
Abstract
Arrestin domain-containing 3 (ARRDC3) is a tumor suppressor whose expression is either lost or suppressed in basal-like breast cancer (BLBC). However, the mechanism by which BLBC suppresses ARRDC3 expression is not established. Here, we show that expression of ARRDC3 in BLBC cells is suppressed at the transcriptional level. Suppression of ARRDC3 expression in BLBC cells involves epigenetic silencing as inhibitors of class III histone deacetylases (HDACs) significantly restores ARRDC3 levels in BLBC cells. SIRT2, among class III HDACs, plays a major role in epigenetic silencing of ARRDC3 in MDA-MB-231 cells. Acetylation levels of the ARRDC3 promoter in BLBC cells is significantly lower than that of other sub-types of BC cells. Chromatin immunopreciptitation analysis established SIRT2 binding at ARRDC3 promoter in BLBC cells. Our studies indicate that SIRT2 dependent epigenetic silencing of ARRDC3 is one of the important events that may contribute to the aggressive nature of BLBC cells.
Similar content being viewed by others


Introduction
Breast cancer is a heterogeneous disease with distinct morphology and behavior1,2,3. Microarray gene expression analysis defines four distinct sub-types of breast cancer, including hormone receptor (HR) positive luminal A and B, human epidermal growth receptor 2 (HER2/neu)-enriched and basal-like breast cancer (BLBC)4,5. Among these breast cancer sub-types, BLBC represents up to 37% of all breast cancers and is one of the most aggressive breast cancer sub-types with poor prognosis6,7,8,9. Approximately 80% of BLBC lacks expression of hormone (estrogen and progesterone) receptors and human epidermal growth receptor 2 (HER-2), which has been a conventional target of breast cancer therapy10,11,12. Consequently, there is no targeted therapy available for patients with the aggressive BLBC subtype. Therefore, dissecting the basic mechanism behind BLBC's aggressive behavior is essential to develop novel target-specific therapy.
In an attempt to establish a better biological mechanism of BLBCs, earlier studies focused on discovering novel therapeutic target genes13,14. The analyses using proteomic, genomic or gene expression profiling revealed potential candidate oncogenes and tumor suppressor genes associated with BLBCs15,16,17. Moreover, combined genome copy number analysis and gene expression profiles showed that the loss of chromosomal regions such as 4p, 5q, 17p and 8p is associated with down-regulation of several tumor suppressor genes in BLBCs16. Another mechanism of the aberrant gene losses in BLBC could be interconnected with epigenetic alterations18. Recent studies showed that epigenetic alterations occur frequently in many human malignancies18. For example, DNA hypermethylation by DNA methyltransferases (DNMTs) and histone deacetylation by histone deacetylases (HDACs) within promoters of tumor suppressor genes leads to undesirable gene silencing19,20,21. Among the mammalian HDACs, SIRT2, an NAD+-dependent protein deacetylase belongs to class III HDACs22. SIRT2 has been shown to be involved in cell survival through deacetylation of α-tubulin, p53, p65, Foxo-1 and -322,23,24,25,26,27 in mammalian cells. However, the role of SIRT2 in cancer has not been established.
One of the tumor suppressor genes whose levels are either low or lost in BLBC is α-Arrestin domain containing 3 (ARRDC3)16. A recent report showed that ARRDC3 negatively regulates integrin β4 signaling by inducing degradation of this integrin in MDA-MB-231 cells28. Another study showed that ARRDC3 suppresses activated β2-adrenergic receptors through the ubiquitination of this receptor by its recruitment with E3 ligase, NEDD4, which further supports the role of ARRDC3 as a tumor suppressor29. Therefore, it is possible to reason that low levels of ARRDC3 in BLBC could contribute to malignancy. However, the mechanisms by which BLBC cells suppress ARRDC3 expression remain to be established. Here, we demonstrate that ARRDC3 is epigenetically silenced in BLBC cells due to its promoter deacetylation via SIRT2. Our studies suggest that SIRT2 dependent epigenetic silencing of ARRDC3 provides one of the molecular signatures that make BLBC aggressive.
