
Abstract
Among vertebrate gastrointestinal microbiome studies, complete representation of taxa is limited, particularly among reptiles. Here, we provide evidence for previously unrecognized host-microbiome associations along the gastrointestinal tract from the American alligator, a crown archosaur with shared ancestry to extinct taxa, including dinosaurs. Microbiome compositional variations reveal that the digestive system consists of multiple, longitudinally heterogeneous microbiomes that strongly correlate to specific gastrointestinal tract organs, regardless of rearing histories or feeding status. A core alligator gut microbiome comprised of Fusobacteria, but depleted in Bacteroidetes and Proteobacteria common to mammalians, is compositionally unique from other vertebrate gut microbiomes, including other reptiles, fish and herbivorous and carnivorous mammals. As such, modern alligator gut microbiomes advance our understanding of archosaur gut microbiome evolution, particularly if conserved host ecology has retained archosaur-specific symbioses over geologic time.
Similar content being viewed by others


Introduction
Vertebrate gastrointestinal (GI) tracts are distinct microenvironments formed by conserved ecological interactions and ancient evolutionary processes between a host and resident microbial flora. Interest in unraveling the ecologies and evolutionary histories of host-microbe symbiotic associations has surged following the advent of next-generation sequencing and ‘-omics’ approaches1,2,3,4. However, reconstruction of vertebrate-microbe evolutionary histories has been limited because some host groups are underrepresented among microbiome studies. Specifically, despite mammals representing only ~10% of known vertebrate taxa5, the majority of GI tract microbiome studies to date focus on mammalian, predominately human, health and disease (e.g. obesity)6 or microbiome acquisition7. In contrast, reptilian taxa (e.g. crocodylians, tuatara, turtles, squamates)8,9,10 represent ~17% of known, extant vertebrate species5. Except for evaluation of fecal coliform bacteria11, which typically do not reflect the actual GI tract microbial diversity12 and from culture-based studies focusing on Salmonella spp.13, almost nothing is known about modern crocodylian GI tract microbiome diversity and ecology, despite its importance14 to advance respiratory and cardiovascular system development studies15.
Here, we expand the current knowledge of GI tract microbiomes from the semi-aquatic, predominantly freshwater American alligator (Alligator mississippiensis), indigenous to the southeastern United States. From wild and farm-raised alligators from fasting and feeding seasons, we identify distinct bacterial communities partitioned as a function of organ type along the length of the GI tract and the presence of a shared or “core” bacterial community. Because the modern alligator has been used as an analogue to understand and infer physiological16 and biomechanical17 traits of extinct aquatic and terrestrial forms, including carnivorous theropod dinosaurs17, as well as to characterize extinct archosaur thermoregulation18, feeding19, respiration16 and mobility17, we propose that the alligator microbiome also provides insight into the evolutionary history of predator digestive systems14,20,21 and archosaur microbiome symbioses22 based on conserved ecological niche and extant symbiotic associations2,23,24.
Results
Wild A. mississippiensis generally fast from October to March25. We hypothesized that protracted fasting would yield the composition of the indigenous symbiotic gut microflora, based on findings from the Burmese python that captured postprandial remodeling in microbiome composition8. During fasting25, the wild alligator stomach contents (<15 mL) consisted of viscous, semi-opaque yellow gastric juices with pH 2.1 to 3.0 (average 2.6; n = 5) and that were occasionally mixed with green, fluid-like material reminiscent of vegetation. Alligators routinely ingest vegetation as a consequence of capturing prey along shorelines and prey for wild alligators includes small mammals, birds, crustaceans, fish, amphibians and other reptiles26. During feeding, wild alligator stomach contents consisted of residual plant material, cutaneous fragments of partially digested crawfish and whole crawfish, with a stomach pH 1.6 to 3.7 (average 2.8; n = 10). From animal “N”, there were 17 whole, 2.5 cm long crawfish recovered and from animal “D”, nematodes, common in wild alligators, were recovered. In contrast, the farm-raised animals were fed a commercially available, dry, pelleted alligator ration (manufactured by Cargill) that consisted primarily of crude protein (45–56%), fat (9–12%) and fiber (3–4%) derived from animal protein products (dried blood meal, meat and bone), wheat, soy, corn and animal fat. Stomach contents from farm-raised individuals were viscous, opaque, brown to yellow fluids and stomach pH ranged from 1.7 to 6.6 (average 3.3; n = 5) during fasting and from 3.0 to 5.9 (average 4.4; n = 5) while feeding.
