
Abstract
Excess nitric oxide (NO) induces apoptosis in some cell types, including macrophages. Heat shock protein of 70 kDa (hsp70) has been reported to protect cells from various stresses, including apoptosis-inducing stimuli. Several mammalian cytosolic DnaJ homologs, partner chaperones of hsp70 family members, have been identified. We asked if a DnaJ homolog is required to prevent NO-mediated apoptosis. When mouse macrophage-like RAW 264.7 cells were treated with an NO donor, SNAP, apoptosis occurred. This apoptosis could be prevented by pretreatment of the cells with heat or a low dose of SNAP. Under these conditions, levels of hsc70 (an hsp70 member) remained unchanged, whereas hsp70 was markedly induced. Of the DnaJ homologs dj1 (hsp40/hdj-1) was strongly induced and dj2 (HSDJ/hdj-2) was moderately induced. In transfection experiments, hsp70, hsc70, dj1 or dj2 alone was ineffective in preventing NO-mediated apoptosis. In contrast, both dj1 and dj2, in combination with hsc70 or hsp70, prevented the cells from apoptosis. The hsp70-DnaJ chaperone pairs exerted their anti-apoptotic effects upstream of caspase 3 activation, and apparently upstream of cytochrome c release from mitochondria.
Similar content being viewed by others


Introduction
Nitric oxide (NO) is an important multifunctional biomolecule involved in a variety of physiological and pathological processes.1,2,3,4 In pathological processes, NO has bactericidal or tumoricidal effects and acts as a host defence agent.4 However, excess NO production is toxic to the host,5 and is thought to be the cause of diseases such as septic shock, autoimmune disease, cerebral infarction, and diabetes mellitus. NO-mediated apoptosis is often observed in such cases.6,7 NO has several cytotoxic effects, including reactions with proteins and nucleic acids. The main targets of NO in proteins are the SH group and Fe of active sites, especially Fe2+ in heme. In the nucleus, NO has been shown to cause mutations of genes and inhibition of DNA repair enzymes and to mediate DNA strand breaks. In several types of cells, including macrophages, chondrocytes, and neurons, NO has been shown to mediate apoptosis.7
Heat shock proteins are induced in response to various stresses and to protect cells from such stresses.8,9 The major heat shock proteins can be divided into several groups based on both size and function. Some heat shock proteins are constitutively expressed and act as molecular chaperones. Members of the hsp70 family are involved in the folding and intracellular transport of newly synthesized proteins.10,11 To protect cells from various stresses, hsp70s bind to damaged and misfolded proteins, and facilitate their refolding or target severely damaged proteins for degradation.8 hsp70 also protects cells from the apoptosis induced by various stresses and agents including NO, oxidative stress, tumor necrosis factor, anti-cancer drugs, ceramide and radiation.8 However, the mechanism of the anti-apoptotic effect of hsp70 is not fully understood. DnaK, the hsp70 homolog of Escherichia coli, is regulated by other molecular chaperones, DnaJ and GrpE.11,12 Several mammalian DnaJ homologs (hsp40 family members) have been identified,13 and it seems likely that hsp70 family members are regulated by these homologs. Major DnaJ homologs in mammalian cytosol are dj1 (hsp40/hdj-1) and dj2 (HSDJ/hdj-2). dj2, but not dj1, has the CaaX prenylation motif at its COOH terminal and is farnesylated.14 One report suggested the refolding of luciferase by hdj-1 (dj1)-hsc70 (heat shock cognate 70),15 whereas another report indicated that hsp40 (dj1)-hsc70 prevented thermally-denatured luciferase from insoluble aggregate.16 In contrast, we found that dj2-hsc70, rather than dj1-hsc70, facilitates mitochondrial protein import and luciferase refolding.17
Hence, we asked if a DnaJ homolog is required to prevent NO-mediated apoptosis, and if so, which member is required. We found that dj1-hsp70 and dj2-hsp70 chaperone pairs are both effective in preventing NO-mediated apoptosis in RAW 264.7 macrophages and that they prevent apoptosis at the upstream of caspase 3 activation, and apparently at the upstream of cytochrome c release from mitochondria.