Results
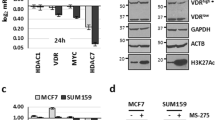
A previous report that ARRDC3 expression inversely correlates with integrin β4 expression by inducing degradation of phosphorylated integrin β4 suggest its role as a tumor suppressor28. However, underlying molecular mechanism by which ARRDC3 expression is regulated in breast cancer cells has yet to be defined. To address this issue, we screened integrin β4 and ARRDC3 expression levels in various sub-types of breast carcinoma cell lines by Western blot. As shown in Figure 1a, ARRDC3 level in BLBC cell lines is significantly lower than those of luminal or Her2 enriched subtype of breast carcinoma cells. In contrast, the level of integrin β4 is much higher in BLBC cells compared to other sub types (Fig. 1a). To assess the mechanisms by which BLBC cells inhibit the expression of ARRDC3, we examined the possibility that ARRDC3 expression is regulated at the transcriptional level. We performed quantitative real-time PCR and found that the endogenous ARRDC3 mRNA level is significantly lower (~3 fold) in BLBC cell line (MDA-MB-231) than that in the luminal BC cell line (MCF-7) (Fig. 1b). We next evaluated the transcriptional activity of ARRDC3 by using a luciferase reporter assay. The transcriptional activity of ARRDC3 promoter (−25 to −2040) is significantly lower (~10 fold) in BLBC cell line (MDA-MB-231) compared with that in Luminal BC cell line (MCF-7) (Fig. 1c). These results suggest that the difference of ARRDC3 levels between BLBC and luminal BC cell lines is likely due to transcriptional regulation.

ARRDC3 expression is suppressed at the transcription level in BLBC cell lines.
(a) Whole cell lysates were prepared from the indicated cell lines. Equal amounts of extracts from each sample were used for Western blot analysis by using antibodies against β4 integrin and ARRDC3. β-Actin was used as loading control. Full-length blots are presented in Supplementary Fig. (b) Total RNAs were isolated from the indicated cell lines. Qquantitative real-time RT-PCR was performed to determine relative changes in ARRDC3 mRNA transcripts. Data were analyzed using CFX Manager™ Software (Bio-Rad). Samples were normalized with GAPDH as reference gene. Values represent mean ± SD of three independent experiments performed in duplicate in all experiments. The statistical analysis was done using Student's t test. *, P < 0.05 (c) Luciferase reportor assay was performed in MDA-MB-231 and MCF-7 cells transiently transfected with pGL4-empty and pGL4-P (−2040/−25) plasmid. The luciferase activities were normalized with β-galactosidase activities. Columns, mean of three representative experiments performed in triplicate; bars, SD. The statistical analysis was done using Student's t test. *, P < 0.05; **, P < 0.01 compared with the results of the control cells (pGL4-empty).
We then tested whether suppression of ARRDC3 transcription in BLBC cells is related to epigenetic silencing. DNA methylation and histone deacetylation are two major epigenetic mechanisms that lead to gene silencing in cancer21,30. To address this possibility, MDA-MB-231cells were treated with the histone deacetylase (HDAC) inhibitors (nicotinamide (NIA) and trichostatin A (TSA)) or the DNA methyltransferase (DNMT) inhibitor (5-aza-2′-deoxycytidine (5-Aza)). Inhibition of class III HDACs (NIA), but not class I and II HDACs (TSA) restored the expression of ARRDC3 in MDA-MB-231 cells (BLBC cell line) (Fig. 2a). Neither of these inhibitors affected the ARRDC3 level in MCF-7 cells (luminal BC cell line) whose basal ARRDC3 level is already high (Fig. 2a). To investigate the involvement of DNA methylation in epigenetic silencing of ARRDC3, MDA-MB-231 cells were treated with 5-Aza in various concentrations (Fig. 2b). 200 nM of 5-Aza resulted in a mild increase of ARRDC3 expression in MDA-MB-231 cells (Fig. 2b). In contrast to the weak restoration of ARRDC3 expression by 5-Aza alone, ARRDC3 level was synergistically restored by combination of 5-Aza with NIA, but not with TSA (Fig. 2c). These data suggest that class III HDAC(s) play a major role in epigenetic silencing of ARRDC3 and DNA methylation may have a supportive role for class III HDAC function.

ARRDC3 expression is suppressed epigenetically in BLBC cells.