General patterns of bacterial phylum-level and proteobacterial class-level relative abundances for each alligator microbiome correlated to seasonal fasting or feeding based on nonparametric multivariate analysis of variation (NPMANOVA) (Fig. 1; Supplementary Tables 1 and 2). Wild fasting (winter) microbiome compositions shifted from communities dominated by Proteobacteria to Firmicutes- and Fusobacteria-dominated communities after renewed feeding in the spring (NPMANOVA P = 0.001, F = 8.17). Gammaproteobacteria comprised ~30% of all wild (fasting) tissue communities, whereas Firmicutes and Fusobacteria comprised 67% and 25%, respectively, for wild spring-collected tissues. Firmicutes represented 17.6% of the winter wild microbiome amplicons, but 66.9% upon renewed feeding and active fat deposition27 (NPMANOVA P < 0.001, F = 16.75). In contrast, Firmicutes comprised a significant portion of both winter farm-raised (35.5% of recovered amplicons) and spring farm-raised (48.5% of recovered amplicons) gut communities (NPMANOVA P = 0.002, F = 3.66). Animal rearing history (i.e. wild or farmed), sex and overall length (as a proxy for age and size) did not significantly explain the observed bacterial variability (NPMANOVA P > 0.05). Similarly, non-metric multidimensional scaling (NMDS) plots showed that wild and farm-raised bacterial communities responded similarly to scaling dimensions in NMDS space (Supplementary Fig. 1). Among all the alligators, seasonal feeding/fasting status explained the significant differences in bacterial compositions (NPMANOVA P < 0.001, F = 6.09).

Phylum-level, gastrointestinal bacterial community representation (>2% of retrieved amplicons) from wild and farm-raised, winter and spring, A. mississippiensis individuals and schematic sample locations.
Data are divided based on rearing history (wild vs. farmed) and subdivided based on season (winter and spring, blue and pink backgrounds, respectively) (Supplementary Table 1). Significant differences in bacterial composition between downstream tissues or organs (paired Student's t-test) are denoted by asterisks: P = 0.05 − 0.01 (*), 0.01 − 0.001 (**) and P < 0.001 (***) (full t-tests in Supplementary Table 7).
But, for each alligator, significant changes in bacterial community compositions were attributed to specific organ or tissue type (i.e. in reference to changes in epithelial tissue along the alimentary canal) (NPMANOVA P < 0.001, F = 2.42; Fig. 1). The mouths had the richest α-diversity based on the number of operational taxonomic units (OTUs). As alligators frequently open their mouths for thermoregulation28, rich α-diversity potentially reflects frequent inoculation with transient environmental bacteria. The upper GI tract communities (e.g. stomach tissue, fluids, duodenum) had the lowest richness and diversity (Supplementary Fig. 2), likely as a consequence of low pH. Intermediate richness and α-diversity values were obtained for the lower GI tract tissues (colon and feces; Supplementary Fig. 1). From the oral and upper GI tract only (i.e. stomach, duodenum), 94% of the amplicons were affiliated with Proteobacteria (Betaproteobacteria and Gammaproteobacteria), Bacteroidetes and Firmicutes, whereas 73% of the amplicons from the lower GI tract (i.e. ileum, colon and feces when present) were affiliated with Fusobacteria, Firmicutes and Bacteroidetes. In general, Fusobacteria dominated the lower GI tract, particularly feces (NPMANOVA P = <0.001, F = 23.5), but were a minor group (4%) when present in the rest of the GI system. Unique community ordination according to tissue type in NMDS space (Fig. 2) showed that communities retrieved from the colon and ileum occupied the largest NMDS ordination space compared to other tissue communities that did not overlap (i.e. feces and tongue). This was supported by β-diversity analyses of shared bacterial OTUs that also revealed a potentially high level of endemism to each organ. The greatest similarity levels in community composition were only among physiologically adjacent organs or from samples derived from the same organ (i.e. stomach and gastric juices; Supplementary Table 3).

Euclidean-based NMDS plot of bacterial diversity as a function of tissue type from all individuals, regardless of rearing history or feeding status.
Gastrointestinal samples proximal to each other had more similar bacterial communities. The plot was constructed using normalized log abundances and the parameters season (spring or winter), animal length, rearing history (farmed or wild) and sex (male or female). Lines denote vector overlays on NMDS ordination, indicating the directionality and strength of change (line length) for a specific parameter.
