
Abstract
Nasopharyngeal carcinoma (NPC) is a common cancer in Southern China and Southeast Asia. The disease is a poorly differentiated carcinoma without effective cure, and the mechanism underlying its development remains largely unknown. Of several factors identified in NPC aetiology in recent years, Epstein-Barr virus (EBV) infection has emerged to be most important. In almost all NPC cells, EBV uses several intracellular mechanisms to cause oncogenic evolution of the infected cells. One such mechanism by which EBV infection induces cellular immortalization is believed to be through the activation of telomerase, an enzyme that is normally repressed but becomes activated during cancer development. Studies show that greater than 85% of primary NPC display high telomerase activity by mechanisms involving EBV infection, consistent with the notion that EBV is commonly involved in inducing cell immortalization. More recently, different EBV proteins have been shown to activate or inhibit the human telomerase reverse transcriptase gene, by modulating intracellular signalling pathways. These findings suggest a new model with a number of challenges towards our understanding, molecular targeting and therapeutic intervention in NPC.
Similar content being viewed by others


Introduction
Nasopharyngeal carcinoma (NPC) is a malignancy of the head and neck. It occurs commonly in the Southern China, Southeast Asia and the Mediterranean basin, but rarely in Europe and North America (<3 cases/100 000 persons/year). Of the endemic areas, highest-incidence foci are in Guandong and Guangxi provinces of South China (25-40 cases/100 000 persons/year). Median risk areas include the Philippines, Indonesia, Vietnam and several countries of North and West Africa, with incidence of 4-8 cases/100 000 persons/year. Based on its histopathology, NPC is classified into three categories, World Health Organization (WHO) type I, type II and type III. Most cases have minimal epithelial maturation and are classified as undifferentiated (WHO type III) or poorly differentiated non-keratinizing (WHO type II) tumours. Some cases are differentiated keratinizing squamous-cell carcinomas (WHO type I) 1, 2, 3.
The mechanisms underlying the development of NPC remain largely unknown 1, 2, 4. Several factors have been identified in its aetiology in recent years, including infection of the Epstein-Barr virus (EBV), environmental risk factors and genetic susceptibility 1, 2, 5. Of these aetiological factors, EBV infection appears to be the most important one 1, 2, 3, 4, 6. As one of the best examples of a human solid tumour consistently associated with virus infection, NPC is associated with EBV constantly, and this is particularly the case in advanced diseases. Importantly, EBV is present in the NPC cells early in the carcinogenic process, before expansion of the malignant cell clone 7. EBV may cause the NPC cell oncogenic clonal evolution by interfering with several intracellular mechanisms in the infected cells 1, 7, 8, 9, 10, 11. Recently, evidence suggests that the enzyme telomerase required for cell immortalization is activated in NPC and the EBV proteins are involved in regulating telomerase activity 12, 13, 14, consistent with the findings that EBV proteins also play a key role in immortalization of infected B-lymphocyte 15, 16, 17. Here we review the recent progress in our understanding of regulation of telomerase activity by EBV proteins, a critical step in cell immortalization.
EBV proteins – latent membrane protein (LMP)1, LMP2A and EBV nuclear antigen (EBNA)2
EBV is a ubiquitous herpesvirus that preferentially infects oropharyngeal epithelial cells and B-lymphocytes in humans. The virus is present in all NPC often including pre-invasive nasopharyngeal lesions 7, 18. Although EBV may cause symptoms with a long latency following infection, and the virus can persist as an asymptomatic infection for decades, the virus causes diseases ranging from mild self-limited illness to infectious mononucleosis. While infected epithelial cells may undergo clonal expansion and immortalization to develop NPC, B-lymphocytes with latent infection are predisposed to transformation with propensity to develop lymphomas (such as Burkitt's lymphoma, post-transplant B-cell lymphoma and Hodgkin's disease) 6, 9. Little is known of the mechanisms underpinning the different processes of EBV infection and why the EBV-infected NPC cells undergo increased proliferation 19. Cytogenetic studies have shown that there are many chromosomal abnormalities in NPC tissues suggesting that genomic instability is a common feature in NPC development 20, 21.