(a) MDA-MB-231(top panel) and MCF-7 cells (bottom panel) were treated with DMSO or EtOH (control) and HDAC inhibitors (NIA or TSA) for 24 hr at indicated concentrations. (b) MDA-MB-231(upper panel) and MCF-7cells (lower panel) were treated with DMSO (control) and DNMT inhibitor (5-Aza-dC) for 48 hr at indicated concentrations. (c) MDA-MB-231cells were pre-treated with DMSO or DNMT inhibitor, 5-Aza-dC (200 nM and 500 nM) for 24 hr, followed by HDAC inhibitors (5 mM NIA and 1 μM TSA) treatment for 24 hr. The lysates from cells mentioned above were prepared and used for Western blotting analysis with anti-ARRDC3 and β-actin (loading control) antibodies. All western blot results presented were carried out at least 3 times.
Restoration of ARRDC3 level by nicotinamide (NIA) in MDA-MB-231 cells suggests that class III HDACs are likely contributors of ARRDC3 suppression in BLBC cells. Class III HDACs includes seven sirtuin family members in mammalian cells (SIRT1 through SIRT722). To narrow down the candidates of class III HDACs that regulate ARRDC3 expression, we treated MDA-MB-231 cells with cambinol, which selectively inhibits both SIRT1 and SIRT231. Treatment with cambinol restores the levels of ARRDC3 in MDA-MB-231 cells (Fig. 3a), indicating that either SIRT1 or SIRT2 or both could be involved in epigenetic suppression of ARRDC3. Then, we stably knockdown expression of SIRT1 or SIRT2 by shRNA and found that knockdown of SIRT2, but not SIRT1, restores ARRDC3 levels in MDA-MB-231 cells (Fig. 3b). The knockdown of SIRT2 by shRNA also significantly reduced the integrin β4 expression levels by 40% (Fig. 3b). The results led to the conclusion that SIRT2 is one of the determinants that regulates the expression of ARRDC3 in MDA-MB-231 cells. We then tested the role of SIRT2 in pro-invasive migration of BLBC cells. As shown in Fig. 3c, inhibition of SIRT2 activity by cambinol or reduction of SIRT2 by shRNA or ARRDC3 overexpression significantly blocks cell motility towards LPA in MDA-MB-231 cells, suggesting that SIRT2 could be an important therapeutic target of BLBC.

SIRT2 plays a major role in epigenetic silencing of ARRDC3 expression in BLBC cells.
(a) MDA-MB-231 cells were treated with DMSO (control) and SIRT1/2 inhibitor, cambinol for 24 hr at the indicated concentrations. Equal amounts of extracts from each sample were used for Western blot analysis using antibodies against SIRT1, SIRT2, ARRDC3 and β-actin (loading control). (b) Whole cell lysates were prepared from MDA-MB-231 cells expressing GFP or SIRT1 (left panel) and SIRT2 (right panel) shRNA. The expression level of SIRT1, SIRT2 b4 integrin and ARRDC3 was detected by Western blot analysis. β-actin levels were used as loading control. All results are representative of three independent experiments. The number given underneath gel image represents fold change compared with control cells. Densitometric analysis was performed to measure the relative intensity of the bands from Western blotting analysis and presented in arbitrary units (right). Statistical analysis was done using Student's t test. **, P < 0.01 comparing to the control (GFP shRNA cells). (c) The ability of cells to migrate toward 100 nM LPA was measured using a transwell cell motility assay. Migration was quantified by counting the cells that migrated to the lower surface of the membrane per square milliliter using bright-field optics. Columns, mean of three representative experiments performed in triplicate; bars, SD. The statistical analysis was done using Student's t test. *, P < 0.05; **, P < 0.01 compared with the results of the control cells.
