
Abstract
A novel mineral-based composite phase change materials (PCMs) was prepared via vacuum impregnation method assisted with microwave-acid treatment of the graphite (G) and bentonite (B) mixture. Graphite and microwave-acid treated bentonite mixture (GBm) had more loading capacity and higher crystallinity of stearic acid (SA) in the SA/GBm composite. The SA/GBm composite showed an enhanced thermal storage capacity, latent heats for melting and freezing (84.64 and 84.14 J/g) was higher than those of SA/B sample (48.43 and 47.13 J/g, respectively). Addition of graphite was beneficial to the enhancement in thermal conductivity of the SA/GBm composite, which could reach 0.77 W/m K, 31% higher than SA/B and 196% than pure SA. Furthermore, atomic-level interfaces between SA and support surfaces were depicted and the mechanism of enhanced thermal storage properties was in detail investigated.
Similar content being viewed by others

Introduction
Thermal energy storage with phase change materials (PCMs) offers a high thermal storage density with a moderate temperature variation1 and has acquired growing attention due to its important role in solar thermal application2,3,4,5, indoor thermal management and humidity control6,7 and demand-side management8. The organic PCMs, such as paraffin, fatty acids and polyethylene glycol, have a good potential application in thermal energy storage and have been intensively investigated by many researchers9,10,11. However, these organic PCMs still have set of shortages besides indisputable advantages, such as the leakage in the melted state and lower thermal conductivity (around 0.2 W/m K) which restricted their wide application8. Therefore, various methods have been developed to overcome these problems. The most potential and practically useful method is to prepare form-stable composite PCMs12. The porous inorganic materials or silicate minerals have been used as supports to stabilized organic PCMs, such as porous silica13,14,15, expanded perlite16,17,18, attapulgite19, diatomite20, halloysite21,22,23, montmorillonite or bentonite24,25 and expanded vermiculite26,27.
To resolve the low thermal conductivity problem, some high conductive materials have been introduced into the form-stable composite PCMs and enhance the rate of thermal storage and release during the melting and freezing cycles. Sarı and Karaipekli17 used expanded perlite to stabilize the lauric acid and successfully prepare the form-stable composites. To improve the thermal conductivity, the expanded graphite was added to the composite and the thermal conductivity of the composite was largely increased from 0.07 to 0.13 W/m K. But the thermal storage capacity of composite was reduced by 7% due to the adding the expanded graphite. This phenomenon also appeared in their related works18,20. Wang et al.28 for the first time used β-Aluminum nitride to enhance the thermal conductivity of PEG/SiO2 form-stable composite which was reported in their previous work15. The thermal conductivities of the composite were successfully enhanced from 0.3847 to 0.7661 W/m K with the increase of mass ratio of β-Aluminum nitride from 5% to 30%, whereas, the thermal storage capacities of the composite decreased from 4% to 20% correspondingly. Therefore, a specific strategy must be developed for this problem, namely the thermal storage capacity of composite could be improved simultaneously while the thermal conductivity of composite be enhanced.
This study aims to provide a technique to achieve this purpose and a novel form-stable SA/GBm composite PCMs was prepared using vacuum impregnation method assisted with microwave-acid treatment of the graphite and bentonite mixture (GB). To begin with, the bentonite with thermal conductivity of 1.0 W/m K was used to stabilize the SA with the maximum loadage of 37.4 wt.% SA (SA/B). Then, the graphite was introduced to enhance the thermal conductivity of the composite and the SA/GB was prepared with the maximum loadage of 36.3 wt.% SA. To increase the maximum loadage of SA and improve the thermal storage capacity of the composite, the GB was subtly implemented by microwave-acid treatment and SA/GBm with the maximum loadage of 45.5 wt.% SA was obtained. It is of interest that the thermal storage capacity of the SA/GBm is 84.6 J/g, which is 62% larger than that of SA/GB and 75% than that of SA/B. The thermal conductivity of the SA/GBm is 0.77 W/m K, which is 31% higher than that of SA/B and 196% than that of pure SA. Finally, we try to depict the atomic-level interfaces between the SA and support surfaces and investigate the mechanisms of enhanced thermal storage properties of the form-stable SA/GBm composite PCMs.