Results
Heat pretreatment prevents NO-mediated apoptosis in RAW 264.7 cells
When mouse macrophage-like RAW 264.7 cells were treated with E. coli lipopolysaccharide (LPS) and interferon-γ (IFN-γ) for 12 h, inducible NO synthase (iNOS) was highly induced and a large amount of NO was produced.18 Under these conditions, morphological changes characteristic of apoptosis occurred (Figure 1A,B). Round-shaped cells and apoptotic bodies were observed in phase-contrast images, and chromatin condensation and nuclear fragmentation were seen in case of Hoechst dye 33258 staining (Figure 1A, c and d). These apoptotic changes were strongly prevented by adding an NO scavenger carboxy-PTIO (Figure 1A, i and j).18 Therefore, LPS/IFN-γ induces apoptosis in RAW 264.7 cells, and this apoptosis is mediated by NO.

NO-mediated apoptosis in RAW 264.7 cells is prevented by heat pretreatment. (A) Cells were untreated or heated at 42°C for 2 h, allowed to recover at 37°C for 1 h, and then treated with or without 150 μg/ml LPS and 100 U/ml IFN-γ for 12 h. At the time of the addition of LPS/IFN-γ, carboxy-PTIO (300 μM) was added to the medium, where indicated. After fixation, the cells were stained with a DNA-specific fluorochrome Hoechst dye 33258. Phase-contrast images (upper panels) and fluorescence images (lower panels) of the same fields are shown. Original magnifications: ×400. Bars, 10 μm. (B) The percentages of apoptotic cells were quantified and are shown as means±S.E. (n=3). More than 200 cells in each dish were analyzed for apoptosis. (C) Cells were untreated or heated at 42°C for 2 h, allowed to recover at 37°C for 1 h, and then treated with or without 150 μg/ml LPS and 100 U/ml IFN-γ for 18 h. Then, NO2− plus NO3− in the medium was measured. The results are shown as means±S.E. (n=4)
Heat treatment induces heat shock proteins, including hsp70, in cells. It has been reported that hsp70 protects cells from the apoptosis induced by various stresses and agents.8 Therefore, we asked whether heat treatment would protect cells from LPS/IFN-γ-induced apoptosis. When RAW 264.7 cells were heated at 42°C for 2 h before LPS/IFN-γ treatment, apoptosis was prevented (Figure 1A, e and f). This anti-apoptotic effect by heat treatment is not due to the suppression of NO production (Figure 1C). Heat treatment did not alter NO production, as based on NO2− plus NO3− in the culture medium.
Heat pretreatment prevents apoptosis induced by SNAP
We next examined whether the anti-apoptotic effect by heat treatment would also be effective in preventing the apoptosis induced by exogenously added NO (Figure 2). Addition of SNAP, an NO donor, induced apoptosis in RAW 264.7 cells (Figure 2A, c and d), and this apoptosis was prevented by carboxy-PTIO (Figure 2A, i and j). Heat pretreatment effectively prevented the apoptosis induced by SNAP (Figure 2A, e and f). Formation of a DNA ladder, characteristic of apoptotic cells, was observed when the cells were treated with SNAP (Figure 2C), and this ladder formation was prevented by heat pretreatment.

Apoptosis in RAW 264.7 cells induced by SNAP is prevented by heat pretreatment. (A) Cells were untreated or heated at 42°C for 2 h, and allowed to recover at 37°C for 1 h, and then treated with or without SNAP (1.5 mM) for 12 h. At the time of the addition of SNAP, carboxy-PTIO (300 μM) was added to the medium, where indicated. After fixation, the cells were stained with Hoechst dye 33258. Phase-contrast images (upper panels) and fluorescence images (lower panels) of the same fields are shown. Original magnifications: ×400. Bars, 10 μm. (B) The percentages of apoptotic cells were quantified and are shown as means±S.E. (n=3). (C) Cells were treated as indicated on the top. DNAs were isolated, resolved on an agarose gel, stained with ethidium bromide, and visualized for DNA fragmentation by UV transillumination
Heat treatment induces heat shock proteins in RAW 264.7 cells
To determine if a heat shock protein(s) is involved in the anti-apoptotic effect seen with heat treatment, we first examined expression of heat shock proteins by immunoblot analysis (Figure 3). The cells were heated at 42°C for 2 h, recovered at 37°C for 1 h and further cultured for 6 h at 37°C in the presence or absence of SNAP. Among the hsp70 family members, hsc70 was present in control cells and was little changed by heat treatment. In contrast, hsp70 which was not detected prior to heat treatment, was markedly induced by treatment. This induction was partly suppressed by the addition of SNAP. Among the DnaJ homologs, dj1 was present prior to heat treatment and was induced strongly by treatment. Addition of SNAP after heat treatment somewhat suppressed the induction of dj1, however, the level of dj1 was still higher than seen in control cells or cells treated with SNAP alone. dj2 was also present in control cells and was weakly induced by heat treatment. Addition of SNAP reduced the level of dj2 and this reduction was prevented by heat treatment.