Based on the result that ARRDC3 level in MDA-MB-231 cells is synergistically restored by HDAC III and DNMT inhibitors (Fig. 2c), we compared the levels of methylation and acetylation of ARRDC3 promoters in MDA-MB-231 (BLBC) and MCF-7 (luminal BC) cells (Fig. 4a). We first measured the methylation status within CpG islands of the ARRDC3 promoter by using methylation-specific PCR. We were not able to detect methylation in the ARRDC3 promoter region examined in both of these cell lines (Fig. 4a). This result suggests that epigenetic silencing of ARRDC3 is not directly related to hypermethylation of its promoter. We then assessed acetylation status of the ARRDC3 promoter by using chromatin immunoprecipitation (ChIP) assays (Fig. 4b). The acetylation level of histone H4 (Lys16) was examined as this residue has been reported to be a major target of histone deacetylation by SIRT232. We found that the level of acetylated lysine 16 of histone H4 is significantly lower in MDA-MB-231 cells compared to that of MCF7 cells (Fig. 4b). Next, we investigated the role of SIRT2 in deacetylation of the ARRDC3 promoter. We first compared the SIRT2 protein levels between BLBC and luminal BC cells and found that SIRT2 levels are significantly higher in BLBC cells (MDA-MB-231 and MDA-MB-468) than that of luminal BC cells (MCF-7 and HCC-1419) (Fig. 4c). We further demonstrated that SIRT2 binding to the ARRDC3 promoter is also higher in BLBC cells than that in the luminal BC cell lines (Fig. 4d). Cambinol inhibition of SIRT2 decreased the interaction of SIRT2 with ARRDC3 promoter and increased the acethylation at Lys16 of histone H4 in ARRDC3 promoter (Fig. 4e). Taken together, these data suggest that histone H4 deacetylation of ARRDC3 promoter in BLBC cells is mediated by SIRT2 whose expression is upregulated in BLBC cells.

Deactylation of ARRDC3 promoter occurs in a SIRT2 dependent manner in BLBC cells.
(a) Methylation-specific PCR (MSP) was performed on bisulphate converted DNA of MDA-MB-231 and MCF-7 cells. Specific primers were used for the detection of methylated (M) and unmethylated (U) ARRDC3 promoter sequences. (b) Chip assays were performed with AcH4K16 antibody. Samples were analyzed by PCR with specific primer for ARRDC3 promoter region. (c) Whole cell lysates were prepared from BLBC cells (MDA-MB-231 and MDA-MB-468) and luminal BC cells (HCC-1494 and MCF-7). The expression level of SIRT2 was detected by Western blot analysis. (d) Chip assays were performed with SIRT2 antibody. Samples were analyzed by PCR with specific primer for ARRDC3 promoter region. (e) MDA-MB-231 cells were treated with Cambinol (Cam) or DMSO (D) for 24 hr and Chip assay was performed using SIRT2 or AcH4K16 anibody. Samples were analyzed by PCR with specific primer for ARRDC3 promoter region. Input chromatin was included as a positive control; immunoprecipitations with IgG antibody were the negative control. (L; 2-log DNA ladder). All results presented were carried out at least 3 times.
Discussion
In the current study, we report a novel regulatory mechanism that leads to suppression of the tumor suppressor, ARRDC3 in BLBC cells. Our data demonstrate that ARRDC3 expression is epigenetically silenced in BLBC cells in comparison to that of other sub types of breast carcinoma cells. We further show that epigenetic silencing of ARRDC3 is mainly mediated by SIRT2. Our results support the hypothesis that epigenetic deregulation of tumor suppressor genes contributes to the aggressive nature of BLBC.
ARRDC3 is part of a cluster in chromosome 5, which is deleted in 17% of BLBC. Therefore, we expected no expression of ARRDC3 at least from some of BLBC cell lines. However, we were able to detect ARRDC3 expression (albeit in low levels) in all five of BLBC cell lines that we analyzed, suggesting that SIRT2 driven epigenetic silencing could represent a major mechanism to explain down-regulation of ARRDC3 over chromosomal deletion in BLBC. SIRT2 binding at ARRDC3 promoter in BLBC cells is likely due to high SIRT2 levels although BLBC specific post-translational modification of SIRT2 or the presence of ancillary protein to mediate SIRT2 binding to ARRDC3 promoter could present another possibility. It remains to be determined how BLBC cells up-regulates SIRT2 expression.
Based on synergistic effects of DNMT inhibitor and class III HDAC inhibitor on restoration of ARRDC3 level, we anticipated the collaboration between DNA methylation and histone deacetylation in epigenetic silencing at the ARRDC3 promoter by serving DNMT complex as a mediator of SIRT2 binding to ARRDC3 promoter. However, we were not able to detect hypermethylation in CpG island of ARRDC3 promoter, suggesting that inhibition of DNA methylation is likely to affect SIRT2 activity or expression independent of its binding to ARRDC3 promoter.