Results
Crystallization characteristics
The montmorillonite (2θ = 5.9°, 19.8°, 35.0° and 61.9°) and the quartz (2θ = 20.8° and 26.6°) are the major phase of the bentonite, indicating from the XRD pattern of bentonite in Fig. 1a. In the XRD pattern of GB, the peaks at 19.8° and 35.0° are caused by the bentonite and the XRD peak at 26.6° are combined from bentonite and graphite due to the bentonite and graphite have the same peak at this position (see Fig. S1). After microwave-acid treatment, the characteristic peaks of the bentonite and graphite appear in the XRD pattern of GBm, which means that the GBm maintain the intrinsic crystal structure of graphite and bentonite. It can be further demonstrated by the XRD results of single bentonite or graphite before and after microwave-acid treatment (Fig. S1). The characteristic peaks of SA appear at 21.5° and 23.8° (Fig. 1b). After the SA was impregnated into the supports, the diffraction peaks of SA and the supports are observed in the XRD pattern of the composites, suggesting that the SA was successfully loaded into the supports. Furthermore, the peaks of the SA in the SA/GBm is basically the same as that of SA in the SA/B and SA/GB, meaning that adding the graphite and microwave-acid treatment of the GB did not affect the crystal structure of SA.

Crystallization of the samples.
(a) XRD patterns of the supports (Q: quartz, M: montmorillonite and G: graphite). (b) XRD patterns of the SA and the composites.
Morphological analysis
The platelet morphology of bentonite is observed from Fig. 2a and some of the platelets are formed aggregates. After the impregnation, the surfaces of bentonite were occupied entirely by SA in the SA/B and the bentonite platelets are surrounded by the continuous PCMs (Fig. 2b). Interestingly, some platelets of SA/B emerge with the thickness and the extent of approximately 0.2 ~ 0.3 μm and range from 0.5 ~ 2 μm, respectively. The bentonite aggregates are relatively independent from graphite while introducing the graphite, indicating from Fig. 2c, which can be proofed by the EDS patterns in Fig. 2d (C1 is mainly graphite while C2 is mainly bentonite). After microwave-acid treatment (Fig. 2e and Fig. 2f), the bentonite aggregates are exfoliated and adhered to the surface of the flaky graphite which has bigger flakes and obvious edges (E1 is mainly graphite while E2 is mainly bentonite). The sizes of bentonite platelet become smaller under microwave-acid treatment (see Fig. S2), also the flaky graphites are exfoliated by this treatment (see Fig. S3). For the SA/GB and SA/GBm (Fig. 2g and Fig. 2h), the SA maybe adsorbed onto the bentonite platelets and then these were adhered to the flaky graphite by SA chains. By comparing Fig. 2g with Fig. 2h, it can be concluded that the exposed platelets in Fig. 2h are smaller than that in Fig. 2g. Also the platelet morphology of all the composites could be seen distinctly (Fig. 2b, Fig. 2g and Fig. 2h), so the attachment but not absorbent SA was removed via thermal filter, that is to say, the loadage in this condition was the maximum.

Morphologies of the samples.
SEM images of the (a) B, (b) SA/B, (c) GB and the corresponding EDS patterns (d), (e) GBm and the corresponding EDS patterns (f), (g) SA/GB and (h) SA/GBm. Tapping-mode AFM image of the SA/GBm, (i) the amplitude image and (j) the corresponding height image and (k) the cross section analysis along the line.
In order to explore the exquisite surface of the SA/GBm, the atomic force microscope is implemented. The amplitude AFM image reveals the rough surface of SA/GBm (Fig. 2i), the “hills” are adhered on the graphite flake (the obvious edges can be seen from large field image, Fig. S4) and two representative “hills” are marked by green and red line, respectively. From the height AFM image (Fig. 2j&k), the thickness and the extent of two “hills” are 0.2 μm and 0.7 μm, 0.25 μm and 0.9 μm, respectively. These results indicated that the “hills” are the SA/B platelets. And the “hills” are adhered to the flaky graphite by SA chains.
Thermal capacities and conductivities
Thermal capacities (latent heat and onset temperature) of SA and composites are determined by the DSC thermal analysis20 (Fig. 3). Thermal capacities of SA, SA/B, SA/GB and SA/GBm are given in Table 1. The curves of pure SA shows a melting temperature (Tm) at 53.12°C in the endothermic curve and a freezing temperature (Tf) in the exothermic curve at 51.84°C. The latent heats of melting (ΔHm) and freezing (ΔHf) of pure SA are 200.2 and 198.4 J/g, respectively.

Thermal capacities of the samples.
DSC curves of the SA, SA/B, SA/GB and SA/GBm.