Heat treatment induces hsp70 and DnaJ family members in RAW 264.7 cells. Cells were treated as indicated on the top and cell extracts (30 μg of protein) were subjected to immunoblot analysis for hsc70, hsp70, dj1 and dj2 proteins. The molecular mass markers on the right were bovine serum albumin (66 kDa) and ovalbumin (46 kDa)
Pretreatment with a low dose of SNAP induces heat shock proteins and prevents SNAP-induced apoptosis in RAW 264.7 cells
Endogenous or exogenous NO was reported to protect cultured hepatocytes from tumor necrosis factor-α-induced apoptosis by inducing hsp70.19 Therefore, we asked whether pretreatment with a low dose of SNAP protects SNAP (high dose)-induced apoptosis in RAW 264.7 cells (Figure 4A,B). Treatment of RAW 264.7 cells with 0.1 mM SNAP did not induce apoptosis. When cells were treated with 0.1 mM SNAP for 3 h before 1.5 mM SNAP treatment, apoptosis was prevented.

Pretreatment with 0.1 mM SNAP induces hsp70 and DnaJ family members and prevents 1.5 mM SNAP-induced apoptosis in RAW 264.7 cells. (A) Cells were untreated or treated with 0.1 mM SNAP for 3 h, and then treated with or without 1.5 mM SNAP for 12 h. After fixation, the cells were stained with Hoechst dye 33258. Phase-contrast images (upper panels) and fluorescence images (lower panels) of the same fields are shown. Original magnifications: ×400. Bars, 10 μm. (B) Experiments were performed as in (A) and the percentages of apoptotic cells are shown as means±S.E. (n=3). More than 100 cells in each dish were analyzed. (C) Cells were treated as indicated on the top and cell extracts (30 μg of protein) were subjected to immunoblot analysis for hsc70, hsp70, dj1 and dj2 proteins
We then examined expression of heat shock proteins by immunoblot analysis (Figure 4C). Cells were treated with 0.1 mM SNAP for 3 h, and further cultured for 6 h in the presence of 0.1 or 1.5 mM SNAP. hsc70 remained little changed by SNAP treatment. In contrast, hsp70 was markedly induced by treatment with 0.1 mM SNAP. Among the DnaJ homologs, dj1 was induced strongly by treatment. Addition of 1.5 mM SNAP after treatment with a low dose of SNAP somewhat suppressed the induction of hsp70 and dj1, however, their levels were still higher than those in control cells or cells treated with 1.5 mM SNAP alone. dj2 was decreased by treatment with 1.5 mM SNAP, and this reduction was completely prevented by pretreatment with 0.1 mM SNAP. The induced levels of hsp70, dj1 and dj2 by 0.1 mM SNAP were similar to those after heat pretreatment.
Coexpression of hsp70 and either dj1 or dj2 prevents SNAP-induced apoptosis in RAW 264.7 cells
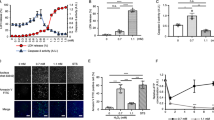
To examine effects of the hsp70 and DnaJ family members on NO-mediated apoptosis, transfection experiments were done. RAW 264.7 cells were transiently transfected with expression plasmids for hsc70, dj1 or dj2 separately or in combination, then the cells were treated with SNAP. hsc70 was first used as an hsp70 family member because hsc70 was found to cooperate with dj2 in facilitating protein refolding and mitochondrial protein import.17 When the cells were treated with 1.5 mM SNAP for 12 h, most cells (about 86%) underwent apoptosis (Figure 5A,B). When the cells were transfected with the hsc70 plasmid alone, SNAP-induced apoptosis was not prevented. Transfection with the dj1 or dj2 plasmid slightly stimulated apoptosis, perhaps because of an unbalance of the chaperone pairs. In contrast, when hsc70 and dj1 plasmids were cotransfected, about 44% (30% over basal level) of the cells escaped from apoptosis and showed normal cell and nuclear structures. Cotransfection of hsc70 and dj2 plasmids was also effective in preventing apoptosis, to a similar extent. Under these conditions, transfection efficiency assessed with a green fluorescent protein (GFP) plasmid, was 20–30%.