The tumor suppressing function of ARRDC3 is likely mediated by linking target substrates such as β-adrenergic receptor and integrin β429 to E3 ligase so that target substrates become ubiquintinated and degraded by the proteasome. The role of integrin β4 in BLBC has been well established33, suggesting that ARRDC3 inhibition of integrin β4 signaling represents one tumor suppressing mechanism missing in BLBC due to epigenetic silencing of ARRDC3. However, it is likely that ARRDC3 has multiple targets other than β-adrenergic receptor and integrin β4 as ARRDC3 contains β-arrestin like domain which could interacts with signaling molecules including small G proteins ARF6 and RhoA34,35, class-6 G-protein couple receptors36 and non-receptor kinases such as c-Src37 and PDE4D38. Therefore, it is ideal to find therapeutic compounds that upregulate the expression of ARRDC3 to maximize its tumor suppressor function in BLBC. In this regard, targeting SIRT2 activity to inhibit BLBC merits consideration as there is no targeted therapy against BLBC yet. Our data that inhibition of SIRT2 activity or expression effectively blocks BLBC cell motility further supports this hypothesis. Considering that a number of pharmacologic agents that specifically inhibit SIRT2 activity were already developed31,39,40, assessing anti-cancer effects of these compounds in vitro as well as in vivo using BLBC model will be an urgent issue.
In conclusion, our studies discovered the novel mechanism of ARRDC3 suppression in BLBC. We identified SIRT2 as a major regulator of epigenetic silencing of ARRDC3 expression in BLBC cells. Our data provide the basis of future studies to understand the molecular signatures of BLBC and to develop BLBC specific therapeutics.
Methods
Cell lines and reagents
HCC-1806 and HCC-1937 breast carcimoma, HCC-1419 and ZR-75-30 breast ductal carcinoma and AU565 breast adenocarcinoma cells were purchased from ATCC (Manassas, VA) and cultured in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin. MCF-7, MDA-MB-231 and MDA-MB-468 breast adenocarcinoma cells were maintained in DMEM with 1 g/L glucose, L-glutamine and sodium pyruvate formulation, supplemented with 10% FBS and 1% penicillin/streptomycin. MDA-MB-231 cells were obtained from the Lombardi Breast Cancer Depository at Georgetown University (Washington, DC). MCF-7 and MDA-MB-486 cells were purchased from ATCC. All cell lines were cultured in humidified incubators at 37°C in 5% CO2. To generate stable SIRT1 or 2 knockdown cell lines, MDA-MB-231 cells were infected with lentiviruses expressing shRNAs (Sigma, St.Louis, MO) targeted against SIRT-1 (target sequence: 5′- CCGGGCAAAGCCTTTCTGAATCTATCTCGAGATAGATTCAGAAAGGCTTTGCTTTTT-3′) or SIRT-2 (target sequence: 5′-CCGGTATGACAACCTAGAGAAGTACCTCGAGGTACTTCTCTAGGTTGTCATATTTTTG-3′) or GFP (as control). The infected cells were then selected by puromycin (20 μg/ml). Transfection was carried out using Lipofectamin LTX-Plus (Life Technology, Grand Island, NY). For pharmacologic inhibition of epigenetic silencing components, nicotinamide (NIA), trichostatin A (TSA), 5-aza-2′-deoxycytidine (5-Aza) and Cambinol were purchased from Sigma (St.Louis, MO). The following antibodies were obtained commercially for Western blotting analysis. Integrin β4 (clone H-101), SIRT1 (clone B-7), SIRT2 (clone H-95) and actin (clone C-11) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). ARRDC3 antibody was obtained from Abcam (Cambridge, MA). For Chip assay, SIRT2 and Lys16-acetylated H4 antibodes were purchased from (EMD Millipore, Billerica, MA).
Western blot analysis
Cells were lysed in cold RIPA-EDTA buffer [50 mM Tris, pH 7.4; 150 mM NaCl; 1% NP-40; 0.5% sodium deoxycholate; 0.1% SDS; and 5 mM EDTA] containing 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4 and protease inhibitor (Thermo Scientific, Rockford, IL). The protein concentrations were determined using the BCA protein assay kit (Thermo Scientific). The samples were separated on 4% to 20% gradient SDS PAGE and transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA). The blots were incubated with primary antibodies in TBS-T or TBS-T with 5% w/v nonfat dry milk and then with appropriate secondary antibodies conjugated to IgG-horseradish peroxidase. Proteins were detected using the pierce ECL Western blotting substrate (Thermo Scientific).