The latent heat is a crucial factor for composites PCMs and demonstrates their thermal capacity. Compared with pure SA, the latent heats of melting and freezing of the composites declined according to the curves of the SA/B, SA/GB and the SA/GBm. The latent heats of melting and freezing are determined as 48.43 and 47.13 J/g for the SA/B, 50.93 and 49.98 J/g for SA/GB and 84.64 and 84.14 J/g for the SA/GBm, which both are higher than that of the SA/B and the SA/GB. And these values less than their theoretic values (for the SA/B, ΔHm: 200.2 J/g × 37.4% = 74.87 J/g, ΔHf: 198.4 J/g × 37.4% = 74.20 J/g; for the SA/GB, ΔHm: 200.2 J/g × 36.3% = 72.67 J/g, ΔHf: 198.4 J/g × 36.3% = 72.02 J/g; for the SA/GBm, ΔHm: 200.2 J/g × 45.5% = 91.09 J/g, ΔHf: 198.4 J/g × 45.5% = 90.27 J/g). The decrease of the latent heats of the composites cannot be attributed to the lower fraction of SA alone. The crystallinity of SA in the composites must be considered on account of the interactions between SA and supports hindered SA from crystallizing and reduced the latent heats of the composites29. The crystallinity of SA (Fc) in the composite is calculated by:

where ΔHcomposite and ΔHpure are the latent heat of the composite and pure SA, respectively and β represents the loadage of SA in the composites. As shown in Table 1, the crystallinity of the SA in the SA/B is 64.7%, while that in the SA/GB is 70.1% and is larger than that of the SA/B. This is why SA/GB has slightly higher latent heats than that of SA/B even has lower loadage than that of SA/B. What is more, SA in the SA/GBm has the largest crystallinity at 92.9%.
As the supports are used for stabilizing PCMs, the interactions (including: capillary forces, surface polarities, hydrogen bonding) occur between SA and supports. For SA/B and SA/GB two composites, SA only interacts with bentonite and graphite. Some of SA are embedded in the pores of bentonite while part of these SA are confined by capillary force and then the movement of the SA during the crystallization will be restricted. In consideration of surface polarities, the surface of the bentonite is polar, but that of graphite is nonpolar. The polarity may enhance the interaction between polar SA and bentonite. For hydrogen bonding, there is hydrogen bonding between SA and bentonite, whereas there is no evidence for hydrogen bonding between SA and graphite (the spectrum of graphite does not exhibit any significant absorption band and it has no feasibility to formed hydrogen bonding with SA, Fig. S5). Thus, under the combined effects of capillary forces, surface polarities and hydrogen bonding, the interaction of the bentonite with SA is much stronger than that of graphite with SA. Some of SA are adsorbed onto the graphite in the SA/GB while introducing the graphite into the SA/B, so the interactions in this sample are weaker than that of SA/B. Consequently, SA in the SA/GB has higher crystallinity than that in the SA/B.
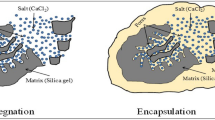
As for the SA/GBm, the maximum loadage of SA is 45.5% and the thermal storage capacity is 84.64 J/g, it is 62.2% larger than that of SA/GB (50.93 J/g). To uncover the reason of improved thermal storage capacity of composites, the pore structures of the supports before and after microwave-acid treatment are measured by nitrogen gas adsorption-desorption. The nitrogen sorption isotherms curves are shown in Fig. 4a, the obtained BET surface areas and pore volumes of supports are listed in Table 2. The BET surface area of GBm (130.34 m2/g, after microwave-acid treatment) is higher than that of GB (71.18 m2/g, before microwave-acid treatment), which means that GBm has more adsorption sites than GB. The total pore volume of GBm (0.18 cm3/g) is larger than that of GB (0.08 cm3/g), indicating that GBm has more hold space than GB. So the GBm possesses more loading capacity than GB due to more adsorption sites and hold space via microwave-acid treatment. The maximum loadage of SA for GBm is 25.3% larger than that of GB and this value is lower than the increasing percentage of thermal storage capacity (62.2%). As we known, the crystallinity of SA in the composite has significant influences on the latent heats of the composite29. So the higher crystallinity of SA in the SA/GBm accounts for residual increasing percentage of thermal storage capacity. The pore size distributions of GB and GBm (inset of Fig. 4a) indicate that they have significantly different pore size distributions. Before microwave-acid treatment, the pore sizes converge on 3.5, 5.5, 9 and 20 nm, respectively. After microwave-acid treatment, the pore sizes distributions shift to 4.5, 13 and 24 nm (green dash line expand to the red full line in the Fig. 4b), respectively, meaning that the GBm has large pore sizes than GB. And then, the vast majority of SA segments in the SA/GBm are easy to move and crystallizable while the movements of some SA segments in the SA/GB are confined (the amorphous SA in the left of blue dash line represents confined SA while the crystallization SA in the right of blue dash line represents crystallizable SA, in Fig. 4b). Therefore, SA in the SA/GBm has higher crystallinity than that in SA/GB, which results in higher latent heats. Combine more SA loading capacity with higher crystallinity, the reasons of improved thermal storage capacity of composites are exploited and the relevant schematic illustration is shown in Fig. 4b.

