiated by Peracid Oxidation: The total synthesis of (±)-rocaglamide J. Am. Chem. Soc. 131, 1607–1616 (2009).
Magnus, P., Freund, W. A., Moorhead, E. J. & Rainey, T. Formal synthesis of (±)-methyl rocaglate using an unprecedented acetyl bromide mediated nazarov reaction J. Am. Chem. Soc. 134, 6140–6142 (2012).
Malona, J. A., Cariou, K., Spencer, W. T., III & Frontier, A. J. Total synthesis of (±)-rocaglamide via oxidation-initiated nazarov cyclization. J. Org. Chem. 77, 1891–1908 (2012).
Chambers, J. M. et al. Total synthesis of 2′′′,5′′′ -diepisilvestrol and its C1′′′ epimer: key structure activity relationships at C1′′′ and C2′′′. J. Nat. Prod. 75(8), 1500–1504 (2012).
Yang, S. M., Fu, W. W., Wang, D. X., Tan, C. H. & Zhu, D. Y., Two new pregnanes from Aglaia perviridis Hiern. J. Asian Nat. Prod. Res. 10, 459–462 (2008).
Zhang, L. et al. Chemical constituents from the leaves of Aglaia perviridis. J. Asian Nat. Prod. Res. 12, 215–219 (2010).
Pan, L. et al. Bioactive flavaglines and other constituents isolated from Aglaia perviridis. J. Nat. Prod. 76, 394–404 (2013).
Hwang, B. Y. et al. Silvestrol and episilvestrol, potential anticancer rocaglate derivatives from Aglaia silvestris. J. Org. Chem. 69, 3350–3358 (2004).
Dumontet, V. et al. New nitrogenous and aromatic derivatives from Aglaia argentea and A. forbesii. Tetrahedron 52, 6931–6942 (1996).
Nugroho, B. W. et al. Insecticidal rocaglamide derivatives from Aglaia elliptica and a. harmsiana. Phytochemistry 49, 1579–1585 (1997).
Nugroho, B. W. et al. An insecticidal rocaglamide derivatives and related compounds from Aglaia odorata (Meliaceae). Phytochemistry 51, 367–376 (1999).
Bacher, M., Hofer, O., Brader, G., Vajrodaya, S. & Greger, H., Thapsakins: possible biogenetic intermediates towards insecticidal cyclopenta[b]benzofurans from Aglaia edulis. Phytochemistry 52, 253–263 (1999).
Salim, A. A. et al. Constituents of the leaves and stem bark of Aglaia foveolata. Tetrahedron 63, 7926–7934 (2007).
Mhaidat, N. M., Wang, Y., Kiejda, K. A., Zhang, X. D. & Hersey, P. M., Docetaxel-induced apoptosis in melanoma cells is dependent on activation of caspase-2. Mol Cancer Ther. 6, 752–761 (2007).
Call, J. A., Eckhardt, S. G. & Camidge, D. R. Targeted manipulation of apoptosis in cancer treatment. Lancet Oncol. 9, 1002–1011 (2008).
Townson, J. L., Naumov, G. N. & Chambers, A. F., The role of apoptosis in tumor progression and metastasis Curr. Mol. Med. 3, 631–642 (2003).
Dolomanov, O. V. et al. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst. 42, 339–334 (2009).
Sheldrick, G. M., A short history of SHELX, Acta Cryst. A64, 112–122 (2008).
Berova, N., Bari L. Di & Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chemical Society reviews 36, 914–931 (2007).
Tomasi, J., Mennucci, B. & Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3093 (2005).
Luo, Z. et al. Cytotoxic alkaloids from the whole plants of Zephyranthes candida. J. Nat. Prod. 75, 2113–2120 (2012).
Lin, Z. M. et al. Diterpenoids from the chinese Liverwort heteroscyphus tener and their antiproliferative effects. J. Nat. Prod. 77, 1336–1340 (2014).
Acknowledgements
This research was supported in part by the National Natural Science Foundation of China (21272275, 81430092), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and by the Program for Changjiang Scholars and Innovative Research Team in University (IRT1193). The theoretical calculations were conducted on the ScGrid of the Supercomputing Centre, Computer Network Information Centre of Chinese Academy of Sciences.
Author information
Authors and Affiliations
Contributions
K.L.Y., L.J. and A.F.L. designed the phytochemical and biological experiments. W.X.B. designed and conducted the density functional theory calculations. A.F.L. conducted the isolation of compounds and analyzed N.M.R. and M.S. data. A.F.L. and L.J. wrote the paper. W.H. and L.Z.R. conducted the biological experiments. Y.M.H measured the NMR data.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
An, FL., Wang, XB., Wang, H. et al. Cytotoxic Rocaglate Derivatives from Leaves of Aglaia perviridis. Sci Rep 6, 20045 (2016). https://doi.org/10.1038/srep20045
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep20045
This article is cited by
-
Comparative phytochemistry of flavaglines (= rocaglamides), a group of highly bioactive flavolignans from Aglaia species (Meliaceae)
Phytochemistry Reviews (2022)
-
Oxo-aglaiastatin-Mediated Inhibition of Translation Initiation
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
