
Abstract
Allele frequencies have long been studied by biologists interested in evolution and speciation. More recently, with the application of molecular markers in human DNA profiling we have also seen the need for reliable population allele frequency estimates for making probabilistic inferences. There is now interest in applying the same DNA profiling technology to identification of plant varieties. HortResearch maintains a large germplasm of horticultural plant species. It is becoming evident that accurate identification of these accessions through DNA fingerprinting is essential for effective utilisation and maintenance of this germplasm. Microsatellites are the markers of choice for this fingerprinting. However, such markers do not reveal the dosage of alleles in a polyploid. Polyploidy is common amongst horticultural plants. Estimating allele frequencies in a polyploid population is, therefore, complicated because of some marker genotypes being phenotypically indistinguishable. For example, in a tetraploid, with four alleles at a locus showing polysomic inheritance, although 35 genotypes are possible, these will fall into only 15 marker phenotypic classes. Furthermore ‘null’ individuals are rarely detected in polyploids. Furthermore, some polyploids can be cryptic exhibiting disomy, instead of the polysomic inheritance. We will discuss the implications of these factors and present an EM-type algorithm for estimating allele frequencies of a polyploid population under certain patterns of inheritance. The method will be demonstrated on simulated data. We also discuss the nature of some of the additional problems that may be encountered with estimating allele frequencies in polyploids for which other solutions still need to be developed.
Similar content being viewed by others