Improved thermal storage capacity of the samples.
(a) Nitrogen adsorption-desorption isotherms and BJH pore size distribution (Inset) of the GB and GBm. (b) Schematic illustration of improving the thermal storage capacity.
In practical applications, the supercooling is an important factor of composite PCMs. The extent of supercooling is calculated as the difference of the melting and freezing temperatures (Table 1). The extent of supercooling in the SA/B (2.51°C) and SA/GB (1.75°C) are slightly larger than that of pure SA (1.28°C) and the extent of supercooling in the SA/GBm is the smallest (0.42°C). This means that the GBm supports SA better than B and GB support SA in reducing the extent of supercooling.
Thermal conductivity is a fundamental factor for composite PCMs and expresses their thermal conduction rate, in which it is governed by phonon propagation due to these composite PCMs are blend of nonmetal and organic. The thermal conductivity of the SA/GBm is 0.77 W/m K and is 31% and 196% higher than that of SA/B (0.59 W/m K) and pure SA (0.26 W/m K), respectively. It means that bentonite improves the thermal conductivity of pure SA and then introducing graphite enhances the thermal conductivity of SA/B.
Thermal storage and release properties
The thermal storage and release performance of SA/GBm is assessed by comparing with that of pure SA. The thermal storage and release curves of the pure SA and the composite are shown in Fig. 5. In the thermal storage process, the temperature of the SA and the composite are the same at 26°C. It takes 580 s for SA to reach the melting temperature, however, for the composite only 335 s. It is obvious that the thermal storage rate of the composite is higher than that of pure SA. And, the composite reach the setup temperature of 80°C at 1640 s, while it is very hard for the SA to reach 80°C and take 1870 s to reach the equilibrium temperature (close to the setup temperature). In the thermal release process, it takes SA 1330 s to reach the freezing temperature compared with 500 s for the composite. It indicates that the thermal release rate of the composite is higher than that of the SA. By comparing the melting and freezing time of SA with that of composite, it is obvious that the thermal storage and release rates were increased by 1.7 and 2.7 times, respectively, which indicates that the thermal storage and release rates are greatly increased by introducing the GBm.

Thermal storage and release properties of the samples.
Thermal storage and release curves of the SA and the SA/GBm.
In addition, Table 3 shows the comparison of the latent heats and the thermal conductivities of the prepared composite PCMs with that of some composite PCMs in literature17,18,20,21,26,30, the SA/GBm composite shows obvious advantages over the reported materials. It is remarkably noted that the prepared SA/GBm composite could become a preferential potential solar thermal energy storage material for the low temperature solar-thermal system with solar passive heating purposes.
Discussion
As can be seen in the FTIR spectra of bentonite (Fig. S6), the characteristic vibration peaks of bentonite are at 3618 cm−1 (O-H antisymmetric stretching)31,32, 3433 cm−1 (O-H symmetric stretching)19,32,33,34, 1634 cm−1 (water bending mode)19,32,35,36, 1035 cm−1 (Si-O-Si antisymmetric stretching)36,37, 914 cm−1 (Al-OH bending)33,38, 798 cm−1 (platy form of tridymite)33, 692 cm−1 (quartz)33, 529 cm−1 (Si-O-Al bending)38 and 465 cm−1 (Si-O bending)35, also all the characteristic peaks reflect in the GB while introducing the graphite to the bentonite. The characteristic peaks of GB are largely consistent with bentonite on account of graphite, which does not exhibit any significant absorption band in the studied region (see Fig. S5). After microwave-acid treatment, these characteristic peaks appear in the spectra of GBm and no new obvious peaks emerge in the FTIR spectrum of single bentonite or graphite before and after microwave-acid treatment (Fig. S5).