RNA preparation and quantitative real-time PCR analysis
Total RNA was isolated from freshly harvested cells with Trizol reagent (Life Technology) and RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. 1 μg of total RNA was DNase I-treated (Qiagen) and reverse transcribed using iScript Advanced cDNA Synthesis Kit (Bio-Rad) following the manufacturer's protocol. For a quantitative real-time PCR assay, DNA transcripts were probed using SsoAdvanced SYBR Green Supermix (Bio-Rad) with PrimerPCR Assay for ARRDC3 (Bio-Rad). Thermocycling was performed with a BioRad CFX96 Real-Time Detection System under the following conditions: activation (95°C/2 m), 40 cycles of denaturation (95°C/5 s), annealing/extension (60°C/30 s) and melt curve (65–95°C/5 s, 0.5°C increment). Reactions were performed in duplicate. The CFX Manager™ Software (Bio-Rad) was used to analyze the qRT-PCR data. Results were compared to a standard curve generated by serial dilutions of input DNA. The relative expression of ARRDC3 was normalized with GAPDH (reference gene) level.
Pharmacologic inhibition assay
For the HDAC inhibition, MDA-MB-231 cells and MCF-7 cells were seeded on 6-well plates. After reaching 70–80% of confluence, cells were treated with either nicotinamide (NIA) or trichostatin A (TSA), respectively at indicated doses overnight. For the DNMTs inhibition, cells were treated with various concentration of 5-Aza -2′-deoxycytidine (5-Aza-dC) for 2 days. For the combination of the two inhibitors, cells were pre-treated with 5-Aza-dC for 1day, followed by 5 mM NIA or 1 μM TSA treatment for 1day. For the SIRT1/2 inhibition, MDA-MB-231 cells were treated with cambinol at indicated doses overnight. After treatments, equal amounts of protein samples were analyzed by western blotting analysis.
ARRDC3 promoter cloning and luciferase assay
Genomic DNA was extracted using Wizard genomic DNA purification kit (Promega, Madison, WI) according to the manufacturer's instruction. Potential promoter sequence of ARRDC3 was predicted and made by using the UCSC genomic sequence tool. The possible region is located approximately within 2000 bp towards the transcription start site (TSS) and the translation start site (ATG). ARRDC3 promoter region was PCR-amplified from the genomic DNA by using Phusion High Fidelity PCR kit (New England BioLabs). The amplified fragment (−2040 to −25) was inserted into XhoI and Bgl II sites in the luciferase reporter vector, pGL4-basic (Promega). The created plasmid was sequenced to confirm the accuracy of the inserts. Cells were transfected with ARRDC3-luciferase reporter plasmid. The luciferase reporter activities were assayed by the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instruction and measured by using luminometer. Data were calculated by normalizing using β-galactosidase activies assay (Promega). After normalization of β-galactosidase activity, the data were expressed as fold activation relative to the empty pGL4 basic control vector.
Cell motility assay
The migration assay was conducted using a transwell cell culture chamber of 8 μm pore size (BD Falcon, Franklin Lakes, NJ) according to the standard procedure. The lower chambers (8 μm pore size) of transwells were coated with collagen overnight at 4°C. After washing with phosphate-buffered saline the next day, cells treated with or without cambinol and SIRT2 knockdown cell lines by shRNA (1 × 105 cells/well) were added to the upper chamber filled with serum free DMEM/bovine serum albumin. The lower chamber was placed in the serum free DMEM/BSA containing a lysophosphatidic acid (LPA) as a chemo-attractant. The chamber was incubated for 2 h at 37°C with 10% CO2. The migrated cells that were attached to the bottom side of the membrane were stained using crystal violet and counted. Assays were performed in triplicate and repeated at least three times.