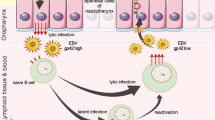
The EBV genome is large exceeding 172 kb of double-stranded DNA and encodes more than 100 genes. At least 10 genes are for EBV replication, six for nuclear antigen proteins (EBNAs) and three for LMPs 22, 23, 24, 25. Several copies of the EBV genome are contained in the nuclei of NPC cells in the form of circular independent DNA molecules (episomes). They give rise to some consistent gene expression for two small untranslated EBV RNAs and viral proteins with oncogenic properties. These viral proteins contribute to cell immortalization and malignant transformation by various signalling mechanisms (Figure 1). While most of the 100 or so EBV genes are silent dictating EBV infection of NPC cells to be mainly latent 25, 26, the transforming effect of EBV on B-lymphocyte is known to involve restricted expression of a subset of latent virus genes. Expression of EBNA1, LMP1, LMP2A and LMP2B has been implicated as important factors for NPC and Hodgkin's disease, while different forms of latency are associated with post-transplant B lymphoma (expression of LMP1 and LMP2) and Burkitt's lymphoma (expression of EBNA1). The LMPs can function as activated receptors in the cell membrane to generate signalling cascades, and the EBNAs are able to move into the cell nucleus to affect gene expression more directly (Figure 1). Among identified viral proteins, LMP1 is best characterized and detected in most NPC patients 18, 27.

Paradigm of the signalling cascades from EBV to cell immortalization and transformation. EBV episomes in NPC cell nucleus express LMPs and nuclear antigens. LMPs intercept and assume constitutive intracellular signalling that is normally mediated by ligand and receptor interactions, while EBNAs can enter the cell nucleus to affect gene expression relatively directly.
LMP1 is an integral plasma membrane protein that consists of a 24 amino acid cytoplasmic N-terminus, six hydrophobic transmembrane domains (TM1–6, separated by five short reverse turns) and a 200 amino acid cytoplasmic C-terminus. LMP1 can assume functions analogous to the constitutively active tumour necrosis factor receptor (TNFR) with the ability to interact with TNFR-associated factors 10. Based on their resemblance to CD40 (a member of the TNFR) and ability to activate the NF-κB transcription factor pathway, two distinct functional domains referred to as COOH-terminal activation regions 1 and 2 have been identified in LMP1 6, 28. LMP1 interacts with members of the TNFR-associated factors family, especially TNFR-associated factor-2 (TRAF2) that serves as an intermediate in the signalling pathway leading to transcriptional activation of NF-κB 10 and thereby potentially human telomerase reverse transcriptase (hTERT) 29. In addition, LMP1 also induces the mitogen-activated protein kinase (MAPK) cascade resulting in the activation of extracellular-regulated protein kinase (ERK), c-Jun NH2-terminal kinase and p38, and stimulates the JAK/signal transducers and activators of transcription pathway and phosphatidylinositol 3-kinase pathway 30, 31. Furthermore, expression of LMP1 has been shown to induce chromosomal aberrations by enhancing spontaneous and bleomycin-induced micronucleus formation, through a mechanism involving a compromised DNA repair capacity in a p53-independent manner in NPC cells 21.
LMP2 is also a transmembrane protein with the N-terminal cytoplasmic tail containing several tyrosine phosphorylation sites. Similar to the B-cell receptor, the immunoreceptor tyrosine-based activation motif (ITAM) of LMP2A recruits and is tyrosine phosphorylated by the src family of protein tyrosine kinases and the syk protein tyrosine kinase. Phosphorylation of the tyrosine residues leads to subsequent activation of phospholipase C-γ2 and calcium/protein kinase C signalling pathways 6. Playing a role in activation of telomerase 32, protein kinase C is implicated in regulating IκB and myc proto-oncogene expression 33. In addition, EBV infection in NPC has been shown to alter the function of p53 and MAPK signalling pathways 8, 10. Thus, EBV LMPs may target multiple intracellular signalling pathways and various sites of cellular machinery important in controlling cell proliferative lifespan during the development of NPC.
By contrast, EBNAs target to the nucleus to affect specific genes more directly. Several nuclear antigens (EBNA1, -2, -3A and -3C) are apparently essential for B-cell immortalization 34, 35. Lacking significant sequence homology to cellular proteins, however, EBNA2 that is transcribed under the W promoter after initial infection of EBV stimulates the bi-directional viral latency C promoter and the LMP2A promoter, thereby transactivating both LMP1 and LMP2A 36. Furthermore, EBNA2 stimulates the expression of cellular genes including c-myc as well as CD21, Hes-1, EBI 1 and 2, and Runx3; but the spectrum of cellular genes directly activated by EBNA2 remains to be fully elucidated 36. Although it does not bind to DNA directly, EBNA2 transactivates target genes by hijacking the DNA-binding protein known as CBF1 (CBF1/RBP-Jk, suppressor of hairless, Lag-1). Under normal cellular conditions, CBF1 transcription factor is bound by intracellular Notch (ICN) – a product of enzymatic cleavage of membrane-bound Notch, and in the presence of ICN, the CBF1 complex recruits co-activators (such as mastermind-like proteins) to become part of a transcriptional activation complex containing p300, CBP and P/CAF histone acetyltransferases. This complex activates various targets including the LMP1 promoter 37, 38. The EBNA2/CBF1 complex can similarly activate gene transcription by recruiting several components of gene transcription machinery using the EBNA2 C-terminal acidic activation domain 38. Thus, through interaction with CBF1, EBNA2 constitutively transduces “Notch” signalling activity.