From the spectra of pure SA, the characteristic vibration peaks of PCMs are at 2920 cm−1 (−CH3 symmetrical stretching), 2846 cm−1 (−CH2 symmetrical stretching), 1705 cm−1 (C = O stretching), 1466 cm−1 (−OH bending), 1300 cm−1 (−OH bending), 936 cm−1 (−OH bending), 726 cm−1 (−OH swinging) and 680 cm−1 (−OH bending)39,40. According to the FTIR spectra of the SA/B, SA/GB and SA/GBm, the characteristic peaks of SA can also be observed in the spectra of the composites except the peak at 936 cm−1. This peak has shifted to a lower wavenumber of 914 cm−1 and forms a broaden peak compared with that of supports. It maybe the formation of hydrogen band formed by -OH of the SA and Al-OH of the supports, as suggested from the report41. The characteristic peaks of supports also appear in the composites. Specially, the peaks at 3433 cm−1 weaken evidently due to the formation of hydrogen band formed by hydrogen of carbon chain of SA and supports.
After contrasting the spectra of the composites with supports and pure SA, it demonstrates that the SA have been absorbed into supports and no bond except hydrogen bond exists between SA and supports and the hydrogen bond may stabilize the SA in the composites and then this interaction avoids the leakage of the melted PCMs from the supports42. Therefore, there is no chemical reaction between the PCMs and supports.
Furthermore, based on the results of AFM and FTIR, we schematically depicted the interfaces of SA and the support surfaces at the atomic level to illustrate the mechanism of enhanced thermal conductivities of composites (Fig. 6). The top view (Fig. 6a) is consistent with AFM results (Fig. 2i&j). The SA/B platelets are adhered to the flaky graphite. From the FTIR results (Fig. S6), the hydrogen bands are formed between the SA and the support. The hydrogen band between SA and bentonite minimize the interfacial phonon scattering and decrease the interface heat resistance by interpenetrating SA and bentonite interface43. The thermal conductivity of bentonite is 1.0 W/m K, which is higher than that of SA. Therefore, the cross-linked three-dimensional network is an efficient pathway for thermal conduction and the SA/B has higher thermal conductivity (0.59 W/m K) than that of pure SA (0.26 W/m K). There is added thermal conduction pathway appearing at the composite PCMs while the high conductive material (graphite, thermal conductivity of 39.5 W/m K) is introduced (Fig. 6b). So, the thermal conductivity of the SA/GBm is 0.77 W/m K and is 31% and 196% higher than that of SA/B and pure SA, respectively.

Atomic-level interfaces between the SA and the support surfaces.
(a) Top view and (b) front view; The white, black, yellow, pink, red, purple spheres represent H, C, Si, Al, O, alkaline atoms, respectively.
In summary, SA/GBm composite with simultaneously enhanced thermal capacity and thermal conductivity was prepared using vacuum impregnation method assisted with microwave-acid treatment of the GB. The SA/GBm composite had higher latent heats of melting and freezing of 84.64 and 84.14 J/g than that of SA/GB of 50.93 and 49.98 J/g and SA/B of 48.43 and 47.13 J/g, respectively. The thermal conductivity of the SA/GBm is 0.77 W/m K, which is 31% higher than that of SA/B (0.59 W/m K) and 196% than pure SA (0.26 W/m K). The thermal storage and release rates of SA/GBm were increased by 1.7 and 2.7 times compared with pure SA, respectively. This as-synthesized composite could be potentially used in solar thermal energy storage systems with solar passive heating purposes.
Methods
Materials
Natural bentonite was obtained from Changde, Hunan, China. Stearic acid (SA, CH3(CH2)16COOH) was supplied by Tianjin Kemiou Chemical Reagent Co., Ltd., China. Graphite was purchased from Qingdao Graphite Co., Ltd., China.
Purification of the natural bentonite
Natural bentonite was mixed with distilled water (the mass ratio of 1:10) at room temperature. This was continuously stirred by a GFJ-0.4 type high-speed dispersion machine with 5000 rpm for 5 min, subsequently pass 0.045 mm sieve. Then this was dispersed with sodium hexametaphosphate (corresponding to a sodium hexametaphosphate mass fraction of 0.1%) and was allowed to stay for 1 h without stirring. Finally the bottom sediments were removed and the water was evaporated. The sample was dried at 105°C for 12 h, labelled as purified bentonite and stored to follow-up experiment.
Synthesis of the supports
The purified bentonite and graphite (the mass ratio of 10:1) were ground for 30 min in a KM-10 type planetary mill with a rotation speed of 500 rpm at room temperature. The mass ratio of balls to sample was 3.5:1. Subsequently, this ground mixture was treated by microwave with hydrochloric acid. 100 g ground sample was added to the 300 g (8 wt.%) HCl solution and the mixture was placed in a microwave oven and irradiated for 5 min at 350 W. After the treatment, the excess of the hydrochloric acid was removed by repetitive washing with distilled water until an AgNO3 test indicated the absence of chlorine ion. The samples were dried at 105°C, ground and labelled as GBm. For comparison, the samples (bentonite, the graphite and bentonite mixture) without microwave-acid treatment were prepared by the same method and labelled as B, GB, respectively. Similarly, the single graphite or bentonite was microwave-acid treated by the same method for supplementary data analysis.