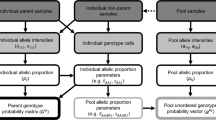
Introduction
Allele frequency data have been used in the past for analysis of phylogeny and population structure of species and of distinct populations of the same species. In more recent years the scope of such studies has increased several fold by the use of polymorphic molecular markers. Markers, such as microsatellites, with their high degree of polymorphism and codominant inheritance provide more information, often sufficient to distinguish individuals within a population. The theory underlying application of DNA profiling-based evidence in forensic science for resolving parentage disputes and alleged suspects of crime is well documented (Evett and Weir, 1998). More recently there has been interest in applying the same DNA technology for identification of plant varieties (Mihalov et al, 2000; Henry, 2001). Irrespective of the application, the use of DNA profiling to identify individuals requires reliable estimates of allele frequencies at the selected marker loci. Generally, allele frequency estimates are obtained from existing databases. Failure to use estimates of high precision can lead to the calculation of incorrect profile probabilities and hence unreliable probabilistic conclusions.
In diploids, the frequencies of codominant alleles can be obtained by simply counting different genotypes in the sample. There is no need to make any assumptions about the population such as random mating. When dominance is present, however, either explicitly or due to the presence of ‘null’ alleles, certain genotypes may be indistinguishable. Then, genotype frequencies cannot be directly translated to allele frequencies. In such cases allele frequencies can only be estimated iteratively by using a Newton–Raphson or EM-type algorithm, after making adequate assumptions about the population. Often this involves the assumption that the population is at Hardy–Weinberg equilibrium (Weir, 1996). One would expect the same techniques to be applicable to polyploids, which have more than two alleles per individual. We will see in the following section that polyploidy, in fact, poses some challenging problems.
Polyploids are common among plant species. For example, one of the horticultural plants of interest to us, kiwifruit, can be a diploid, tetraploid, hexaploid or octaploid, depending on the species or even the selection within a species (Ferguson et al, 1996). According to Otto and Whitton (2000), polyploidy has been one of the more predominant modes of speciation in plants. Polyploids are broadly classified as either allopolyploids that contain more than two distinct genomes, or autopolyploids, which have multiples of the same genome. Allopolyploids with their homologous pairs of chromosomes form bivalents during meiosis, just like any diploid. Following pairing, segregation at a locus in the first division of meiosis can be either reductional or equational (Mather, 1935). Equational separation at a locus is due to crossover between the locus and the centromere, which would result in chromosome pairs with heteroallelic chromatids. In contrast, the separation is said to be reductional when such an event is absent. Irrespective of the type of separation at a locus, the homologous chromosome movement to opposite poles is always disjunctional in the case of bivalents. This type of inheritance often seen in allopolyploids is referred to as disomy.
The autopolyploid segregation is much more complex (Mather, 1936). Both homologous and/or homeologous chromosomes can pair to produce various configurations during meiosis, including multivalents. The type of inheritance that follows multivalent formation is referred to as polysomy. As in the case of disomy, crossovers can happen between the locus and spindle attachment leading to equational separation at the locus. However, in a multivalent configuration, any two chromosomes of a locus separating equationally can either be attached to the same spindle or do a different one. Consequently, the resulting separation of chromosomes can be either disjunctional or nondisjunctional. Nondisjunctional separation can produce gametes with duplicate copies of the same allele, even though the parental alleles were all distinct. This process is called the ‘double reduction’ (Mather, 1935). Double reduction depends on the occurrence of three events in sequence: equational separation, genetical nondisjunction, and finally the resulting heteroallelic chromosomes lining up with the same allele facing the same direction in anaphase II of meiosis. Hence, the segregation pattern in autopolyploids differs from allopolyploids, in that it is likely to vary depending on the locus. In particular, when a marker locus is further away from the centromere, more crossovers can occur resulting in double reduction gametes and consequently a higher proportion of homozygote individuals in progeny.
While bivalent pairing and multivalent pairing are the two extremes, a number of polyploids represent intermediate stages displaying a combination of both pairing behaviours. There are several recent papers on this subject, both from a cytogenetic and a mathematical modelling point of view (Sybenga, 1994, 1995, 1996, 1999; Jackson and Jackson, 1996). The evidence suggests that these situations should be characterised by a general polyploid inheritance model with no complete preference to homologous or homeologous paring (Wu et al, 2001). The degree of homologous over homeologous paring is often described by the ‘preferential pairing factor’, which may be measured from multivalent frequencies (Sybenga, 1994).
The presence of more than two alleles in an individual and the complexity of inheritance pattern, described above, pose several problems when estimating allele frequencies in polyploids. Firstly, in polyploids even with codominant markers, the dosage of alleles cannot be deduced with certainty for some marker phenotypes, since current technology does not allow for estimating allele dosage from observed band intensities. As an example, for a tetraploid with four distinct codominant alleles showing polysomic inheritance 35 genotypes are possible but these fall under only 15 marker phenotypes. Of these marker phenotypes, genotypes can be deduced fully only in case of monoallele and quadriallele classes. Consequently, polypoid allele frequencies cannot be calculated directly. As second complication arises if ‘null’ alleles are truly present in a population. Their presence may not be obvious: unlike the case of diploids, null individuals extremely will be rare, even when the null allele frequency is moderately high.
Allele frequencies are generally estimated with sample data of unrelated individuals from a population. In such instances, the use of an EM type algorithm for estimating allele frequencies in the case of dominance is well established. Broman (2001) describes methods for estimating allele frequencies with data on sibships. When estimating allele frequencies it is often necessary to describe and assume a population structure. Ronfort et al (1998) has re-examined the population structure parameters commonly used in diploid species for tetrasomic inheritance in autotetraploids. Construction of linkage maps in autotetraploids has been studied by Hackett and Luo (2003). Luo et al (2004) have presented a theoretical basis for linkage analysis in autotetraploids. Although inheritance patterns in polyploids are covered well in the literature, there is very little or no information about methods for estimating their allele frequencies. We believe the topic is not only of interest to molecular biologist and plant breeders who wish to use DNA fingerprinting for characterisation of cultivars, but to the plant ecologist who could be studying populations from an evolutionary point of view.