We used average-neighbor cluster analyses of amplicons recovered from each organ or tissue type to assess the potential for a core microbiome. Defining a host-specific or environment-specific core microbial community can be used to develop a screening tool that could identify host health, immunity and disease29,30 and to deduce symbiotic evolutionary relationships through time31. The number of unique OTUs were not significantly different between feeding and fasting in wild (two-tailed, paired t-test P = 0.17) and farmed (P = 0.25) animals. More unique OTUs were recovered from oral samples; however, for individuals where tongue tissues were unavailable and esophageal tissues were utilized, the greatest number of unique OTUs were from the lower GI (colon or feces; Supplementary Table 4). The number of unique OTUs specific to wild and farm-raised organ or tissue type ranged from 2.7% up to 86.6% of all recovered OTUs from that sample type (average of 26.2% unique OTUs per organ or tissue; Supplementary Table 3). The largest number of shared OTUs based on average-neighbor cluster analysis was observed in individuals from the same season (e.g. winter vs. spring; Supplementary Table 4). A maximum of 13 OTUs were shared among all wild individuals (Fig. 3; Supplementary Table 5). Although it may be too stringent given potential error incurred with pyrosequencing32, a 96% sequence similarity cutoff was used to denote species- to genus-level associations33 among the OTUs. One OTU from the GI tracts of all wild and farmed individuals was assigned to the Enterobacteriales. The number of shared OTUs increased by lowering the sequence similarity cut-offs to 95% and 91%, which represented higher taxonomic associations32,33 (Table 1). Resulting OTUs identified at this level included representatives of Bacteroidetes (Bacteroidales), Firmicutes (Clostridia, Clostridiaceae, Clostridiales), Fusobacteria (Fusobacteriaceae) and Gammaproteobacteria (Enterobacteriales).

Schematic average neighbor cluster analyses for shared bacterial OTUs retrieved from wild and farm-raised alligators.
OTU taxonomy was based on BLAST results (Supplementary Table 5) and the value shown in brackets refers to the number of OTUs at phylum- or class-level. Cluster analysis at 96% sequence similarity (solid black lines) resulted in 63 shared OTUs, one of which was recovered from all wild and farmed individuals (Enterobacteriales). Reducing the similarity threshold to 95% (dashed lines) and 91% (grey lines), reduced the number of OTUs to 59 and 41, respectively. At 95%, two shared OTUs among all animals formed.
Discussion
Microbiome acquisition occurs over evolutionary time and reflects complex feedbacks between the host34, environment35, diet36, immune response and microbe-microbe community interactions37 that change as new lineages invade new ecological niches. However, reptilian taxa are currently underrepresented among gut microbiome studies, thereby resulting in limited potential to reconstruct the evolutionary history of vertebrate-microbe symbioses. Based on previous studies that emphasize host feeding status controls microbiome composition8 and because alligators have distinctive physiological and biochemical differences compared to other animal hosts, particularly with respect to seasonal fasting38, we expected that alligator GI tract microbiomes would differ as a function of rearing history. Our results reveal longitudinally heterogeneous microbiomes along the alligator GI tract, with compositional differences significantly correlating to organ or tissue and also to the farm-raised versus wild alligator diets.
The dissimilarity between our alligator microbiome compositional findings and the conclusions drawn in earlier GI tract studies is striking. This is likely because the previous work, including from mammalian hosts, relies predominately on colonic biopsies12,29,30,31, feces9,23,31,39, gastric fluids40, cecal contents3, or intestines1,29,35 to define host microbiomes. However, these limited samples may not provide the insight intended12. From our analyses, distinct microbial communities were detected between the alligator mucosa and lumenal contents, as has also been observed in mice41. Also, the fecal microbial community composition did not represent the composite alligator GI tract microbiome. Representatives from the Fusobacteria phylum were most abundant from farm-raised and wild feces, but were minor members of GI tract communities (Supplementary Table 1). In contrast, other undetected phyla in the feces (e.g. Actinobacteria) had significant representation in other regions of the GI tract. If alligator feces alone were used to represent the gut microbiome, or at least the lower GI tract, then Bacteroidetes would be considered the most prevalent microbial group in the alligator microbiome. But, four OTUs total were affiliated with Bacteroidetes, which comprised only 10% of the composite microbiome from all of the digestive organs (Table 1). The relative abundances of Bacteroidetes were considerably lower than what has been reported in other vertebrate microbiome studies40,42, which again may be due to the use of fecal, cecal, or intestinal samples alone. Additionally, it is possible that increasing the number of individual hosts in a study could alter interpretations of inferred composite microbiomes. However, at least from our results, the observed pattern of microbial community variation along the length of the alimentary canal should remain because of the underlying function of each organ for digestion and the significant correlation between community composition and organ or tissue type. Future studies should utilize representative samples of the entire gut if the GI tract is considered to be a heterogeneous ecosystem with organ-specific microbiomes.