Preparation of the composites
The composites were prepared using vacuum impregnation method (Fig. S7): 30 g support and 70 g SA were placed inside a conical flask and a device of preventing of backward suction was used to connect the conical flask with vacuum pump. The vacuum of conical flask was evacuated to −0.1 MPa for 5 min. Then, the conical flask was placed in thermostatical water bath at 95°C for 20 min. The vacuum pump was then turned down and allowed the air to enter the flask again, with ultrasonic heating at 80°C for 5 min. After cooling, the composite was thermally filtered to remove superfluous SA at 80°C for 48 h, finally obtained composites and named by SA/B, SA/GB and SA/GBm corresponding to B, GB and GBm, respectively. The maximum loadages of SA for SA/B, SA/GB and SA/GBm were determined by ignition loss method and were 37.4 wt.%, 36.3 wt.% and 45.5 wt.%, respectively.
Characterization
Differential scanning calorimetry (DSC) analysis of SA and composites was made by TA instruments DSC Q10 (V9.9 Build 303) (the accuracy of apparatus was ± 0.10°C in phase change temperatures and ± 1% in latent heat capacities, respectively.) at a heating rate of 5°C/min under a constant stream of argon at atmospheric pressure. Liquid nitrogen was used for cooling around the sample during the freezing period. X-ray diffraction (XRD) was carried out using a Rigaku D/max-rA analyzer (Cu-Kα) under the following conditions: voltage: 40 kV; current: 40 mA, scan range from 5° to 80° and step size of 0.02°. The thermal conductivities were tested by TPS 2500 Hot Disk Thermal Constant Analyser. The microstructures of the samples were investigated by a JEOL JSM-6360LV scanning electron microscopy (SEM). Fourier transformation infrared spectroscopy (FTIR) spectra were recorded using a Thermo Electron Corporation Nicolet 6700 FTIR spectrometer in the range of 4000–400 cm−1. Atomic force microscopy (AFM) images of the SA/GBm deposited on mica substrates were obtained using a Nanoscope V Multimode AFM (Digital Instruments). To test the thermal storage/release performance of composites, thermal cycling test was conducted using the experimental set-up in Fig. S8. The samples were separately filled into identical test tubes. One thermocouple with a precision of ± 0.1°C was placed at the centers of each tube for temperature measurement. Firstly, the test tubes were put inside in the thermostatical water bath at 25°C for temperature equilibrium. Later, the test tubes were put into the thermostatical water bath at 80°C immediately. After the temperature of samples reached 80°C, the test tubes were put into the thermostatical water bath at 25°C again. The temperature variations of samples measured by thermocouple were recorded automatically by using data logger. To compare the pore structures, nitrogen gas adsorption-desorption isotherms of the graphite and bentonite mixture without and with microwave-acid treatment were measured at 77 K using an ASAP 2020 unit.
References
Zhou, D., Zhao, C. Y. & Tian, Y. Review on thermal energy storage with phase change materials (PCMs) in building applications. Appl. Energy 92, 593–605 (2012).
Wang, T., Mantha, D. & Reddy, R. G. Novel low melting point quaternary eutectic system for solar thermal energy storage. Appl. Energy 102, 1422–1429 (2013).
Tian, Y. & Zhao, C. Y. A review of solar collectors and thermal energy storage in solar thermal applications. Appl. Energy 104, 538–553 (2013).
Nithyanandam, K. & Pitchumani, R. Computational studies on a latent thermal energy storage system with integral heat pipes for concentrating solar power. Appl. Energy 103, 400–415 (2013).
Rao, Z., Wang, S. & Peng, F. Self diffusion of the nano-encapsulated phase change materials: A molecular dynamics study. Appl. Energy 100, 303–308 (2012).
Rao, Z., Wang, S. & Zhang, Z. Energy saving latent heat storage and environmental friendly humidity-controlled materials for indoor climate. Renew. Sust. Energ. Rev. 16, 3136–3145 (2012).
Zhou, G., Yang, Y. & Xu, H. Performance of shape-stabilized phase change material wallboard with periodical outside heat flux waves. Appl. Energy 88, 2113–2121 (2011).
Arteconi, A., Hewitt, N. J. & Polonara, F. State of the art of thermal storage for demand-side management. Appl. Energy 93, 371–389 (2012).