Telomeres, telomerase and cell immortalization
Telomeres are specialized structures at the ends of eukaryotic chromosomes. They are comprised of extended arrays of repeated G- and C-rich hexanucleotides and their binding proteins. The functions of telomeres include protecting chromosome ends from recombination, fusion and degradation, and regulating cell proliferative lifespan 39. Telomeres shorten with each round of DNA replication due to the inability of conventional DNA polymerases to replicate the extreme ends of chromosomes 39. Hence, cell division is accompanied by gradual reduction in telomere length. Characteristic of replicative senescence, short telomere triggers cells to exit the cell cycle permanently. To overcome telomere shortening, activation of the specialized reverse transcriptase telomerase maintains telomeres, which enables cells to divide continuously 39, 40.
Telomerase is a large ribonucleoprotein complex responsible for the maintenance of telomeres and continuous proliferation of germ line, stem and neoplastic cells. The core telomerase enzyme contains an RNA moiety and a protein subunit. Human telomerase RNA (hTERC) comprises 450 nucleotides, whereas the protein component hTERT is a polypeptide of 1132 amino acid residues 41, 42, 43, 44. Being the catalyst interacting with both hTERC and telomeric DNA, hTERT contains four major regions: an N-terminal regulatory (R) domain, RNA-binding (RB) domain, reverse transcription (RT) domain and C-terminal dimerization domain 45. Several motifs in the R and RB domains of hTERT are conserved in different species, important for various molecular interactions 41, 46, 47, 48. As the rate-limiting component of telomerase, hTERT induces immortalization of a number of cell types in culture 49 (reviewed in 50). Recently, hTERT has also induced an immortalized nasopharyngeal epithelial cell line from non-malignant nasopharyngeal biopsies, which is accompanied by other genetic alterations seen in NPC including homozygous deletion of the p16INK4A 12. Consistently, hTERT is below detectable levels in most normal human somatic cells, but is expressed concomitantly with telomerase activity during cellular immortalization 40. This endorses the notion that hTERT gene expression is the first, rate-limiting step of telomerase activation in cellular immortalization 49.
Molecular mechanisms regulating telomerase activity
The regulation of telomerase activity has been investigated with a focus on the rate-limiting component hTERT. Studies have shown that hTERT is controlled from gene expression (epigenetic, transcriptional and splicing) to post-translational modification (phosphorylation and protein-protein interactions). Most noteworthy of these levels of regulation is hTERT transcription. The hTERT promoter encompasses binding sites for various transcriptional regulators including c-myc, Sp1, p53, ETS and oestrogen (E2) receptor 50. Complex interactions between distinct transcription factors and the hTERT gene promoter enable activation of the hTERT gene transcription and thereby telomerase activation in response to a wide variety of environmental factors and signalling pathways (Figure 2). These include MAPK, transforming growth factor β (TGF-β) and NF-κB signalling pathways. Signals transmitted through MAPK family can increase or decrease hTERT transcription depending on different stimuli 51, 52, 53, 54, 55, whereas TGF-β inhibits hTERT gene transcription by Smad 3 signaling 56, 57, 58. In addition, hTERT is also targeted for activation by several viral oncogenes including human papillomavirus (HPV) type-16 E6/E7 and simian virus 40 T antigens 59, 60.

Schematic illustration of molecular regulation of the hTERT gene promoter by intracellular signalling pathways. The multifactorial regulation of the hTERT gene includes E2, oestrogen receptor (ER) and its antagonist tamoxifen (TM), TGF-β and its downstream signalling protein Smad3, and the signalling molecule ceramide (CM). The action of the proto-oncogene c-myc is regulated by TGF-β, oestrogen, CM and MAX/MAD network. The EBV protein LMP1 transduces signals through recruiting TNFR-associated factors such as TRAF2 that may stimulate NF-κB-mediated activation of hTERT (see Figure 3). The EBV protein LMP2A may recruit Syk tyrosine kinase to probably induce AP-1-mediated inhibition of the hTERT gene through ERK (see Figure 3). Stimulatory effects are in blue lines and inhibitory actions are in red dot lines.