Significant differences in the alligator GI tract microbiome community composition according to organ or tissue type may be due to the host's metabolism and physiology (e.g. carnivore, herbivore, or omnivore) and metabolic capabilities of members of the gut microbiome. But, differences between farm-raised and wild alligator gut bacterial community compositions could also be attributed to host diet. Firmicutes dominated the farm-raised alligator GI tract microbiomes throughout the year, comparable to microbiomes of other captive-bred and raised reptiles8,9. However, Firmicutes only dominated wild alligator microbiomes during seasonal feeding. The farm-raised alligators had significantly greater fat deposits compared to their wild counterparts. Obesity in farm-raised alligators has been observed previously26 due to a diet high in carbohydrates and saturated fats38 to increase animal size for commercial meat and skin production. Obesity and excess fatty tissue have been correlated to shifts in microbiome composition in humans6,42. In other mammalian taxa, including mice42, Firmicutes are also prevalent members of microbiome communities from obese individuals. The intriguing association between increased fat deposition and the predominance of Firmicutes in gut microbiomes from feeding alligators, as well as from obese vertebrates, suggests that diet has the potential to modify gut microbiome physiology when the ecological niche occupied by the wild counterpart changes.
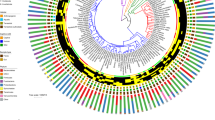
From the alligator GI tract microbiomes, a core community of Fusobacteria, Bacteroidetes, Firmicutes and Proteobacteria is strikingly different from the core microbiomes inferred for other vertebrates, including mammals23,43, birds3,4,39 and even other reptiles8,9,10 (Fig. 4, Supplementary Tables 7, 8). As all other vertebrates sampled to date possess representatives of Bacteroidetes, Firmicutes and Proteobacteria, regardless of host taxonomy, the presence of Fusobacteria in all alligators sampled makes the alligator gut microbiome unique. Fusobacteria represented ~12% of the OTUs in the core alligator microbiome and dominated the lower GI tract tissue bacterial communities and fecal material (Table 1). In contrast, Fusobacteria have been retrieved in low abundances from composite gut microbiomes from human (<10% of all oral sequences)43, fish (<13% of sequences)35 and chicken (<3.0% of sequences)4 hosts, with Fusobacteria recovered from human oral cavities playing a critical role in initial biofilm development44. In these other hosts, Firmicutes and Bacteroidetes represent the overwhelming majority of recovered microbiome OTUs.

Euclidean-based cluster analysis of phylum-level bacterial diversity of representative vertebrate taxa.
Colors correspond to major divisions of host phylogenetic affiliation (mammals, reptiles, birds and fish) and letters reflect diet. Data used for alligator are based on average feces bacterial composition (Supplementary Table 8). Feces were selected as the majority of microbiome studies utilize fecal material. Phylum-level bacterial assignments were selected as the published data generally did not include lower taxonomic assignments.
Our results raise a fundamental question: why are Fusobacteria prevalent in the American alligator GI tract? Fusobacteria are a poorly-studied phylum comprised of approximately 32 described species, with an overall uncertain phylogenetic history. Some researchers place the phylum at a basal position45 based on both rRNA and core protein analyses with the hyperthermophilic Aquificae and Thermotogae phyla, but others consider Fusobacteria to have closer phylogenetic affiliation with Bacteroidetes and Firmicutes44. Placement as a basal phylum has led to an inferred divergence ~3.5 billion years ago45. However, because of high horizontal gene transfer within Firmicutes and because Fusobacteria are frequently recovered with Firmicutes, a divergence no earlier than 400 Myr ago is also proposed, which coincides with the origin of the vertebrate digestive tract44. The almost complete lack of environmental studies that describe the presence, diversity and functional role of Fusobacteria45 in microbiomes confounds the uncertain phylogenetic position. Although Fusobacteria are associated with oral diseases in humans44, their prevalence in alligators can be interpreted as specialized, perhaps critical to host health or nutrient acquisition, because alligators possess innate immune compatibility not present in mammalians. Fusobacteria are also known to play a role in biofilm development44, so their presence in the GI tract could affect lumen biofilm development. This would explain the dominance of Fusobacteria in alligator feces. As other vertebrate microbiome studies rarely sample epithelial tissue (although see46,47 for exceptions), it is possible that Fusobacteria have been undersampled in microbiome studies until now. Nevertheless, based on what data are available, we speculate that Fusobacteria in the lower GI tract of alligators may occupy a functional role in digestive organ development and nutrient acquisition that precedes a similar ecological niche that is now occupied by Firmicutes and Bacteroidetes in mammals based on a combination of strong selective pressures driven by host genetics, a conserved diet, niche occupation and unique biochemical adaptations48.
Microbiomes have the potential to provide a window into an ancestral condition that has been retained over geologic time. Others postulate that vertebrate gut microbiome evolution may correlate to tetrapod evolution2,23. But, the diversity of bacterial communities residing in vertebrate guts is far from explored, as evidenced from our study that reveals a distinct alligator gut microbiome. An analysis of current microbiome data, albeit based on feces that likely represent biased microbiome compositional information (Fig. 4), still shows that microbiome compositions do not reflect host phylogenetic affiliations for all Vertebrata. Placement of sister taxa based on microbiome composition alone may lead to non-biologically meaningful information (i.e. placement of gorillas as sister taxa to zebrafish rather than to other hominids included in the dataset). Consequently, what is known from mammals is likely not representative of tetrapods as a whole, although successful attempts have been made to reconstruct the evolutionary history of mammalian digestive systems2,23,24.