Coexpression of hsp70, and dj1 or dj2 protected RAW 264.7 cells from NO-mediated apoptosis. (A) Cells were transfected with insertless pCAGGS (Vector), pCAGGS-hsc70, pCAGGS-dj1, or pCAGGS-dj2, or their combination, as indicated on the top. Twenty-four hours after transfection, SNAP (1.5 mM) was added to the medium and cells were cultured for 12 h, fixed, analyzed for apoptosis by the TUNEL method and then stained with Hoechst dye 33258. Phase-contrast images (upper panels) and fluorescence images (middle and lower panels) of the same fields are shown. Arrows indicate cells rescued from apoptosis. Original magnifications: ×400. Bars, 10 μm. A portion of the cells were detached from coverslips by treatment with SNAP. (B) Experiments were performed as in (A) and the percentages of apoptotic cells are shown as means±S.E. (n=3). More than 100 cells in each dish were analyzed. (C) Cells were cotransfected with pCAGGS-hsc70 and pCAGGS-dj1 and cultured for 24 h (lower panels). The cells were then fixed and immunostained with a monoclonal antibody against hsc70 or an antiserum against dj1 as described in Materials and Methods. Upper panels show untreated control. Fluorescence images for hsc70 (left panels) and dj1 (right panels) of the same fields are shown. Arrows show transfected cells. Original magnifications: ×1000. Bar, 10 μm. (D) Cells were transfected with insertless pCAGGS (Vector) or pCAGGS-hsp70 alone or in combination with pCAGGS-dj1 or pCAGGS-dj2, as indicated on the top. Twenty-four hours after transfection, SNAP (1.5 mM) was added to the medium and cells were cultured for 12 h, fixed, and then stained with Hoechst dye 33258. Phase-contrast images (upper panels) and fluorescence images (lower panels) of the same fields are shown. Arrows indicate cells rescued from apoptosis. Original magnifications: ×400. Bars, 10 μm. A portion of the cells were detached from coverslips by treatment with SNAP. (E) Experiments were performed as in (D) and the percentages of apoptotic cells are shown as means±S.E. (n=3). More than 100 cells in each dish were analyzed
Co-expression of hsc70 and dj1 was confirmed by double immunostaining (Figure 5C). When control cells were immunostained with an antibody against hsc70 or dj1, all cells were weakly positive. On the other hand, when the hsc70 and dj1 plasmids were cotransfected, some cells (about 25%) were strongly positive for both hsc70 and dj1.
We next compared the effect of hsp70 (inducible form) with hsc70 in transfection experiments (Figure 5D,E; see also Figure 5A,B). Transfection of hsp70 alone was not effective in preventing SNAP-induced apoptosis in RAW 264.7 cells. In contrast, when hsp70 and dj1 or dj2 plasmids were cotransfected, about 30% of cells escaped from apoptosis. Therefore, we conclude that both hsp70 and hsc70 are effective in preventing SNAP-induced apoptosis in combination with DnaJ homologs.
hsc70-DnaJ chaperone pairs prevent apoptosis upstream of caspase 3 activation
In the cascade of apoptosis, caspase proteases play a key role.5 There are several subgroups in caspases, among which, caspase 3 activation is positioned downstream of the caspase activation cascade, and is thought to be the point of no return in the process of cell death. To clarify the mechanism of prevention of apoptosis by the chaperones, we examined caspase 3 activity in two ways. Figure 6A shows caspase 3 activity of the extract of RAW 264.7 cells. SNAP treatment markedly induced the activity. This activation was strongly prevented by heat pretreatment. Heat treatment alone did not induce caspase 3 activation.