At post-translational levels, we and others have shown that hTERT is a phosphoprotein and that phosphorylation by protein kinase C and Akt up-regulates telomerase activity 61, 62, 63. Conversely, protein phosphatase 2A dephosphorylates hTERT and inhibits telomerase activity 61. In addition, several other proteins have been implicated in playing direct and indirect roles regulating human telomerase activity (reviewed by Smogorzewska and de Lange 64). These proteins include heat-shock proteins p23 and Hsp90, PinX1 and p53, with p23 and Hsp90 being implicated in facilitating hTERT folding and telomerase holoenzyme assembly 65, 66, 67, 68. PinX1 as a telomere-binding protein associates with hTERT/hTERC complex in vitro and inhibits telomerase activity 69, 70, 71. Moreover, p53 also associates with telomeres and inhibits telomerase 72, 73, 74, 75. Thus, these multitudes of regulation facilitate tight controls of hTERT and telomerase in various scenarios and milieus.
Regulation of telomerase activity by the EBV proteins
Several recent studies have shown that EBV causes cell immortalization by employing its viral proteins to acquire control mechanisms over the hTERT gene. In both primary human nasopharyngeal epithelial cell culture and NPC cell lines, telomerase is stimulated by expression of LMP1 in association with increased gene expression of c-myc 14, 15. Withdrawal or suppression of LMP1 expression decreases telomerase activity and c-myc transactivation, suggesting a causative relationship between LMP1 and hTERT 14, 15. Blocking c-myc gene expression or mutating the c-myc-responsive E-box elements in the minimal core of hTERT promoter inhibits the hTERT expression induced by LMP1, pointing to a c-myc-dependent transactivation of the hTERT gene by LMP1 14 (Figure 3). Moreover, LMP1 has recently been shown to modulate telomerase activity by inducing a direct binding of NF-κB p65 subunit to hTERT and thereby facilitating translocation of both proteins from the cytoplasm to the nucleus in NPC cells (Figure 3). These effects are blocked by NF-κB inhibitor, (benzylcarbonyl)-Leu-Leu-phenylalaninal, or a dominant-negative mutant of NF-κB 13.

Model of molecular regulation of the hTERT by LMP1 and LMP2A in EBV-induced cell immortalization. (A) Regulations of hTERT and its gene by LMP1. The EBV LMP1 assumes constitutive TNF signalling by recruiting TNFR-associated factors such TRAF2 to induce NF-κB signalling cascade. Although substantially different from TNF signalling, activation of IκB kinase leads to phosphorylation of the inhibitor IκB to release NF-κB, which enters the nucleus to induce NF-κB signalling, including activation of the c-myc gene that in turn stimulates hTERT gene transcription. On the other hand, NF-κB p65 has also been shown to facilitate hTERT translocation into the nucleus through protein-protein interaction. (B) Negative regulation of the hTERT gene by EBV LMP2A. LMP2A has been shown to use its long cytoplasmic region to recruit Syk and Lyn tyrosine kinases, following tyrosine phosphorylation. Syk has been implicated in ERK activation, and ERK induces phosphorylation and activation of the transcription factor AP-1 that has been recently shown to repress the hTERT gene directly.
These findings together suggest that LMP1 influences telomerase with at least two mechanisms: (i) to transactivate the hTERT gene; and (ii) to promote the hTERT protein nuclear entry, both of which are predominantly mediated by the NF-κB signalling pathway. This hypothesis is consistent with previous findings that hTERT protein interacts directly with NF-κB p65, an effect reversed by the IKK inhibitor PS-1145 or NF-κB nuclear translocation inhibitor SN-50 in human multiple myeloma MM.1S cells 76. It is also in line with the findings that NF-κB transactivates c-myc to stimulate hTERT promoter activity, which can be induced by human T-cell lymphotropic virus type-I in primary T cells in vitro and in adult T-cell leukemia/lymphoma samples ex vivo 77. However, what is not yet clear includes the protein motifs that mediate the molecular interactions between the hTERT and NF-κB p65 proteins, and the biological consequence of LMP1 signalling in the maintenance of telomeres. Nonetheless, recent studies attempting to target LMP1 have shown that silencing LMP1 by siRNA induces apoptosis in EBV-positive lymphoma cells in association with telomerase inhibition, suggesting that molecular targeting of LMP1 may be of therapeutic value in EBV-associated tumours 15. These concepts may be reinforced by the data that expression of hTERT alone immortalizes non-malignant nasopharyngeal epithelial cells from clinical biopsies 12.