With a similar approach, the alligator microbiomes can be used conservatively to reconstruct the ecology and evolutionary history of crocodylian and perhaps even archosaur, digestive systems20. Pseudosuchia and Avemetatarsalia, lineages within Archosauria, diverged in the Early Triassic (approx. 250 Myr ago), giving rise to modern crocodylians and avians, respectively49. The order Crocodylia is comprised of 23 extant species of caiman, alligators, crocodiles and gharials within the Alligatoridea, Crocodyloidea and Gavialoidea families50. Recent phylogenetic reconstructions from extant taxa and the robust crocodylian fossil record, which extends from the Late Cretaceous (70–84 Myr ago), highlight that modern crocodylians have had extensive, dynamic evolutionary histories that inform us about the ecology, physiology and biochemistry of the ancient lineages18,50. For instance, the biochemically unique crocodylian blood serum with its antibacterial properties was likely an ancestral trait that originated in response to aggressive behavior associated with frequent injuries and occupation of potentially pathogen-rich wetlands48. Fossil evidence indicates that numerous crocodylian taxa had similar trophic statuses and occupied comparable ecological niches to the modern51, generally functioning as top predators and scavengers within a narrow aquatic to semi-aquatic niche (e.g. wetlands and floodplains). We do not know when modern archosaur symbioses became established in the geologic past and what the ancestral condition may have been. However, it is possible that ancestral crocodylian and extinct archosaur GI tract microbiomes would have been more similar to modern alligators than to other vertebrates studied to date, assuming comparable trophic status, diet and niche occupation through time. Future research should include a robust evaluation of the ecological status (i.e. aquatic, semi-aquatic or terrestrial), feeding habits and phylogenetic position of archosaurs49,52 to test this hypothesis. Nevertheless, the implications from the modern alligator gut microbiome comprised of Bacteroidetes, Firmicutes, Proteobacteria and Fusobacteria, the dominance of which is unique among vertebrates, advance our understanding of archosaur gut microbiome evolution and host-microbe symbioses.
Methods
Sample and dataset acquisition
Organs and tissues sampled along the length of the gastrointestinal tract (Fig. 1) from eight salvaged A. mississippiensis juveniles (114–180 cm, snout to tail lengths) were acquired within hours of death from individuals sacrificed for other studies in December 2009, May/April, August and December 2010 at the Rockefeller Wildlife Refuge, Louisiana, USA (under the Louisiana Natural Heritage Program, permit LNHP-10-009). In the winter, two farm-raised but fasted (140 and 160 cm, snout to tail length; fasted 6 weeks) and two wild fasting (114 and 180 cm) alligators were used. In the spring, two fed farm-raised (130 and 131 cm) and two presumed feeding (which was later confirmed from the presence of prey items in the stomachs) wild alligators (132 and 140 cm) were used. As the alligators were salvaged, not all tissues were available for sampling every time (e.g. esophageal tissue was collected from a wild and a farmed animal during the spring because tongue tissue was unavailable due to skull removal by a different researcher). Within hours of death, several grams of epithelial tissue were aseptically excised or scraped from along the digestive tract with sterile scalpel blades and stored in 100% molecular grade ethanol at −20°C. Tissue scrapings ensured complete evaluation of the biofilms lining the GI tract, as well as any microbes adhering to or within the mucosa. Gastric fluid was collected by making a small (6–10 cm length) incision into the stomach with sterile scalpel blades to provide direct access to the contents. Liquids were collected using sterile pipettes and solid material was collected using sterilized forceps. Stomach contents were then transferred to sterile 50 mL Falcon tubes. Following the collection of gastric contents, tissue scrapings were collected. Fecal samples were collected directly from the colon aseptically with sterile scalpel blades by making a small (6–10 cm) incision into the colon. As with gastric tissue samples, colon scrapings were collected following collection and removal of fecal material. In triplicate, tissue (~1 cm3), gastric fluids (200 μl) and fecal material (~1 cm3 when present) were homogenized in 150 to 200 μl 1x TE buffer using a hand-held Kontes pellet pestle and total nucleic acids were extracted using the Qiagen DNeasy kit, following manufacturer instructions.