Heat pretreatment or coexpression of hsc70 and dj1 or dj2 prevents NO-mediated caspase 3 activation in RAW 264.7 cells. (A) Cells were treated as indicated, extracts were prepared, and caspase 3 activity was measured, as described in Materials and Methods. The results are shown as means±S.E. (n=3). (B) Cells were transfected with insertless pCAGGS (a–f), pCAGGS-hsc70 plus pCAGGS-dj1 (g and h), or pCAGGS-hsc70 plus pCAGGS-dj2 (i and j). 24 h after transfection, SNAP (1.5 mM) was added to the medium then the cells were cultured for 12 h. In e and f, the cells were heated at 42°C for 2 h, allowed to recover at 37°C for 1 h and SNAP was then added. Caspase 3 activity was detected in situ, as described in Materials and Methods. Phase contrast images (upper panels) and caspase 3-fluorescence images (lower panels) of the same fields are shown. Arrows indicate cells negative for caspase 3 activity. Original magnifications: ×400. Bars, 10 μm. (C) Experiments were performed as in (B) and the percentages of caspase 3 activity negative cells are shown as means±S.E. (n=3). More than 100 cells in each dish were analyzed
Figure 6B,C show in situ detection of caspase 3 activity. Almost all cells became caspase 3 activity-positive by SNAP treatment. When the cells were heat-treated prior to SNAP addition, cells were negative or were only weakly positive for caspase 3 activity. When the hsc70 and dj1 expression plasmids were co-transfected, about 40% of the cells were caspase 3 activity-negative. These cells were morphologically normal as seen in phase-contrast microscopy. Cotransfection of hsc70 and dj2 plasmids was also effective in preventing caspase 3 activation. These results indicate that hsc70 and dj1 or dj2 chaperone pairs prevent apoptosis upstream of the caspase 3 activation.
Heat pretreatment prevents apoptosis upstream of cytochrome c release from mitochondria
While the cascade of NO-mediated apoptosis has not been fully clarified, cytochrome c release from mitochondria is thought to be involved in the process of caspase activation.5 Therefore, we asked whether cytochrome c release from mitochondria would be prevented by heat pretreatment (Figure 7). After heat and SNAP treatments, RAW 264.7 cells were fractionated into soluble fraction and particulate fraction containing mitochondria, using digitonin, and immunoblot analysis was performed. In untreated cells, cytochrome c was recovered exclusively in the particulate fraction, while it was recovered mostly in the soluble fraction after SNAP treatment. When the cells were heat-treated prior to SNAP addition, cytochrome c was almost completely recovered in the particulate fraction. Under these conditions, a mitochondrial matrix protein hsp60 was recovered exclusively in the particulate fraction, whereas a cytosolic protein glyceraldehyde 3-phosphate dehydrogenase (G3PDH) was recovered exclusively in the soluble fraction. Therefore, we propose that cytochrome c release occurs in NO-mediated apoptosis in RAW 264.7 cells and that this release is prevented by heat pretreatment, most likely by induction of hsp70-DnaJ chaperone pairs.

Heat pretreatment prevents NO-mediated cytochrome c release in RAW 264.7 cells. Cells were heated at 42°C for 2 h, allowed to recover at 37°C for 1 h, and then treated with 1.5 mM SNAP for 12 h, as indicated on the top. Fractionation of the cells was done as described in Materials and Methods. Distributions of total protein in the soluble (S) fraction and the particulate (P) fraction were 65 and 35%, respectively. The soluble fraction and the particulate fraction (10 μg of protein) were subjected to immunoblot analysis for cytochrome c, hsp60 and G3PDH
Discussion
hsp70 protects cells from various stresses.8 Although anti-apoptotic effects of hsp70 have been reported in several systems, related mechanisms are unclear. In a rat insulinoma cell line, overexpression of hsp70 was reported to prevent NO-induced cell death, but its mechanism is unknown.20 In heat-induced cell death, hsp70 was reported to inhibit the activation of the stress-activated protein kinase SAPK/JNK (c-Jun N-terminal kinase) and the processing of caspase 3.21 On the other hand, the site of hsp70 action was suggested to be downstream of caspase 3 in tumor necrosis factor-α (TNFα)-induced apoptosis.22 In addition, the overexpression of hsp70 prevented heat-induced apoptosis, but not Fas-mediated apoptosis in a Jurkat T cell line.23 In these studies, however, participation of a DnaJ family member(s) was not given attention. In the present study, we found that hsp70-dj1 or dj2 pairs prevent NO-mediated apoptosis in RAW 264.7 cells. Overexpression of hsp70 alone was not effective in preventing apoptosis.