However, LMP2A has recently been shown to inhibit the hTERT gene 17 (Figure 3). Overexpression of LMP2A results in a significant reduction in hTERT mRNA and the hTERT gene promoter activity in human tumour cell lines 17. The effect seems to be mediated by a mechanism involving the phosphorylation sites in the ITAM motif of LMP2A. Moreover, it has been shown that the effect is not simply secondary to a slow proliferation of the cells, as LMP2A expression does not affect cell cycle progression 17. The exact role of LMP2A inhibition of telomerase activity is still unknown in EBV-associated cell immortalization. The inhibitory effect is consistent with the current view that LMP2A is dispensable in EBV-induced cell immortalization in B cells, but seems contradictory against the notion that LMP2 promotes survival and inhibits apoptosis of Burkitt's lymphoma cells and in gastric carcinoma HSC-39 cell line by a mechanism of inducing Akt phosphorylation and activation 78, 79. It has been speculated that LMP2A inhibits telomerase activity to suppress B-cell activation in order to maintain viral latency 17. Further investigation is required to characterize the mechanisms by which LMP2A may regulate hTERT gene expression in both human epithelial cells and B-lymphocytes.
Since LMP2A recruits Syk tyrosine kinase and induces ERK signalling, ERK might be involved in mediating the hTERT gene repression (Figure 3). ERK repression of the hTERT gene has recently been shown to be mediated by AP1 nuclear transcription repressor activity 51, 80. However, previous studies have also shown that AP-1 activity is increased in parallel to that of NF-κB in NPC 81; this is reminiscent of HPV infection in which E6 stimulates the hTERT gene, whereas E2 inhibits it 82. Together, these results suggest that the relative ratio of the gene expression levels may determine cell proliferation, immortality or death. Measuring the different levels of LMP proteins might provide further information, and targeting LMP1 might tilt the balance to favour repression of the hTERT gene and arrest of cell proliferation and immortalization.
Since EBNA2 transactivates both LMP1 and LMP2A after initial infection of EBV, by stimulating the bi-directional viral latency C promoter and the LMP2A promoter 36, and EBNA2 also stimulates the expression of cellular genes including c-myc 36, 83, it may be possible that EBNA2 is involved in stimulating the hTERT gene (Figure 4). Being a nuclear antigen that does not bind to DNA directly, EBNA2 may transactivate target genes by hijacking the DNA-binding protein known as CBF1; it has been shown that after EBV infection, the viral EBNA2 binds to and activates CBF1 constitutively in the place of ICN 38. CBF1 transcription factor is bound by ICN under normal conditions, which recruits co-activators to form a transcriptional complex containing p300, CBP and P/CAF histone acetyltransferases. This complex activates various genes including c-myc and LMP1 that are both involved in activation of telomerase 37, 38 (Figure 4).

Hypothetical models of transcriptional activation of the hTERT gene by EBNA2. (A) EBNA2 assumes the Notch signalling constitutively by direct binding to and activating the transcription factor CBF1, inducing transactivation of c-myc that in turn activates the hTERT gene. (B) EBNA2 transactivates the LMP1 gene, and LMP1 on plasma membrane recruits TNFR-associated factor TRAF2, inducing NF-κB signalling, which results in c-myc and hTERT gene transcription.
Perspectives
As a predominant aetiological factor of NPC, EBV viral proteins assume significant liabilities on NPC cell immortalization and transformation. Although considerable information has been learnt about EBV gene expression and protein action in malignancies, further studies are required to squarely delineate the dynamic, temporal and spatial actions of EBV proteins. The findings of EBV stimulation of the hTERT gene and the use of siRNA approaches to intercept the pathway from EBV to telomerase may provide an exciting paradigm for strategic consideration in NPC therapeutic studies. Further investigations into the interactions of EBV proteins with local cellular proteins and nucleic acids will provide more defined foundations and targets for intervention. Investigations into genetic traits and susceptible factors in high-risk families and populations are required directly. The relationship between EBV infection, cancer cell transformation, and immune surveillance and elimination of the tumour antigens also remains to be deciphered. With the increased understandings of the environmental, genetic, cellular and molecular mechanisms, development of immune approaches such as vaccines are also urgently required in prophylactics and medication. It is inexorable that prevention of viral infection and elimination of the infected cells will lead to eradication of NPC and related diseases in the near future.
References
Young LS, Rickinson AB . Epstein-Barr virus: 40 years on. Nat Rev Cancer 2004; 4:757–768.
Busson P, Keryer C, Ooka T, Corbex M . EBV-associated nasopharyngeal carcinomas: from epidemiology to virus-targeting strategies. Trends Microbiol 2004; 12:356–360.
Vokes EE, Liebowitz DN, Weichselbaum RR . Nasopharyngeal carcinoma. Lancet 1997; 350:1087–1091.