Purified DNA from each gastrointestinal sample (n = 48) was sent to the Research and Testing Laboratories in Lubbock, TX (USA), for 454 tag-encoded FLX titanium amplicon pyrosequencing of the V1–V3 region of bacterial 16S rRNA genes from nucleic acids extracted from gastric fluids, fecal material and epithelial lining from along the digestive tract. Samples from two individuals (SKA_09D_W and SKA_09D_F) were sequenced in January 2010 and the remaining samples from six additional individuals were sequenced in January/February 2011. After purification of DNA using previously described methods53, amplicon pyrosequencing was done using the forward primer 28f (5′-GAGTTTGATCNTGGCTCAG-3′) and the reverse primer 519r (5′-GTNTTACNGCGGCKGCTC-3′)53.
Dataset processing and statistical analysis
Amplicon trimming, removal of primers, barcodes and low quality reads, screening for chimera, alignments, taxonomic assignments, assessment of α- and β-diversity and statistical analyses were consistent with previous studies3,30,54,55. Additional screening, OTU definitions at 96% sequence identity, average-neighbor cluster analysis, rarefaction curves and statistical calculations were run with mothur, version 1.28.054. The datasets were used to calculate rarefaction curves (Supplementary Fig. 3), revealing high to moderate sample coverage, as well as bacterial diversity indices (Supplementary Fig. 2) and richness values (Supplementary Fig. 2). The Ribosomal Database Project (RDP) was utilized for sequence trimming, alignment and taxonomic assignment3,30. Processing included screening to trim reads less than 150 bp in length, removing primers, barcodes and low quality reads (Q < 20) using RDP. Following alignment in RDP, potentially chimeric reads were identified using the uchime command in mothur54. Reads identified as potentially chimeric were visually examined, compared to known reads using BLAST and removed. Additional screening following chimera removal using mothur included removing sequences with homopolymeric regions (>8 bp), removing ambiguous reads and ensuring uniform start positions for all reads and a minimum length of 150 bp55. After filtering, reads were uploaded to RDP for taxonomic assignment. Subsequent average-neighbor clustering, rarefaction curve generation and statistical analyses were performed using mothur. A core microbiome was obtained by clustering reads using variable OTU cut-offs (96%, 95%, 91% sequence similarity). Raw pyrosequence files were deposited in the GenBank Short Read Archive (accession numbers SRA023831 and SRA062824). Student's t-tests and NPMANOVA were performed in PAST56 to test the significance of bacterial community composition, number of recovered OTUs and percentages of OTUs (α = 0.05). NPMANOVA based on Euclidean distances of log-transformed data was utilized to evaluate the environmental variable(s) that accounted for the observed variations in bacterial community composition.
References
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
Ley, R. E., Lozupone, C. A., Hamady, M., Knight, R. & Gordon, J. I. Worlds within worlds: evolution of the vertebrate gut microbiota. Nat. Rev. Microbiol. 6, 776–788 (2008).
Matsui, H. et al. Microbial diversity in ostrich ceca as revealed by 16S ribosomal RNA gene clone library and detection of novel Fibrobacter species. Anaerobe 16, 83–93 (2010).
Qu, A. et al. Comparative metagenomics reveals host specific metavirulomes and horizontal gene transfer elements in the chicken cecum microbiome. PLoS One 3, e2945 (2008).
Barnosky, A. D. et al. Has the Earth's sixth mass extinction already arrived? Nature 471, 51–57 (2011).
Ley, R. E. Obesity and the human microbiome. Curr. Opin. Gastroenterol. 26, 5–11 (2010).
Buddington, R. K., Williams, C. H., Kostek, B. M., Buddington, K. K. & Kullen, M. J. Maternal-to-infant transmission of probiotics: concept validation in mice, rats and pigs. Neonatology 97, 250–256 (2010).
Costello, E. K., Gordon, J. I., Secor, S. M. & Knight, R. Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 4, 1375–1385 (2010).
Hong, P. Y., Wheeler, E., Cann, I. K. O. & Mackie, R. I. Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galápagos Islands using 16S rRNA-based pyrosequencing. ISME J. 5, 1461–1470 (2011).
Keenan, S. W. Freshwater vertebrate animal metagenomics, alligatorinae. Encyclopedia of Metagenomics. Nelson, K. (ed.) (Springer-Verlag, 2013).
Johnston, M. A., Porter, D. E., Scott, G. I., Rhodes, W. E. & Webster, L. F. Isolation of faecal coliform bacteria from the American alligator (Alligator mississippiensis). J. Appl. Microbiol. 108, 965–973 (2010).
Zoetendal, E. G., Cheng, B., Koike, S. & Mackie, R. I. Molecular microbial ecology of the gastrointestinal tract: from phylogeny to function. Curr. Issues Intest. Microbiol. 5, 31–47 (2004).
Obwolo, M. J. & Zwart, P. Prevalence of Salmonella in the intestinal tracts of farm-reared crocodiles (Crocodylus niloticus) in Zimbabwe. J. Zoo Wildlife Med. 24, 175–176 (1993).