Our motivation for this study comes from HortResearch's interest in managing and utilizing its collections of horticultural plant species to best advantage, particularly those of apple and kiwifruit. Many horticultural species, including kiwifruit, blueberry and citrus are polyploids. It is becoming evident that accurate identification of these germplasm accessions through DNA fingerprinting is essential. Furthermore, if DNA profiling is to be used for identification of newly developed varieties, we need to estimate allele frequencies reliably from existing allele databases. We develop an allele frequency estimation method for a general, but even-numbered ploidy level and mating under polysomic or disomic inheritance. The theoretical framework is based on a polyploid population in which a fixed fraction of individuals in any generation are selfing and others are mating at random. We assume that the population has reached equilibrium genotype frequencies after a number of generations of mating. We consider the simple case where with polysomy the inheritance is only by random chromosome segregation. Under these circumstances we derive equations that describe allele frequencies of the population. We present an EM-based algorithm for estimating allele frequencies from sample data. A programme to implement the algorithm is written in SAS/IML® (SAS, 2001). We demonstrate and verify the method on simulated datasets.
The problem description
Table 1 shows the number of genotypes and phenotypes expected for an autotetraploid with u codominant alleles per locus. In an autotetraploid up to four alleles (i, j, k and l) are present in any one individual. Autotetraploids would most likely form tetravalents during meiosis, and if random chromosome segregation occurs six different gametes would be formed, in equal proportions: ij, ik, il, jk, jl, kl. These combine randomly to give four possible phenotypic classes: mono, bi-, tri- and quadriallelic. Of these, there is one-to-one agreement between the phenotype and the genotype only in case of mono and quadriallelic classes. The biallelic class, for example could be a simplex (iiij or ijjj) or a duplex (iijj), and in the triallelelic class any of the three alleles could be the paired one, ie iijk, ijjk and ijkk. Therefore, in an autotetraploid the four phenotypic classes in fact arise from eight different genotypic classes. Of course the actual number of phenotypes and genotypes will depend on u, the number of alleles per locus (Table 1). Even with u=4, the number of phenotypes expected is less than half the number of genotypes (15 against 35, Table 1).
As noted earlier the segregation in an allotetraploid will resemble that of a diploid and we expect the genotypic and phenotypic classes in progeny to be different from that of an autotetraploid. For an individual carrying four alleles (i, j, k and l) there is now preferential pairing such that say, i∣j and k∣l. We will refer to the two ‘pseudoloci’ of the homologous pairs of chromosomes at the same locus as homeologous loci. An allotetraploid will, therefore, form only four different gametes: ik, il, jk and jl. These pair at random to form four progeny genotype classes: ijkl, iikl, ijkk, iikk. It is noted that if alleles at the two homeologous loci are nonoverlapping, a monoallelic genotypic class will not be possible. Furthermore, if we know which alleles belong to which homeologous locus, the genotypes can be distinguished fully by their phenotypes. However, often this is not the case and several combinations of the allelic distribution need to be tested against the observed frequencies of phenotypes to make any reliable conclusions. Also, the presence of ‘null’ alleles would make the problem more complicated even with disomic inheritance.
The presence of a null (unidentifiable) allele will make more genotypes hidden. For example, for the autotetraploid case if one of the four alleles is a null we would still expect 35 genotypes, but only eight phenotypes (Table 1). Null individuals will be rarer in polyploids than diploids. As an example, if the null allele is present at 20% in an autotetraploid population, assuming random chromosome segregation the null individuals are expected to occur at 0.16%, that is, less than 2 in 1000. In a diploid with the same allele frequency, it is expected at 4%. Thus at higher ploidy levels even with large samples absence of null individuals is not good evidence of the absence of a null allele. Consequently, the null allele should be always included in any estimator of allele frequencies in polyploids.
Statistically, the estimation of allele frequencies in polyploids is one of incomplete data. We will need to make certain assumptions about the genetic structure of a population in order to proceed. The usual assumption in the case of diploids is that the population is random mating and is at Hardy–Weinberg equilibrium. Since we are primarily interested in plant species, selfing needs to be considered as a possibility. Many plants reproduce by a mixture of random mating and selfing. With random mating Hardy–Weinberg proportions are attained after a single generation. In contrast, each generation of selfing will increase the number of homozygotes at the expense of heterozygotes. In a mixed mating population the two opposing effects of outcrossing and selfing will reach equilibrium asymptotically. Obviously, with more selfing, the number of generations required to reach these equilibrium genotype frequencies is greater. One approach to derive the outcome of mixed mating is to consider the population as a mixture of two populations, that is, one mating by random and the other only by selfing. This is equivalent to saying that each plant reproduces a fraction, s, of time by selfing and the remainder of time by random mating.
Throughout this paper we will use the following notation: m=polyploidy level, that is, number of alleles per individual per locus; u=number of different alleles per locus in the population excluding the null; pi=frequency of ith allele; Pijkl=frequency of ijklth genotype in a given generation; Rijkl=frequency of ijklth genotype following random mating only; s=selfing fraction per generation.
We seek to develop an estimator of allele frequency for a general (but even numbered) polyploid population, which has mixed mating and where the inheritance pattern is either polysomic or disomic.
The polyploid inheritance model
We will first develop a theoretical model that describe allele frequencies in a tetraploid population and later show how it could be generalised to any even numbered ploidy level. We make the following assumptions about the population: (1) inheritance is either by polysomy or disomy and not by a mixture of both; (2) only random chromosome segregation occur in meiosis, that is, no crossovers; (3) a mixed mating system, that is, individuals mate by selfing by a fixed fraction, s, in each generation and the remaining mating is random; (4) unidentified alleles are present and classified together as ‘null’; (5) there is no selective advantage for any genotype, which implies allele frequencies remain unchanged between parents and their progeny; (6) the population is at equilibrium.
Polysomic inheritance
Ignoring nulls for the moment, let the parent allele frequency vector be p = (p0, p1, pi … pu). With polysomic inheritance, the expected genotype frequencies in progeny after a generation of random mating should follow a multinomial distribution,