Rozanna, D., Chuah, T. G., Salmiah, A., Choong, T. S. Y. & Sa'ari, M. Fatty acids as phase change materials (PCMs) for thermal energy storage: A review. Int. J. Green Energy 1, 495–513 (2004).
Sharma, A., Tyagi, V. V., Chen, C. R. & Buddhi, D. Review on thermal energy storage with phase change materials and applications. Renew. Sust. Energ. Rev. 13, 318–345 (2009).
Sarier, N. & Onder, E. Organic phase change materials and their textile applications: An overview. Thermochim. Acta 540, 7–60 (2012).
Kenisarin, M. M. & Kenisarina, K. M. Form-stable phase change materials for thermal energy storage. Renew. Sust. Energ. Rev. 16, 1999–2040 (2012).
Zhou, X., Xiao, H., Feng, J., Zhang, C. & Jiang, Y. Preparation and thermal properties of paraffin/porous silica ceramic composite. Compos. Sci. Technol. 69, 1246–1249 (2009).
Zhou, X., Xiao, H., Feng, J., Zhang, C. & Jiang, Y. Pore structure modification of silica matrix infiltrated with paraffin as phase change material. Chem. Eng. Res. Des. 88, 1013–1017 (2010).
Wang, W., Yang, X., Fang, Y. & Ding, J. Preparation and performance of form-stable polyethylene glycol/silicon dioxide composites as solid–liquid phase change materials. Appl. Energy 86, 170–174 (2009).
Karaipekli, A. & Sarı, A. Capric-myristic acid/expanded perlite composite as form-stable phase change material for latent heat thermal energy storage. Renew. Energy 33, 2599–2605 (2008).
Sarı, A., Karaipekli, A. & Alkan, C. Preparation, characterization and thermal properties of lauric acid/expanded perlite as novel form-stable composite phase change material. Chem. Eng. J. 155, 899–904 (2009).
Sarı, A. & Karaipekli, A. Preparation, thermal properties and thermal reliability of capric acid/expanded perlite composite for thermal energy storage. Mater. Chem. Phys. 109, 459–464 (2008).
Li, M., Wu, Z. & Kao, H. Study on preparation, structure and thermal energy storage property of capric–palmitic acid/attapulgite composite phase change materials. Appl. Energy 88, 3125–3132 (2011).
Karaman, S., Karaipekli, A., Sarı, A. & Biçer, A. Polyethylene glycol (PEG)/diatomite composite as a novel form-stable phase change material for thermal energy storage. Sol. Energy Mater. Sol. Cells 95, 1647–1653 (2011).
Mei, D., Zhang, B., Liu, R., Zhang, Y. & Liu, J. Preparation of capric acid/halloysite nanotube composite as form-stable phase change material for thermal energy storage. Sol. Energy Mater. Sol. Cells 95, 2772–2777 (2011).
Mei, D., Zhang, B., Liu, R., Zhang, H. & Liu, J. Preparation of stearic acid/halloysite nanotube composite as form-stable PCM for thermal energy storage. Int. J. Energy Res. 35, 828–834 (2011).
Zhang, J., Zhang, X., Wan, Y., Mei, D. & Zhang, B. Preparation and thermal energy properties of paraffin/halloysite nanotube composite as form-stable phase change material. Sol. Energy 86, 1142–1148 (2012).
Sarier, N., Onder, E., Ozay, S. & Ozkilic, Y. Preparation of phase change material–montmorillonite composites suitable for thermal energy storage. Thermochim. Acta 524, 39–46 (2011).
Li, M., Wu, Z., Kao, H. & Tan, J. Experimental investigation of preparation and thermal performances of paraffin/bentonite composite phase change material. Energy. Conv. Manag. 52, 3275–3281 (2011).
Karaipekli, A. & Sarı, A. Capric-myristic acid/vermiculite composite as form-stable phase change material for thermal energy storage. Sol. Energy 83, 323–332 (2009).
Karaipekli, A. & Sarı, A. Preparation, thermal properties and thermal reliability of eutectic mixtures of fatty acids/expanded vermiculite as novel form-stable composites for energy storage. J. Ind. Eng. Chem. 16, 767–773 (2010).
Wang, W., Yang, X., Fang, Y., Ding, J. & Yan, J. Enhanced thermal conductivity and thermal performance of form-stable composite phase change materials by using β-Aluminum nitride. Appl. Energy 86, 1196–1200 (2009).
Wang, C. et al. Shape-stabilized phase change materials based on polyethylene glycol/porous carbon composite: The influence of the pore structure of the carbon materials. Sol. Energy Mater. Sol. Cells 105, 21–26 (2012).