McDermott AL, Dutt SN, Watkinson JC . The aetiology of nasopharyngeal carcinoma. Clin Otolaryngol 2001; 26:82–92.
Feng BJ, Huang W, Shugart YY, et al. Genome-wide scan for familial nasopharyngeal carcinoma reveals evidence of linkage to chromosome 4. Nat Genet 2002; 31:395–399.
Thorley-Lawson DA . Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 2001; 1:75–82.
Kathrin Herrmann GN . Epstein-Barr virus-associated carcinomas: facts and fiction. J Pathol 2003; 199:140–145.
Farrell PJ . Signal transduction from the Epstein-Barr virus LMP-1 transforming protein. Trends Microbiol 1998; 6:175–177; discussion 177–178.
Griffin BE, Xue SA . Epstein-Barr virus infections and their association with human malignancies: some key questions. Ann Med 1998; 30:249–259.
Eliopoulos AG, Young LS . LMP1 structure and signal transduction. Semin Cancer Biol 2001; 11:435–444.
Murray PG, Young LS . The role of the Epstein-Barr virus in human disease. Front Biosci 2002; 7:d519–d540.
Li HM, Man C, Jin Y, et al. Molecular and cytogenetic changes involved in the immortalization of nasopharyngeal epithelial cells by telomerase. Int J Cancer 2006; 119:1567–1576.
Ding L, Li LL, Yang J, et al. Epstein-Barr virus encoded latent membrane protein 1 modulates nuclear translocation of telomerase reverse transcriptase protein by activating nuclear factor-kappaB p65 in human nasopharyngeal carcinoma cells. Int J Biochem Cell Biol 2005; 37:1881–1889.
Yang J, Deng X, Deng L, et al. Telomerase activation by Epstein-Barr virus latent membrane protein 1 is associated with c-Myc expression in human nasopharyngeal epithelial cells. J Exp Clin Cancer Res 2004; 23:495–506.
Mei YP, Zhu XF, Zhou JM, et al. siRNA targeting LMP1-induced apoptosis in EBV-positive lymphoma cells is associated with inhibition of telomerase activity and expression. Cancer Lett 2006; 232:189–198.
Peng R, Moses SC, Tan J, et al. The Epstein-Barr virus EBNA-LP protein preferentially coactivates EBNA2-mediated stimulation of latent membrane proteins expressed from the viral divergent promoter. J Virol 2005; 79:4492–4505.
Chen F, Liu C, Lindvall C, et al. Epstein-Barr virus latent membrane 2A (LMP2A) down-regulates telomerase reverse transcriptase (hTERT) in epithelial cell lines. Int J Cancer 2005; 113:284–289.
Zhong BL, Zong YS, Lin SX, et al. Epstein-Barr virus infection in precursor lesions of nasopharyngeal carcinoma. Ai Zheng 2006; 25:136–142.
Lin CT, Kao HJ, Lin JL, et al. Response of nasopharyngeal carcinoma cells to Epstein-Barr virus infection in vitro. Lab Invest 2000; 80:1149–1160.
Huang DP, Ho JH, Chan WK, et al. Cytogenetics of undifferentiated nasopharyngeal carcinoma xenografts from southern Chinese. Int J Cancer 1989; 43:936–939.
Liu M-T, Chen Y-R, Chen S-C, et al. Epstein-Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene 2004; 23:2531–2539.
Smith P . Epstein-Barr virus complementary strand transcripts (CSTs/BARTs) and cancer. Semin Cancer Biol 2001; 11:469–476.
Rowe M . Cell transformation induced by Epstein-Barr virus – living dangerously. Semin Cancer Biol 2001; 11:403–405.
Rickinson A . Epstein-Barr virus. Virus Res 2002; 82:109–113.
Roy NS, Nakano T, Keyoung HM, et al. Telomerase immortalization of neuronally restricted progenitor cells derived from the human fetal spinal cord. Nat Biotechnol 2004; 22:297–305.
Raab-Traub N . Epstein-Barr virus in the pathogenesis of NPC. Semin Cancer Biol 2002; 12:431–441.
Burgos JS . Involvement of the Epstein-Barr virus in the nasopharyngeal carcinoma pathogenesis. Med Oncol 2005; 22:113–121.
Lam N, Sugden B . CD40 and its viral mimic, LMP1: similar means to different ends. Cell Signal 2003; 15:9–16.
Yin L, Hubbard AK, Giardina C . NF-kappa B regulates transcription of the mouse telomerase catalytic subunit. J Biol Chem 2000; 275:36671–36675.