Zanno, L. E. & Makovicky, P. J. Herbivorous ecomorphology and specialization patterns in theropod dinosaur evolution. Proc. Natl. Acad. Sci. USA 108, 232–237 (2011).
Barboza, P. S. et al. Digestive challenges for vertebrate animals: microbial diversity, cardiorespiratory coupling and dietary specialization. Physiol. Biochem. Zool. 83, 764–774 (2010).
Schachner, E. R., Farmer, C. G., McDonald, A. T. & Dodson, P. Evolution of the dinosauriform respiratory apparatus: new evidence from the postcranial axial skeleton. Anat. Rec. 294, 1532–1547 (2011).
Rayfield, E. J. Finite element analysis and understanding the biomechanics and evolution of living and fossil organisms. Annu. Rev. Earth Pl. Sc. 35, 541–576 (2007).
Seymour, R. S., Bennett-Stamper, C. L., Johnston, S. D., Carrier, D. R. & Grigg, G. C. Evidence for endothermic ancestors of crocodiles at the stem of archosaur evolution. Physiol. Biochem. Zool. 77, 1051–1067 (2004).
Farmer, C. G., Uriona, T. J., Olsen, B. D., Steenblik, M. & Sanders, K. The right-to-left shunt of crocodilians serves digestion. Physiol. Biochem. Zool. 81, 125–137 (2008).
Hone, D., Tsuihiji, T., Watabe, M. & Tsogtbaatr, K. Pterosaurs as a food source for small dromaeosaurs. Palaeogeogr. Palaeocl. 331, 27–30 (2012).
Varricchio, D. J. Gut contents from a Cretaceous Tyrannosaurid: implications for theropod dinosaur digestive tracts. J. Paleontol. 75, 401–406 (2001).
Farlow, J. O. Speculations about the diet and digestive physiology of herbivorous dinosaurs. Paleobiology 13, 60–72 (1987).
Ley, R. E. et al. Evolution of mammals and their gut microbes. Science 320, 1647–1651 (2008).
Moeller, A. H. et al. Chimpanzees and humans harbour compositionally similar gut enterotypes. Nat. Commun. 3, 1179 10.1038/ncomms2159 (2012).
Joanen, T. & McNease, L. Alligator farming research in Louisiana. Wildlife Management: Crocodiles and Alligators. Webb, G. J. W., Manolis, S. C. & Whitehead, P. J. (eds.), 329–340 (Surrey Beatty & Sons, 1987).
Elsey, R. M., McNease, L., Joanen, T. & Kinler, N. Food habits of native wild and farm-released juvenile alligators. Proc. Annu. Conf. SEAFWA 46, 57–66 (1992).
Wang, T., Hung, C. C. Y. & Randall, D. J. The comparative physiology of food deprivation: from feast to famine. Annu. Rev. Physiol. 68, 223–251 (2006).
Spotila, J. R., Terpin, K. M. & Dodson, P. Mouth gaping as an effective thermoregulatory device in alligators. Nature 265, 235–236 (1977).
Roeselers, G. et al. Evidence for a core gut microbiota in the zebrafish. ISME J. 5, 1595–1608 (2011).
Zhang, H. S. et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 106, 2365–2370 (2009).
Ochman, H. et al. Evolutionary relationships of wild hominids recapitulated by gut microbial communities. PLoS Biol. 8 (2010).
Kunin, V., Engelbrektson, A., Ochman, H. & Hugenholtz, P. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ. Microbiol. 12, 118–123 (2010).
Huse, S. M., Ye, Y., Zhou, Y. & Fodor, A. A. A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7 (2012).
Zoetendal, E., Akkermans, A. D., Akkermans-van, V., de Visser, J. A. G. M. & De Vos, W. M. The host genotype affects the bacterial community in the human gastrointestinal tract. Microb. Ecol. Health D. 13, 129–134 (2001).
Wu, S. G. et al. Composition, diversity and origin of the bacterial community in grass carp intestine. PLoS One 7 (2012).
Wu, G. D. et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 333, 105–108 (2011).
Faust, K. & Raes, J. Microbial interactions: from networks to models. Nat. Rev. Microbiol. 10, 538–550 (2012).
Lance, V. A., Morici, L. A., Elsey, R. M., Lund, E. D. & Place, A. R. Hyperlipidemia and reproductive failure in captive-reared alligators: vitamin E, vitamin A, plasma lipids, fatty acids and steroid hormones. Comp. Biochem. Phys. B 128, 285–294 (2001).
Lu, J. R. & Domingo, J. S. Turkey fecal microbial community structure and functional gene diversity revealed by 16S rRNA gene and metagenomic sequences. J. Microbiol. 46, 469–477 (2008).