Following (1), for a tetraploid the expected frequencies of marker genotype classes in progeny after random mating are: monoallele  biallele (simplex)
biallele (simplex)  biallele (duplex)
biallele (duplex)  triallele
triallele  and quadriallele
and quadriallele  .
.
Now we take the situation where the mating is only by selfing. To deal with selfing we need to follow through each parent genotype rather than the allele frequencies of the parent population. With polysomic inheritance a selfing tetraploid forms up to six different gametes and these combine in pairs at random. Hence, the progeny resulting from selfing of each parent genotype class are:

By collecting terms of each progeny genotype from the right hand side of (2) we can write out the new genotype frequencies after one generation of selfing as:

where the sums are over all subscripts not in the variable on the left. It is a simple case now to extend this to a population that is mixed mating by taking a proportion s of individuals mating by selfing and the remaining (1–s) mating at random. Again, for the case of a tetraploid showing polysomic inheritance we get:

where P′… and P… are respectively genotype frequencies in the new progeny and the parent generation, and R… are genotype frequencies generated by random mating of parents as given by (1). Note that R… is a function of parental allele frequencies.
With mixed mating genotype frequencies will come to equilibrium only gradually. At the steady-state genotype frequencies in the new generation equal the frequencies in the old, so these equations can be solved for P… in terms of R… A tidier approach, however, is to write the set of Equation (4) in matrix form as,

where A is a ng × ng matrix we call the ‘selfing matrix’, with ng being the number of genotypes. Each element of the selfing matrix is the proportion of the genotype represented by column formed by selfing of the genotype indicated by the row. Note that for a given set of alleles, and hence the genotypes, the selfing matrix is known in advance. At equilibrium the two vectors P′ and P are equal, hence (5) can be rearranged to give:

where E[P] is now the vector of expected equilibrium genotype frequencies.
Disomic inheritance
We have seen that disomic differs from polysomic inheritance simply in the way the alleles at a given locus are selected to form gametes. In the case of an allotetraploid – instead of random selection from a set of four chromosomes, the set is now divided into two ‘homeologous loci’ with two chromosomes in each, and one chromosome is chosen from each locus to form gametes. For a hexaploid, there would be three homeologous loci each containing two chromosomes etc. Here we refer to many ‘loci’ within one ‘location’, but the mathematics should be identical to the case of diploid inheritance, that is, observing nonconnected loci showing diploid inheritance together in multiples (two-at-a-time in case of a tetraploid). Here, we consider only the case of ‘disjoint’ or ‘nonoverlapping’ allele sets. There is also the possibility that allele sets are ‘overlapping’, that is, one or more alleles are common across different pairs.
As with polysomic inheritance, consider the case of a tetraploid. We use the same notation but restricting i and j as subscripts for the first homeologous locus, k and l as subscripts for the second. The corresponding frequency vector for the first and second loci is: p=(p0, p1, pi … pu1) and q=(q0, q1, qk … qu2). First ignoring the nulls, with random mating the resulting genotype class frequencies will be given by:

Given the disjoint assumption, monoallelic individuals are only possible if one of the two loci contains a null allele.
As before, we now consider the genotype frequencies following selfing. Once again, for simplicity we initially ignore the nulls, so there are only four genotypic classes, all individually observable. Selfing leads to the following in the next generation:

Hence, with selfing the genotypes frequencies in the new progeny generation will be,

Putting the proportion s of selfing from (9) together with the proportion (1–s) of random mating from (7) we get a set of equations similar to that of (4). Now assuming steady state, so the genotypic proportions in the new generation (P′…) are the same as in the old (P…), we can solve the equations to give P… as functions of the allele frequencies. As with the polysomic case, putting it in matrix form yields the same form of expression for E[P].
Effect of null alleles
We note that the situation for a null allele is the same as that for a recessive marker allele. Hence, any solution we come up for the null can equally be applied to dominant/recessive markers. We will investigate the effect of null alleles on observed phenotype frequencies by taking as an example the disomic case with m=4, and with nonoverlapping alleles. We note that in the absence of null alleles, only the bi-, tri- and quadriallele phenotypic classes are possible (7). If a null allele is present at each homeologous locus, in addition to the above, five new genotypic classes are possible:  with i=0 and k=0 are the phenotypic null individuals (note: P.(P) denote the phenotypic frequency); monoallelic at the first locus,
with i=0 and k=0 are the phenotypic null individuals (note: P.(P) denote the phenotypic frequency); monoallelic at the first locus,  with j=0 and k=0; monoallelic at the second locus,
with j=0 and k=0; monoallelic at the second locus,  with i=0 and l=0; biallelic with both alleles from the first locus,
with i=0 and l=0; biallelic with both alleles from the first locus,  with k=0; biallelic with both alleles from the second locus,
with k=0; biallelic with both alleles from the second locus,  with i=0. Furthermore, the usual bi- and triallelic phenotypic classes (7) will now include genotypes containing null alleles, such that
with i=0. Furthermore, the usual bi- and triallelic phenotypic classes (7) will now include genotypes containing null alleles, such that  ; etc.
; etc.
In general, when finding which alleles belong to which homeologous locus we may need to consider all possible combinations of alleles at each locus. Hopefully, all except one combination can be excluded based on observed individual phenotypes. It is more likely that a few combinations are all possible and a final decision needs to be made based on maximum likelihood of observed individual frequencies given the fitted parameters of allele frequencies. In the estimation method, which will be described in the next section, we have assumed that allele distribution across homeologous loci are known a priori. In the case of polysomy this is not necessary since all alleles belong to the same locus.
So, including nulls the theory is exactly the same but for the observation process, and the fact that there is now one more allele (u+1) in case of polysomy, and one more allele at each homeologous locus (u1+1, u2+1) for disomy. The observation process can be factored in when a set of genotypes are mapped on to the corresponding set of phenotypes. Computationally, the process involves converting the genotypic frequency vector P to the phenotypic one, P(P). We do this by a conversion matrix, C with dimensions equal to the numbers of phenotypes by genotypes, np × ng such that,

The challenge is now to computationally generate the C matrix for the general case of m ploidy level and any possible number of alleles per locus including the null.
Estimation
Given the observed allelic phenotypes of individuals making up a sample, the objective here is to estimate allele frequencies that maximise the likelihood of the observed outcome. As a result of some marker genotypes are indistinguishable, the estimation problem is one of incomplete data. The Expectation Maximisation (EM) algorithm (Dempster et al, 1977) is an iterative procedure that can lead to maximum likelihood estimation (MLE) of model parameters in situations where observed data are considered to be incomplete. Each iteration consists of an expectation step followed by a maximisation step, and iterations continue until convergence, that is, successive parameter values are very close to each other and further iterations show no significant improvement. For within population analyses, the multinomial distribution is used as a basis for likelihood estimation of allele frequencies (Weir, 1996). If the observed multinomial counts, Ni(i=1, …, np) of phenotypes depend in general on a set of parameters, φj (j=1, …, ν), so that the expected frequencies are functions of φj's, Qi (φ1, …, φν), the likelihood is written as:

When the parameter vector, φ, is equivalent to the allele frequency vector, p it can be shown that MLE of p is just the frequencies found using the estimated genotype frequencies (Weir, 1996). An alternative estimation method is to use a Bayesian approach (Weir, 1996) where a prior distribution is assumed for the parameters, which are the allele frequencies. When there are only two alleles, sampling can be assumed to be binomial and the beta distribution, which is the conjugate to binomial, is the most appropriate prior. Where several alleles are involved and the sampling distribution is multinomial, the Dirichlet prior, which is the conjugate for multinomial can be used. The likelihood is the probability for sample data conditional on the parameters. Given the prior and the likelihood of sample data, a posterior distribution could be calculated.
We take the MLE approach, and use the EM algorithm for estimating the allele frequency parameter vector. In our situation, the EM algorithm consists of the following steps:
-
1)
assign initial values to the allele frequency vector, p
-
2)
E-step:
-
a)
use the value of current vector p(0) with known values of s and the matrix A to calculate expected genotype frequencies using (6), that is, E[P]=(1−s)(I−s AT)−1R
-
b)
now use the E[P] and the observed phenotypic frequency vector, P(PObs) to provide the estimated genotype frequency vector, P̂. Computationally, these steps can be described as follows:

where C is the conversion matrix as defined in Equation (10), and U is a unit vector of dimension ng. Note the symbols ‘*’ and ‘^’ are, respectively, the matrix multiplication and the exponentiation operator, and the same preceded by a ‘.’ are the corresponding elementwise operators;
-
a)
-
3)
M-step: use P̂ to count and calculate the new allele frequency vector,
 where G is a ng × (u+1) matrix for which elements within each row given by 0 or 1 indicate absence or presence of each allele, and m the ploidy level.
where G is a ng × (u+1) matrix for which elements within each row given by 0 or 1 indicate absence or presence of each allele, and m the ploidy level. -
4)
repeat the process until convergence, that is
 , where pi(t) is the ith element of the frequency vector in tth iteration and c the tolerance value which is set to 10−8 in our case.
, where pi(t) is the ith element of the frequency vector in tth iteration and c the tolerance value which is set to 10−8 in our case.