Dawson CW, Tramountanis G, Eliopoulos AG, Young LS . Epstein-Barr virus latent membrane protein 1 (LMP1) activates the phosphatidylinositol 3-kinase/Akt pathway to promote cell survival and induce actin filament remodeling. J Biol Chem 2003; 278:3694–3704.
Wan J, Sun L, Mendoza JW, et al. Elucidation of the c-Jun N-terminal kinase pathway mediated by Epstein-Barr Virus-encoded latent membrane protein 1. Mol Cell Biol 2004; 24:192–199.
Liu JP . Studies of the molecular mechanisms in the regulation of telomerase activity. FASEB J 1999; 13:2091–2104.
Liu J-P . Protein kinase C and its substrates. Mol Cell Endocrinol 1996; 116:1–29.
Cohen P . The structure and regulation of protein phosphatases. Annu Rev Biochem 1989; 58:453–508.
Hammerschmidt W, Sugden B . Genetic analysis of immortalizing functions of Epstein–Barr virus in human B lymphocytes. Nature 1989; 340:393–397.
Gordadze AV, Onunwor CW, Peng R, et al. EBNA2 amino acids 3 to 30 are required for induction of LMP-1 and immortalization maintenance. J Virol 2004; 78:3919–3929.
Strobl LJ, Hofelmayr H, Marschall G, et al. Activated Notch1 modulates gene expression in B cells similarly to Epstein-Barr viral nuclear antigen 2. J Virol 2000; 74:1727–1735.
Wang L, Grossman SR, Kieff E . Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc Natl Acad Sci U S A 2000; 97:430–435.
Blackburn EH . Switching and signaling at the telomere. Cell 2001; 106:661–673.
Shay JW, Wright WE . Senescence and immortalization: role of telomeres and telomerase. Carcinogenesis 2005; 26:867–874.
Nakamura TM, Morin GB, Chapman KB, et al. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997; 277:955–959.
Meyerson M, Counter CM, Eaton EN, et al. hEst2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997; 90:785–795.
Nakayama J, Saito M, Nakamura H, et al. TLP1: a gene encoding a protein component of mammalian telomerase is a novel member of WD repeats family. Cell 1997; 88:875–884.
Harrington L, McPhail T, Mar V, et al. A mammalian telomerase-associated protein. Science 1997; 275:973–977.
Kelleher C, Teixeira MT, Forstemann K, Lingner J . Telomerase: biochemical considerations for enzyme and substrate. Trends Biochem Sci 2002; 27:572–579.
Lai CK, Mitchell JR, Collins K . RNA binding domain of telomerase reverse transcriptase. Mol Cell Biol 2001; 21:990–1000.
Moriarty TJ, Huard S, Dupuis S, Autexier C . Functional multimerization of human telomerase requires an RNA interaction domain in the N terminus of the catalytic subunit. Mol Cell Biol 2002; 22:1253–1265.
Beattie TL, Zhou W, Robinson MO, Harrington L . Polymerization defects within human telomerase are distinct from telomerase RNA and TEP1 binding. Mol Biol Cell 2000; 11:3329–3340.
Bodnar AG, Ouellette M, Frolkis M, et al. Extension of life-span by introduction of telomerase into normal human cells. Science 1998; 279:349–352.
Bayne S, Liu JP . Hormones and growth factors regulate telomerase activity in ageing and cancer. Mol Cell Endocrinol 2005; 240:11–22.
Takakura M, Kyo S, Inoue M, et al. Function of AP-1 in transcription of the telomerase reverse transcriptase gene (TERT) in human and mouse Cells. Mol Cell Biol 2005; 25:8037–8043.
Maida Y, Kyo S, Kanaya T, et al. Direct activation of telomerase by EGF through Ets-mediated transactivation of TERT via MAP kinase signaling pathway. Oncogene 2002; 21:4071–4079.
Goueli BS, Janknecht R . Upregulation of the catalytic telomerase subunit by the transcription factor ER81 and oncogenic HER2/Neu, Ras, or Raf. Mol Cell Biol 2004; 24:25–35.
Wang Z, Kyo S, Takakura M, et al. Progesterone regulates human telomerase reverse transcriptase gene expression via activation of mitogen-activated protein kinase signaling pathway. Cancer Res 2000; 60:5376–5381.
Alfonso-De Matte MY, Yang H, Evans MS, et al. Telomerase is regulated by c-Jun NH2-terminal kinase in ovarian surface epithelial cells. Cancer Res 2002; 62:4575–4578.
Hu B, Tack DC, Liu T, et al. Role of Smad3 in the regulation of rat telomerase reverse transcriptase by TGFbeta. Oncogene 2005; 25:1030–1041.