Pei, C. X. et al. Diversity and abundance of the bacterial 16S rRNA gene sequences in forestomach of alpacas (Lama pacos) and sheep (Ovis aries). Anaerobe 16, 426–432 (2010).
Nava, G. M., Friedrichsen, H. J. & Stappenbeck, T. S. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 5, 627–638 (2011).
Ley, R. E. et al. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 102, 11070–11075 (2005).
Costello, E. K. et al. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009).
Mira, A., Pushker, R., Legault, B. A., Moreira, D. & Rodriques-Valera, F. Evolutionary relationships of Fusobacterium nucleatum based on phylogenetic analysis and comparative genomics. BMC Evol. Biol. 4, 50 (2004).
Battistuzzi, F. U. & Hedges, S. B. A major clade of prokaryotes with ancient adaptations to life on land. Mol. Biol. Evol. 26, 335–343 (2008).
Zoetendal, E. G. et al. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl. Environ. Microbiol. 68, 3401–3407 (2002).
Wang, X., Heazlewood, S. P., Krause, D. O. & Florin, T. H. J. Molecular characterization of the microbial species that colonize human ileal and colonic mucosa by using 16S rDNA sequence analysis. J. Appl. Microbiol. 95, 508–520 (2003).
Merchant, M. E. et al. Comparisons of innate immune activity of all known living crocodylian species. Comp. Biochem. Phys. B 143, 133–137 (2006).
Nesbitt, S. J. The early evolution of archosaurs: relationships and the origin of major clades. B. Am. Mus. Nat. Hist. 1–288 (2011).
Brochu, C. A. Phylogenetic approaches toward crocodylian history. Annu. Rev. Earth Pl. Sc. 31, 357–397 (2003).
Noto, C. R., Main, D. J. & Drumheller, S. K. Feeding traces and paleobiology of a Cretaceous (Cenomanian) crocodyliform: example from the Woodbine Formation of Texas. Palaios 27, 105–115 (2012).
Pol, D., Turner, A. H. & Norell, M. A. Morphology of the Late Cretaceous crocodylomorph Shamosuchus djadochtaensis and a discussion of neosuchian phylogeny as related to the origin of Eusuchia. B. Am. Mus. Nat. Hist. 1–103 (2009).
Dowd, S. E. et al. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol. 8 (2008).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541 (2009).
Schloss, P. D., Gevers, D. & Westcott, S. L. Reducing the effects of PCR amplificaiton and sequencing artifacts on 16S rRNA-based studies. PLoS One 6 (2011).
Hammer, Ø., Harper, D. A. T. & Ryan, P. D. PAST: paleontological statistics software pacakge for education and data analysis. Palaeontol. Electron. 4, 1–9 (2001).
Acknowledgements
P. “Scooter” Trosclair, III supplied salvaged alligator tissues. C. Will, B. Headd, B. Hopkins, M. Osborne, J. Hicks, B. Donnelly, R. Teruyama assisted with sample collection. L. Anderson, J. Nevarez, G. Byerly, V. Lance, C. Sumrall, C. Fedo, L. Kah and M. Radosevich provided feedback on the manuscript. S. Cone assisted with data analysis. D. Hines provided feed samples and discussion of feed manufacturing. M. Tellez provided identifications of crocodylian parasites. M. Merchant provided discussion on crocodylian biochemistry. S.W.K. was supported by the Marathon Oil Corporation Geoscience Diversity Enrichment Program (GeoDE) in the Department of Geology and Geophysics (LSU). Continued support has been provided by the Jones Endowment fund (UTK) and the Department of Earth & Planetary Sciences (UTK).
Author information
Authors and Affiliations
Contributions
S.W.K. and A.S.E. designed the sampling protocol, collected samples, analyzed and interpreted data and wrote the paper. R.M.E. assisted with sample collection and contributed to the text.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Keenan, S., Engel, A. & Elsey, R. The alligator gut microbiome and implications for archosaur symbioses. Sci Rep 3, 2877 (2013). https://doi.org/10.1038/srep02877
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02877
This article is cited by
-
Wild microbiomes of striped plateau lizards vary with reproductive season, sex, and body size
Scientific Reports (2022)
-
Gut microbiota in two recently diverged passerine species: evaluating the effects of species identity, habitat use and geographic distance
BMC Ecology and Evolution (2021)
-
The gut microbiome and metabolome of Himalayan Griffons (Gyps himalayensis): insights into the adaptation to carrion-feeding habits in avian scavengers
Avian Research (2021)
-
Differences in the gut microbiomes of dogs and wolves: roles of antibiotics and starch
BMC Veterinary Research (2021)
-
Taxonomy, not locality, influences the cloacal microbiota of two nearctic colubrids: a preliminary analysis
Molecular Biology Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