 where G is a ng × (u+1) matrix for which elements within each row given by 0 or 1 indicate absence or presence of each allele, and m the ploidy level.
where G is a ng × (u+1) matrix for which elements within each row given by 0 or 1 indicate absence or presence of each allele, and m the ploidy level. , where pi(t) is the ith element of the frequency vector in tth iteration and c the tolerance value which is set to 10−8 in our case.
, where pi(t) is the ith element of the frequency vector in tth iteration and c the tolerance value which is set to 10−8 in our case.We have written a computer programme in SAS/IML® that can estimate allele frequencies from sample phenotypic data from a population, which shows polysomic inheritance. It would be possible to extend it to the disomic case. The system information, assumed to be known is, the ploidy (m, but even numbered), number of alleles (u), and the selfing fraction (s). The SAS programme was written as a series of subroutines and functions. Here we do not attempt to give details of computations, but only name some of these subroutines/functions with their specific functionalities: GENLIST, sets up the list of genotypes; PHENLIST, sets up the phenotypic array; INDEXG, returns the index value of a genotype from the genotypic array; INDEXP, returns same from the phenotypic array; RANMUL, sets up multipliers for genotype frequencies under random mating; SELFMAT, sets up the selfing matrix, A; CONVMAT, sets up the conversion matrix, C; GPROBS, computes the expected equilibrium genotype frequencies given the selfing fraction and allele frequencies; SIMSAMPLE, simulates a random sample from the idealised population; EXPECTATION and MAXIMISATION, compute expectation and maximisation of the EM algorithm, respectively.
Simulation study
We performed several simulation experiments to verify our estimation method. We generated simulated data of varying sample size n from an infinite population with ploidy level m, having a locus containing a number u of distinct alleles (plus the null) with a frequency vector p=(p0, p1, pi … pu), and mating by a mixture of selfing (s) and random mating (1−s). Note the first element of the allele frequency vector represents the ‘null’ allele. It was assumed that inheritance was by polysomy and the population had reached equilibrium genotype frequencies. In the first set of simulations, we set: u=4, s=0.5, p=(0.2, 0.1, 0.2, 0.3, 0.2), but varied the sample size n=50, 100 or 500. For each sample size we generated 1000 simulations and estimated allele frequencies for each simulated sample by applying the proposed method. The mean of estimated allele frequency vectors, p̂ over the 1000 simulated samples and its S.E. are shown in Table 2. Clearly, the estimation method has performed extremely well with no significant bias at any of the sample sizes used. The estimated SE of the estimator, except that of the null allele, p̂0 closely agreed with SE expected for a completely random mating population, that is, p(1−p)/(2n). Compared with alleles 2 and 4 which have the same frequency as the null allele (p=0.2), it is apparent that the null was estimated at a much lower precision (Table 2). This would be expected because, in contrast to others, the null allele is observed phenotypically only if null individuals are present in the sample. The median number of iterations for convergence was ∼40.
We also tested the method for situations where allele frequencies were more unequal, for example p=(0.2, 0.02, 0.05, 0.08, 0.65), Table 2. These results also indicate the method performed consistently well, except for an apparent slight downward bias on the null allele frequency with small sample sizes. For the null allele when allele frequencies were more unequal the precision was lower (Table 2). The median number of iterations for convergence for this set of samples was ∼80.
In order to be confident that the method works well with other ploidy levels we conducted the above simulation experiment with the same parameter values, p=(0.2, 0.1, 0.2, 0.3, 0.2), but for a hexaploid (m=6), (Table 2). The number of iterations for convergence was close to double that required for the tetraploid. We also verified the estimation method for a range of selfing fractions (Table 3). The estimated frequencies agreed very closely with the actual.
Discussion
Even with codominant alleles, estimation of allele frequencies in polyploids is complicated because a high proportion of genotypes are indistinguishable. An estimation method for the general case of any even ploidy level with either polysomic or disomic inheritance has been presented here. When tested against simulated data, the estimation algorithm written in SAS/IML® provided quick convergence and unbiased estimates under different ploidy levels and varying sample sizes, giving a method for easily estimating allele frequencies. However, with real data, the reliability of the estimator would depend very much on the validity of the assumptions made in deriving our inheritance model.
A key assumption that might be violated in a real population is that of genotype frequency equilibrium. This assumption may be quite reasonable in population studies in ecology where mating within and between individuals in the population has gone on for many generations. However, horticultural and agricultural germplasm collections are seldom single populations that have reached such equilibria. We hope to investigate further the effect of lack of genotype frequency equilibrium on our allele frequency estimator. The inheritance model proposed assumed either disomic or polysomic inheritance. As noted earlier, in reality, inheritance patterns in polyploids can be much more complex. While polysomy and disomy are the two extremes, many polyploids actually exhibit a combination of both types. However, it may be argued that a given marker located on a certain chromosome would consistently behave one way or the other. Our model also assumed that there were no crossovers between the centromere and marker locus. When a marker locus is distant from the centromere crossovers can happen which in the case of polysomic inheritance can lead to double-reduction gametes. It would be possible to accommodate this in the model by taking double reduction as the joint probability of four independent events happening in sequence: formation of polyvalents, q; formation of equational heteroallelic chromosomes due to a crossover between locus and centromere, e; nondisjunction in Anaphase I, a; and finally correct orientation in Metaphase II, that is, the two sister chromatids moving to the same pole in Anaphase II, probability=½. For an autotetraploid, assuming the probability of quadrivalent formation q=1 we get the Pr{double reduction}, α=ea/2. If chromosomes pair randomly when moving to the same pole at Anaphase I, then a = for a tetraploid. The probability e will depend on the crossover frequency, which in turn depends on the physical distance between the locus and the centromere. In the extreme case of free recombination, e = [fraction 6 over 7] that is, random chromatid segregation (Mather, 1936) with 1/7 reductional and [fraction 6 over 7] equational separation. This implies that in a typical case of an autotetraploid, one could expect the double reduction events to occur at a frequency of 1/7 This is an aspect we plan to incorporate into our estimation procedure in the future.
In this paper, we have assumed that the proportion of selfing (s) is known, or can be estimated from knowledge of the mating system. In cases where the proportion of selfing is unknown, the EM algorithm outlined above can be extended to incorporate the estimation of s as well, albeit with some reduction in the precision of the allele frequency estimates. An estimator for s can be obtained by rearranging (5) to give