The relationship between hsp70 and DnaJ family has been studied in a protein folding system.11 Nascent polypeptide chains on ribosomes and unfolded polypeptides under stressed conditions are susceptible to aggregation because hydrophobic residues are exposed. Therefore, suppression of aggregation is essential to maintain proteins in a state competent for folding and acquiring active conformation. hsp70 binds to hydrophobic regions of unfolded polypeptides and prevents aggregation. The interaction of hsp70 with polypeptides is regulated by the binding of ATP to ATPase domain of hsp70. hsp70’s actions are facilitated by cochaperones such as members of the DnaJ family. DnaJs stimulate ATPase activity of hsp70. We earlier reported that both hsc70 and dj2 are required for protein folding and for protein import into mitochondria in vitro.17 It was also reported that dj2 is involved in the recognition of misfolded proteins and protein biogenesis in vivo.24,25 In the present study, we found that dj1 or dj2 is also needed for the anti-apoptotic function of hsp70. The hsp70-DnaJ pair exerts anti-apoptotic functions upstream of caspase 3 activation, and apparently of cytochrome c release. Recently, it was proposed that the maintenance of proper protein intermediate conformation might also be important for normal functions of many proteins, particularly those that participate in the assembly of transient complex structures and those that participate in signaling pathways.13 Therefore, the hsp70-DnaJ chaperone system may be involved in the assembly of an anti-apoptotic protein(s). The target molecule of the anti-apoptotic actions of hsp70-dj1 or dj2 pair remains to be determined.
Materials and Methods
Plasmids
pCAGGS-hsc70 and pCAGGS-hsp70, the mammalian expression plasmids for human hsc70 and hsp70 respectively, was constructed by inserting the full-length cDNA fragment for human hsc70 or hsp70 into the XhoI site of the plasmid pCAGGS26 after linker attachment. pCAGGS-dj1, a plasmid for human dj1, was constructed by inserting the full-length dj1 cDNA fragment of pQE-9/Hsp4016 into the XhoI site of pCAGGS after linker attachment. pCAGGS-dj2, a human dj2 (HSDJ) expression plasmid, was as described.14
Materials
A monoclonal antibody against human/mouse hsc70 was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Monoclonal antibodies against human hsp70 and human hsp60 were obtained from StressGen Biotechnologies Corp. (Victoria, Canada). Anti-human dj1 and dj2 antibodies were those described previously.17 Monoclonal antibodies against human/mouse cytochrome c and rabbit G3PDH were obtained from R & D Systems, Inc. (Minneapolis, MN, USA) and CHEMICON International Inc. (Temecula, CA, USA), respectively.
Cell culture and DNA transfection
Mouse macrophage-like RAW 264.7 cells were grown in Eagle's minimal essential medium supplemented with 10% fetal calf serum. Transfection of the cells with plasmids was carried out using Trans-IT-LT1 Polyamine (PanVera Corp., Madison, WI, USA) according to the protocol provided by the manufacturer.
Treatment of cells
RAW 264.7 cells were pretreated at 42°C for 2 h and allowed to recover at 37°C for 1 h or with a NO donor SNAP (S-nitroso-N-acetyl-DL-penicillamine, 0.1 mM) for 3 h, and then treated with a combination of E. coli LPS (serotype 0127:B8, Sigma Chem. Co., St. Louis, MO, USA, 150 μg/ml) and mouse IFN-γ (100 U/ml) or SNAP (1.5 mM), for the indicated periods. A NO-scavenger carboxy-PTIO [2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide, 300 μM] was added to the medium concomitantly with the addition of LPS/IFN-γ or SNAP.
Immunoblot analysis
RAW 264.7 cells were homogenized in 20 mM HEPES-KOH (pH 7.5) containing 1% Triton X-100, 20% glycerol, and 1 mM dithiothreitol. After centrifugation at 15 000×g for 10 min, the supernatants served as cell extracts for immunoblot analysis. Immunodetection was performed using ECL kits (Amersham Life Science, Buckinghamshire, UK), according to the protocol provided by the manufacturer. The chemiluminograms were quantified using a MacBAS bio-image analyzer (Fuji Photo Film, Tokyo, Japan).
Subcellular fractionation
Cell fractionation was done essentially as described.27 Briefly, the cell suspension (about 5×107 cells/ml) was mixed with an equal volume of digitonin (1 mg/ml) in phosphate-buffered saline (PBS), incubated at 25°C for 5 min, then kept on ice for 5 min, and centrifuged at 15 000×g for 2 min. The supernatant served as the soluble fraction. The pellet containing mitochondria was dissolved in 0.5% Triton X-100 in PBS, insoluble material was removed by centrifugation, and the supernatant used as the particulate fraction.