Sarı, A. & Biçer, A. Thermal energy storage properties and thermal reliability of some fatty acid esters/building material composites as novel form-stable PCMs. Sol. Energy Mater. Sol. Cells 101, 114–122 (2012).
Bouberka, Z. et al. Adsorption of Supranol Yellow 4 GL from aqueous solution by surfactant-treated aluminum/chromium-intercalated bentonite. J. Hazard. Mater. 162, 378–385 (2009).
Myneni, S. C. B., Traina, S. J., Waychunas, G. A. & Logan, T. J. Vibrational spectroscopy of functional group chemistry and arsenate coordination in ettringite. Geochim. Cosmochim. Acta 62, 3499–3514 (1998).
Benli, B. Effect of borax addition on the structural modification of bentonite in biodegradable alginate-based biocomposites. J. Appl. Polym. Sci. 128, 4172–4180 (2013).
Yang, H., Li, M., Fu, L., Tang, A. & Mann, S. Controlled assembly of Sb2S3 nanoparticles on silica/polymer nanotubes: Insights into the nature of hybrid interfaces. Sci. Rep. 3, 1336 (2013).
Yang, H., Deng, Y., Du, C. & Jin, S. Novel synthesis of ordered mesoporous materials Al-MCM-41 from bentonite. Appl. Clay Sci. 47, 351–355 (2010).
Ma, J. et al. Synthesis and characterization of Ag3PO4 immobilized in bentonite for the sunlight-driven degradation of Orange II. Appl. Catal. B-Environ. 134–135, 1–6 (2013).
Jiang, T., Zhao, Q. & Yin, H. Synthesis of highly stabilized mesoporous molecular sieves using natural clay as raw material. Appl. Clay Sci. 35, 155–161 (2007).
Hu, P. & Yang, H. Insight into the physicochemical aspects of kaolins with different morphologies. Appl. Clay Sci. 74, 58–65 (2013).
Fang, G., Li, H., Chen, Z. & Liu, X. Preparation and characterization of stearic acid/expanded graphite composites as thermal energy storage materials. Energy 35, 4622–4626 (2010).
Ferreira, M., Wohnrath, K., Riul, J. R. A., Giacometti, J. A. & Oliveira, J. R. O. N. Interactions at the molecular level between biphosphine ruthenium complexes and stearic acid in Langmuir and Langmuir−Blodgett films. J. Phys. Chem. B 106, 7272–7277 (2002).
Li, W. et al. Effect of the intermolecular hydrogen bond between carbazole and N,N-dimethylformamide/isopropanolamine on the solubility of carbazole. Energy & Fuels 26, 6316–6322 (2012).
Wang, C. et al. Graphene oxide stabilized polyethylene glycol for heat storage. Phys. Chem. Chem. Phys. 14, 13233–13238 (2012).
O'Brien, P. J. et al. Bonding-induced thermal conductance enhancement at inorganic heterointerfaces using nanomolecular monolayers. Nat. Mater. 12, 118–122 (2013).
Acknowledgements
This work was supported by the National Science Fund for Distinguished Young Scholars (51225403), the Specialized Research Fund for the Doctoral Program of Higher Education (20120162110079) and the Scientific Research Foundation for ROCS of SEM (2011–1139).
Author information
Authors and Affiliations
Contributions
H.M.Y. conceived the project and wrote the final paper. C.C.L. wrote initial drafts of the work. C.C.L. designed the experiments, synthesized and characterized the materials. L.J.F., J.O.Y. and H.M.Y. carried out the first-principle computations. All authors discussed the results and commented on the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, C., Fu, L., Ouyang, J. et al. Enhanced performance and interfacial investigation of mineral-based composite phase change materials for thermal energy storage. Sci Rep 3, 1908 (2013). https://doi.org/10.1038/srep01908
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01908
This article is cited by
-
Thermal Performances of Myristic Acid/Bentonite/Graphene Composite Phase Change Materials
International Journal of Thermophysics (2023)
-
Thermal Transport Properties of MWCNT Based Natural Azerbaijani Bentonite Ceramic Composites
International Journal of Thermophysics (2023)
-
Modified sepiolite stabilized stearic acid as a form-stable phase change material for thermal energy storage
International Journal of Minerals, Metallurgy and Materials (2023)
-
Experimental study of thermal performance on the adsorption of stearic acid into different morphology sepiolite
Journal of Thermal Analysis and Calorimetry (2022)
-
Silica-confined composite form-stable phase change materials: a review
Journal of Thermal Analysis and Calorimetry (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