A least-squares estimator of s is therefore

At the same point in each iteration of the EM algorithm where new genotype frequencies are estimated using (6), this equation can be used to obtain new estimates of the proportion of selfing (s). It is worth noting that with the selfing estimated using the least-squares approach, the estimates of allele frequency obtained will no longer in general be maximum likelihood.
References
Broman KW (2001). Estimation of allele frequencies with data on sibships. Genetic Epidemiol 20: 307–315.
Dempster AP, Laird NM, Rubin DB (1977). Maximum likelihood from incomplete data via EM algorithm. J R Stat Soc Ser B 39: 1–38.
Evett IW, Weir BS (1998). Interpreting DNA Evidence: Statistical Genetics for Forensic Scientists. Sinauer Associates: MA.
Ferguson AR, Seal AG, McNeilage MA, Fraser LG, Harvey CF, Beatson RA (1996). Kiwifruit. In: Janick J, Moore JN (eds) Fruit Breeding, Vine and Small Fruit Crops. John Wiley & Sons: New York, Vol II, pp 371–471.
Hackett CA, Luo ZW (2003). TetraploidMap: construction of a linkage map in autotetraploid species. J Hered 94: 358–359.
Henry RJ (2001). Plant Genotyping: The DNA Fingerprinting of Plants. CABI Publishing: New York.
Jackson RC, Jackson JW (1996). Gene segregation in autotetraploids: prediction from meiotic configurations. Am J Bot 83: 673–678.
Luo ZW, Zhang RM, Kearsey MJ (2004). Theoretical basis for genetic linkage analysis in autotetraploid species. Proc Natl Acad Sci 101: 7040–7045.
Mather K (1935). Reductional and equational separation of the chromosomes in bivalents and multivalents. J Genet 30: 53–78.
Mather K (1936). Segregation and linkage in autotetraploids. J Genet 32: 287–314.
Mihalov JJ, Marderosian AD, Pierce JC (2000). DNA identification of commercial ginseng samples. J Agric Food Chem 48: 3744–3752.
Otto SP, Whitton J (2000). Polyploid incidence and evolution. Ann Rev Genet 34: 401–437.
Ronfort J, Jenczewski E, Bataillon T, Rousset F (1998). Analysis of population structure in autotetraploid species. Genetics 150: 921–930.
SAS Institute Inc (2001). SAS/IML® Software: Changes and Enhancements, Release 8.2, Cary, NC.
Sybenga A (1994). Preferential pairing estimates from multivalent frequencies in tetraploids. Genome 37: 1045–1055.
Sybenga A (1995). Meiotic pairing in autohexaploid Lathyrus: a mathematical model. Heredity 75: 343–350.
Sybenga A (1996). Chromosome pairing affinity and quadrivalent formation in polyploids: do segmental allopolyploids exist? Genome 39: 1176–1184.
Sybenga A (1999). What makes homologous chromosomes find each other in meiosis? A review and a hypothesis. Chromosoma 108: 209–219.
Weir BS (1996). Genetic Data Analysis II. Sinauer Associates: MA.
Wu R, Gallo-Meagher M, Littell RC, Zhao-Bang Z (2001). A general polyploid model for analysing gene segregation in outcrossing tetraploid species. Genetics 159: 869–882.
Acknowledgements
This work was supported by a grant from the New Zealand Foundation for Research, Science and Technology. We thank the two referees and the Editor for their valuable comments on an earlier version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
De Silva, H., Hall, A., Rikkerink, E. et al. Estimation of allele frequencies in polyploids under certain patterns of inheritance. Heredity 95, 327–334 (2005). https://doi.org/10.1038/sj.hdy.6800728
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.hdy.6800728
Keywords
This article is cited by
-
Sensitivity to habitat fragmentation across European landscapes in three temperate forest herbs
Landscape Ecology (2021)
-
Sequencing depth and genotype quality: accuracy and breeding operation considerations for genomic selection applications in autopolyploid crops
Theoretical and Applied Genetics (2020)
-
Strategies of molecular diversity assessment to infer morphophysiological and adaptive diversity of germplasm accessions: an alfalfa case study
Euphytica (2020)
-
Molecular discrimination and ploidy level determination for elite willow cultivars
Tree Genetics & Genomes (2018)
-
Population assignment in autopolyploids
Heredity (2017)