Immunocytochemical staining
RAW 264.7 cells were cultured on coverslips and transfected with plasmids as described above. The cells were fixed with 4% paraformaldehyde for 40 min at room temperature and treated with PBS containing 1% Triton X-100. The cells were then incubated with a mouse monoclonal antibody against hsc70 and a rabbit antiserum against dj1, and then with goat anti-mouse IgG conjugated with Cy2 and goat anti-rabbit IgG conjugated with Cy3 (Amersham) as second antibodies. The stained cells were observed under a fluorescence microscope.
Detection of apoptosis
To analyze morphological changes of nuclei, the cells were fixed, stained with Hoechst dye 33258 (8 μg/ml) for 5 min, washed with PBS, and observed under a fluorescence microscope. DNA fragmentation was detected by the terminal transferase-mediated dUTP-biotin nick end labeling method (TUNEL method), using an in situ apoptosis detection kit (Takara, Kusatsu, Japan) according to the protocol provided by the manufacturer. To detect DNA ladder formation, DNA was isolated from the cells as described,28 separated on agarose gels, stained with ethidium bromide, and visualized by UV transillumination.
Measurement of caspase 3 activity
Caspase 3 activity was measured using the Caspase Assay System, Colorimetric (Promega, Madison, WI, USA) according to the protocol provided by the manufacturer. Caspase 3 activity-positive cells were detected using the PhiPhiLux-G1D2 kit (OncoImmunin, Inc., College Park, MD, USA) according to the protocol provided by the manufacturer.
Measurement of NO production
Concentration of NO2− plus NO3− in culture supernatants was measured using the Griess reagent with the NO2−/NO3− assay kit-C (Dojindo Labs, Kumamoto, Japan) after reduction of NO3− to NO2− according to the protocol provided by the manufacturer.
Abbreviations
- carboxy-PTIO:
-
2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide
- hsc70:
-
70 kDa heat shock cognate protein
- hsp70:
-
70 kDa heat shock protein
- IFN-γ:
-
interferon-γ
- LPS:
-
lipopolysaccharide
- NO:
-
nitric oxide
- SNAP:
-
S-nitroso-N-acetyl-DL-penicillamine
References
Christopherson KS, Bredt DS . 1997 Nitric oxide in excitable tissues: physiological roles and disease J. Clin. Invest. 100: 2424–2429
Harrison DG . 1997 Cellular and molecular mechanisms of endothelial cell dysfunction J. Clin. Invest. 100: 2153–2157
Mayer B, Hemmens B . 1997 Biosynthesis and action of nitric oxide in mammalian cells Trends Biochem. Sci. 22: 477–481
Nathan C . 1997 Inducible nitric oxide synthase: what difference does it make? J. Clin. Invest. 100: 2417–2423
Brüne B, von Knethen A, Sandau KB . 1998 Nitric oxide and its role in apoptosis Eur. J. Pharmacol. 351: 261–272
Dimmeler S, Zeiher AM . 1997 Nitric oxide and apoptosis: another paradigm for the double-edged role of nitric oxide Nitric Oxide 1: 275–281
Kröncke KD, Fehsel K, Kolb-Bachofen V . 1997 Nitric oxide: cytotoxicity versus cytoprotection how, why, when,and where? Nitric Oxide 1: 107–120
Samali A, Orrenius S . 1998 Heat shock proteins: regulators of stress response and apoptosis Cell Stress Chaperones 3: 228–236
Smith DF, Whitesell L, Katsanis E . 1998 Molecular chaperones: biology and prospects for pharmacological intervention Pharmacol. Rev. 50: 493–514
Johnson JL, Craig EA . 1997 Protein folding in vivo: unraveling complex pathways Cell 90: 201–204
Netzer WJ, Hartl FU . 1998 Protein folding in the cytosol: chaperonin-dependent and -independent mechanisms Trends Biochem. Sci. 23: 68–73
Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B . 1999 Mechanism of regulation of hsp70 chaperones by DnaJ cochaperones Proc. Natl. Acad. Sci. USA 96: 5452–5457
Kelley WL . 1998 The J-domain family and the recruitment of chaperone power Trends Biochem. Sci. 23: 222–227
Kanazawa M, Terada K, Kato S, Mori M . 1997 HSDJ, a human homolog of DnaJ, is farnesylated and is involved in protein import into mitochondria J. Biochem. (Tokyo) 121: 890–895