DNA methylation assay of ARRDC3 promoter
DNA was extracted from cultured cells (MDA-MB-231 and MCF-7) and treated with sodium bisulfite using the EpiTect Plus LyseAll Bisulfite kit (QIAGEN) according to the manufacturer's instruction. After bisulfate modification, the DNA was amplified by Methylation specific PCR (MSP). The CpG islands in ARRDC3 promoter were analyzed by online tool (http://www.cpgislands.com) with default settings (ObsCpG/ExpCpG: 0.6; minimum average percentage of CpG: 50%; minimum length of reported CpG island: 200 bp). The primer for methylation-specific PCR (MSP) was designed by ABI Methyl primer Express v 1.0 and used to amplify with the EpiTect MSP Kit (QIAGEN) according to the manufacture's protocol. The PCR products were visualized by ethidium bromide staining on a 2% agarose gel.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) experiments were performed by using a Magna ChIP Assay kit (EMD Millipore). We followed the manufacturer's protocol with slight modifications. Briefly, the cells were crosslinked with 37% formaldehyde at 37°C for 10 min and then sonicated on ice to shear lengths of 200–1000 bp DNA fragmentsat by using a Branson sonifier (Danbury, CT). The samples containing the soluble chromatin were incubated with magnetic protein A/G beads and immunoprecipitated with antibodies against modified histone (Lys16-acetylated H4) and SIRT2 overnight at 4°C. After washing, the crosslinking between DNA and chromatin was reversed and eluted by incubation with proteinase K for 2 hr at 62°C, followed by incubation at 95°C for 10 min. The antibody-chromatin-DNA complexes were eluted from magnetic beads. DNA was purified by spin columns and subjected to PCR with the appropriated primer pairs. The primers are as follows: for ARRDC3, 5′-ATCCCAGTGTGATGTGACGA -3′ (forward) and 5′-TTTGCAACAGAATCGGAAAA -3′ (reverse); and for GAPDH, 5′-TACTAGCGGTTTTACGGGCG-3′ (forward) and 5′-TCGAACAGGAGGAGCAGAGAGCGA -3′ (reverse). Amplified PCR products were resolved on a 2% agarose gel containing ethidium bromide and quantitated using Image J. For total DNA samples (input), an aliquot of lysate used in the immunoprecipitation was processed along with the rest of the samples. Calculation of the amount of immunoprecipitated DNA in each ChIP was based on the relative signal to the corresponding total DNA signal.
References
Lin, S. X. et al. Molecular therapy of breast cancer: progress and future directions. Nat Rev Endocrinol. 6, 485–493 (2010).
Da Silva, L. & Lakhani, S. R. Pathology of hereditary breast cancer. Mod Pathol. 23 Suppl 2, S46–51 (2010).
Carey, L. A. Through a glass darkly: advances in understanding breast cancer biology, 2000–2010. Clin Breast Cancer. 10, 188–195 (2010).
Prat, A. & Perou, C. M. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 5, 5–23 (2011).
Goodison, S., Sun, Y. & Urquidi, V. Derivation of cancer diagnostic and prognostic signatures from gene expression data. Bioanalysis. 2, 855–862 (2010).
Venkitaraman, R. Triple-negative/basal-like breast cancer: clinical, pathologic and molecular features. Expert Rev Anticancer Ther. 10, 199–207 (2010).
Seal, M. D. & Chia, S. K. What is the difference between triple-negative and basal breast cancers? Cancer J. 16, 12–16 (2010).
Toft, D. J. & Cryns, V. L. Minireview: Basal-like breast cancer: from molecular profiles to targeted therapies. Mol Endocrinol. 25, 199–211 (2011).
Haupt, B., Ro, J. Y. & Schwartz, M. R. Basal-like breast carcinoma: a phenotypically distinct entity. Arch Pathol Lab Med. 134, 130–133 (2010).
Isakoff, S. J. Triple-negative breast cancer: role of specific chemotherapy agents. Cancer J. 16, 53–61 (2010).
Ismail-Khan, R. & Bui, M. M. A review of triple-negative breast cancer. Cancer Control. 17, 173–176 (2010).
de Ruijter, T. C. et al. Characteristics of triple-negative breast cancer. J Cancer Res Clin Oncol. 137, 183–192 (2011).
Carey, L. et al. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 7, 683–692 (2010).
Bosch, A. et al. Triple-negative breast cancer: molecular features, pathogenesis, treatment and current lines of research. Cancer Treat Rev. 36, 206–215 (2010).
Charafe-Jauffret, E. et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 25, 2273–2284 (2006).
Adelaide, J. et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 67, 11565–11575 (2007).
Turner, N. et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 29, 2013–2023 (2010).
Baylin, S. B. & Ohm, J. E. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 6, 107–116 (2006).
Yang, X. J. & Seto, E. HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene. 26, 5310–5318 (2007).
Peserico, A. & Simone, C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. J Biomed Biotechnol. 2011, 371832 (2011).
Vaissiere, T., Sawan, C. & Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 659, 40–48 (2008).