Li H, Xu D, Toh BH, Liu JP . TGF-beta and cancer: Is Smad3 a repressor of hTERT gene? Cell Research 2006; 16:169–173.
Li H, Xu D, Li J, et al. Transforming growth factor beta suppresses human telomerase reverse transcriptase by smad3 interactions with C-Myc and hTERT gene. J Biol Chem 2006; 281:25588–255600.
Kiyono T, Foster SA, Koop JI, et al. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature 1998; 396:84–88.
Veldman T, Horikawa I, Barrett JC, Schlegel R . Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J Virol 2001; 75:4467–4472.
Li H, Zhao L, Yang Z, et al. Telomerase is controlled by protein kinase Calpha in human breast cancer cells. J Biol Chem 1998; 273:33436–33442.
Kang SS, Kwon T, Kwon DY, Do SI . Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem 1999; 274:13085–13090.
Kimura A, Ohmichi M, Kawagoe J, et al. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene 2004; 23:4505–4515.
Smogorzewska A, de Lange T . Regulation of telomerase by telomeric proteins. Annu Rev Biochem 2004; 73:177–208.
Holt SE, Aisner DL, Baur J, et al. Functional requirement of p23 and hsp90 in telomerase complexes. Genes Dev 1999; 13:817–826.
Kawauchi K, Ihjima K, Yamada O . IL-2 increases human telomerase reverse transcriptase activity transcriptionally and posttranslationally through phosphatidylinositol 3'-kinase/Akt, heat shock protein 90, and mammalian target of rapamycin in transformed NK cells. J Immunol 2005; 174:5261–5269.
Forsythe HL, Jarvis JL, Turner JW, et al. Stable association of hsp90 and p23, but not hsp70, with active human telomerase. J Biol Chem 2001; 276:15571–15574.
Haendeler J, Hoffmann J, Rahman S, et al. Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett 2003; 536:180–186.
Zhou XZ, Lu KP . The Pin2/TRF1-interacting protein PinX1 is a potent telomerase inhibitor. Cell 2001; 107:347–359.
Banik SS, Counter CM . Characterization of interactions between PinX1 and human telomerase subunits hTERT and hTR. J Biol Chem 2004; 279:51745–51748.
Poh M, Boyer M, Solan A, et al. Blood vessels engineered from human cells. Lancet 2005; 365:2122–2124.
Stansel RM, Subramanian D, Griffith JD . p53 binds telomeric single strand overhangs and t-loop junctions in vitro. J Biol Chem 2002; 277:11625–11628.
Shats I, Milyavsky M, Tang X, et al. p53-dependent down-regulation of telomerase is mediated by p21waf1. J Biol Chem 2004; 279:50976–50985.
Li H, Cao Y, Berndt MC, et al. Molecular interactions between telomerase and the tumor suppressor protein p53 in vitro. Oncogene 1999; 18:6785–6794.
Cao Y, Li H, Deb S, Liu JP . TERT regulates cell survival independent telomerase activity. Oncogene 2002; 21:3130–3138.
Akiyama M, Hideshima T, Hayashi T, et al. Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res 2003; 63:18–21.
Sinha-Datta U, Horikawa I, Michishita E, et al. Transcriptional activation of hTERT through the NF-kappaB pathway in HTLV-I-transformed cells. Blood 2004; 104:2523–2531.
Scholle F, Bendt KM, Raab-Traub N . Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt. J Virol 2000; 74:10681–10689.
Fukuda M, Longnecker R . Latent membrane protein 2A inhibits transforming growth factor-{beta}1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J Virol 2004; 78:1697–1705.
Li H, Pinto AR, Duan W, et al. Telomerase down-regulation does not mediate PC12 pheochromocytoma cell differentiation induced by NGF, but requires MAP kinase signalling. J Neurochem 2005; 95:891–901.
Deng L, Yang J, Zhao XR, et al. Cells in G2/M phase increased in human nasopharyngeal carcinoma cell line by EBV-LMP1 through activation of NF-kappaB and AP-1. Cell Res 2003; 13:187–194.
Lee D, Kim HZ, Jeong KW, et al. Human papillomavirus E2 down-regulates the human telomerase reverse transcriptase promoter. J Biol Chem 2002; 277:27748–27756.
Kaiser C, Laux G, Eick D, et al. The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2. J Virol 1999; 73:4481–4484.
Acknowledgements
This work was supported by grants from the Australian Research Council and National Health and Medical Research Council of Australia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, JP., Cassar, L., Pinto, A. et al. Mechanisms of cell immortalization mediated by EB viral activation of telomerase in nasopharyngeal carcinoma. Cell Res 16, 809–817 (2006). https://doi.org/10.1038/sj.cr.7310098
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7310098