Freeman BC, Morimoto RI . 1996 The human cytosolic molecular chaperones hsp90, hsp70 (hsc70) and hdj-1 have distinct roles in recognition of a non-native protein and protein refolding EMBO J. 15: 2969–2979
Minami Y, Hohfeld J, Ohtsuka K, Hartl FU . 1996 Regulation of the heat-shock protein 70 reaction cycle by the mammalian DnaJ homolog, Hsp40 J. Biol. Chem. 271: 19617–19624
Terada K, Kanazawa M, Bukau B, Mori M . 1997 The human DnaJ homologue dj2 facilitates mitochondrial protein import and luciferase refolding J. Cell Biol. 139: 1089–1095
Gotoh T, Mori M . 1999 Arginase II downregulates nitric oxide (NO) production and prevents NO-mediated apoptosis in murine macrophage-derived RAW 264.7 cells J. Cell Biol. 144: 427–434
Kim Y-M, deVera ME, Watkins SC, Billiar TR . 1997 Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-α-induced apoptosis by inducing heat shock protein 70 expression J. Biol. Chem. 272: 1402–1411
Bellmann K, Jäättelä M, Wissing D, Burkart V, Kolb H . 1996 Heat shock protein hsp70 overexpression confers resistance against nitric oxide FEBS Lett. 391: 185–188
Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B . 1997 Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis Mol. Cell. Biol. 17: 5317–5327
Jäättelä M, Wissing D, Kokholm K, Kallunki T, Egeblad M . 1998 Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases EMBO J. 17: 6124–6134
Liossis S-NC, Ding XZ, Kiang JG, Tsokos GC . 1997 Overexpression of the heat shock protein 70 enhances the TCR/CD3- and Fas/Apo-1/CD95-mediated apoptotic cell death in Jurkat T cells J. Immunol. 158: 5668–5675
Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY . 1998 Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1 Nature Genet. 19: 148–154
Meacham GC, Lu Z, King S, Sorscher E, Tousson A, Cyr DM . 1999 The Hdj-2/Hsc70 chaperone pair facilitates early steps in CFTR biogenesis EMBO J. 18: 1492–1505
Niwa H, Yamamura K, Miyazaki J . 1991 Efficient selection for high-expression transfectants with a novel eukaryotic vector Gene 108: 193–199
Yano M, Kanazawa M, Terada K, Namchai C, Yamaizumi M, Hanson B, Hoogenraad N, Mori M . 1997 Visualization of mitochondrial protein import in cultured mammalian cells with green fluorescent protein and effects of overexpression of the human import receptor Tom20 J. Biol. Chem. 272: 8459–8465
Ishizawa M, Kobayashi Y, Miyamura T, Matsuura S . 1991 Simple procedure of DNA isolation from human serum Nucleic Acids Res. 19: 5792
Acknowledgements
We thank our colleagues for suggestions and discussion and M Ohara for comments on the manuscript. This work was supported in part by Grants-in-Aid (10557020 to M Mori and 11770064 to T Gotoh) from the Ministry of Education, Science, Sports, and Culture of Japan.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by J Stamler
Rights and permissions
About this article
Cite this article
Gotoh, T., Terada, K. & Mori, M. hsp70-DnaJ chaperone pairs prevent nitric oxide-mediated apoptosis in RAW 264.7 macrophages. Cell Death Differ 8, 357–366 (2001). https://doi.org/10.1038/sj.cdd.4400829
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4400829
Keywords
This article is cited by
-
The comparison between curcumin and propolis against sepsis-induced oxidative stress, inflammation, and apoptosis in kidney of adult male rat
Future Journal of Pharmaceutical Sciences (2020)
-
Quantitative proteomics of heat-treated human cells show an across-the-board mild depletion of housekeeping proteins to massively accumulate few HSPs
Cell Stress and Chaperones (2015)
-
Cannabidiol protects oligodendrocyte progenitor cells from inflammation-induced apoptosis by attenuating endoplasmic reticulum stress
Cell Death & Disease (2012)
-
Celecoxib upregulates endoplasmic reticulum chaperones that inhibit celecoxib-induced apoptosis in human gastric cells
Oncogene (2006)
-
hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced apoptosis by inhibiting translocation of Bax to mitochondria
Cell Death & Differentiation (2004)