Taylor, D. M., Maxwell, M. M., Luthi-Carter, R. & Kazantsev, A. G. Biological and potential therapeutic roles of sirtuin deacetylases. Cell Mol Life Sci. 65, 4000–4018 (2008).
Daitoku, H. et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 101, 10042–10047 (2004).
Rothgiesser, K. M. et al. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J Cell Sci. 123, 4251–4258 (2010).
North, B. J. et al. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 11, 437–444 (2003).
Jin, Y. H. et al. Sirt2 interacts with 14-3-3 beta/gamma and down-regulates the activity of p53. Biochem Biophys Res Commun. 368, 690–695 (2008).
Wang, F. et al. Deacetylation of FOXO3 by SIRT1 or SIRT2 leads to Skp2-mediated FOXO3 ubiquitination and degradation. Oncogene. 31, 1546–1557 (2012).
Draheim, K. M. et al. ARRDC3 suppresses breast cancer progression by negatively regulating integrin beta4. Oncogene. 29, 5032–5047 (2010).
Nabhan, J. F., Pan, H. & Lu, Q. Arrestin domain-containing protein 3 recruits the NEDD4 E3 ligase to mediate ubiquitination of the beta2-adrenergic receptor. EMBO Rep. 11, 605–611 (2010).
Jones, P. A. & Baylin, S. B. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 3, 415–428 (2002).
Heltweg, B. et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 66, 4368–4377 (2006).
McClure, J. M., Wierman, M. B., Maqani, N. & Smith, J. S. Isonicotinamide enhances Sir2 protein-mediated silencing and longevity in yeast by raising intracellular NAD+ concentration. J Biol Chem. 287, 20957–20966 (2012).
Lu, S., Simin, K., Khan, A. & Mercurio, A. M. Analysis of integrin beta4 expression in human breast cancer: association with basal-like tumors and prognostic significance. Clin Cancer Res. 14, 1050–1058 (2008).
Claing, A. et al. beta-Arrestin-mediated ADP-ribosylation factor 6 activation and beta 2-adrenergic receptor endocytosis. J Biol Chem. 276, 42509–42513 (2001).
Barnes, W. G. et al. beta-Arrestin 1 and Galphaq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem. 280, 8041–8050 (2005).
Kovacs, J. J. et al. Arrestin development: emerging roles for beta-arrestins in developmental signaling pathways. Dev Cell. 17, 443–458 (2009).
Miller, W. E. et al. beta-arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. Role of beta-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J Biol Chem. 275, 11312–11319 (2000).
Perry, S. J. et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 298, 834–836 (2002).
Bonezzi, K. et al. Inhibition of SIRT2 potentiates the anti-motility activity of taxanes: implications for antineoplastic combination therapies. Neoplasia. 14, 846–854 (2012).
Peck, B. et al. SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2. Mol Cancer Ther. 9, 844–855 (2010).
Acknowledgements
This study is supported by American Cancer Society (RSG-09-091-01-CSM: J.C.), NIH-NCI (R01CA163657-01A1: J.C.), NIH-NCI (5 R01 CA155223-02: K.P.).
Author information
Authors and Affiliations
Contributions
Y.H.S. prepared for Figs 1–4 and wrote introduction, results and materials/methods section. K.P. provided the reagents and ideas for Fig. 2–4. J.C. designed and monitored the whole assays listed in the manuscript, wrote abstract and discussion and edited the writing in the entire manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
supplementary info
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Soung, Y., Pruitt, K. & Chung, J. Epigenetic silencing of ARRDC3 expression in basal-like breast cancer cells. Sci Rep 4, 3846 (2014). https://doi.org/10.1038/srep03846
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03846
This article is cited by
-
Landscape of NcRNAs involved in drug resistance of breast cancer
Clinical and Translational Oncology (2023)
-
Protease-Activated Receptor 1 (PAR1) Expression Contributes to HPV-Associated Oropharyngeal Cancer Prognosis
Head and Neck Pathology (2023)
-
Acetylation of conserved DVL-1 lysines regulates its nuclear translocation and binding to gene promoters in triple-negative breast cancer
Scientific Reports (2019)
-
Prediction of miRNA-mRNA associations in Alzheimer’s disease mice using network topology
BMC Genomics (2014)
-
Promoter hypermethylation may be an important mechanism of the transcriptional inactivation of ARRDC3, GATA5, and ELP3 in invasive ductal breast carcinoma
Molecular and Cellular Biochemistry (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
