
Abstract
Specific ligands of the peripheral benzodiazepine receptor (PBR) are known to induce apoptosis and cell cycle arrest in oesophageal cancer cells. However, the underlying mechanisms are still unknown. Here, we investigated the transcriptional alterations and activation of protein kinases in response to PBR-specific ligands. Using cDNA arrays, we examined the transcriptional effects of the PBR-specific ligand FGIN-1-27 in two oesophageal cancer cell lines, KYSE-140 (squamous cell carcinoma) and OE-33 (adenocarcinoma). In oesophageal cancer cells, FGIN-1-27 induced extensive changes in the expression of genes involved in the regulation of apoptosis and cell cycle. Both in oesophageal cancer cell lines (KYSE-140, OE-33) we observed a strong upregulation of the growth arrest and DNA-damage-inducible genes, gadd45 and gadd153, in response to PBR ligands. gadd genes are known to be induced by p38MAPK activation. Using Western blotting we detected a time- and dose-dependent phosphorylation of p38MAPK, which was found to be functionally involved in gadd induction, apoptosis, and cell cycle arrest. In conclusion, our data indicate that PBR-specific ligands cause apoptosis and cell cycle arrest by activation of the p38MAPK pathway and induction of gadd45 and gadd153.
Similar content being viewed by others


Main
Originally, the peripheral benzodiazepine receptor (PBR) was described as another class of binding sites for benzodiazepines distinct from the central benzodiazepine receptor (CBR) (Braestrup and Squires, 1977). It was identified as a mitochondrial 18 kDa protein (McEnery et al, 1992). In contrast to CBR, PBR exhibits high affinity for the isoquinoline carboxamide PK 11195 and the indoleacetamide FGIN-1-27 (Kozikowski et al, 1993). Conversely, clonazepam binds with high affinity to CBR, but has extremely low affinity for PBR (Wang et al, 1984b). In the outer mitochondrial membrane, PBR is associated with the voltage-dependent anion channel and the adenine nucleotide translocator, all of which contribute to the formation of the mitochondrial permeability transition pore (McEnery et al, 1992).
Although present in all tissues to some extent, PBR is highly expressed in steroid-producing tissues (Beurdeley-Thomas et al, 2000). Besides its well-established function in the regulation of steroidogenesis (Papadopoulos et al, 1997), the abundance of PBR in cancers of the colon (Katz et al, 1990b; Maaser et al, 2002a), brain (Cornu et al, 1992), breast (Hardwick et al, 1999), ovary (Katz et al, 1990a), and liver (Venturini et al, 1998) suggests an additional role in tumorigenesis. The proliferation of breast cancer (Beinlich et al, 1999; Carmel et al, 1999), melanoma (Landau et al, 1998), Leydig's cell tumour (Garnier et al, 1993), astrocytoma (Neary et al, 1995), colorectal (Maaser et al, 2001), and oesophageal carcinoma (Sutter et al, 2002) was shown to be inhibited by PBR-specific ligands. In haematopoetic and epithelial cells, the antiproliferative effects of PBR-specific ligands were mediated by the induction of apoptosis and cell cycle arrest (Tanimoto et al, 1999; Maaser et al, 2001; Sutter et al, 2002). However, the underlying mechanisms of PBR-ligand-mediated apoptosis are far from being understood. Several reports have been published on the potential mechanisms contributing to PBR-ligand-mediated apoptosis. Recently, we have shown that mitochondrial membrane permeabilisation is a necessary and early step in PBR-ligand-mediated apoptosis of oesophageal cancer cells (Sutter et al, 2002). In other cell types, a mitochondria-dependent mechanism has been suggested (Fischer et al, 2001; Maaser et al, 2001). Additionally, PBR-ligand-induced apoptosis of thymocytes was found to be sensitive to actinomycin D, cycloheximide, and the protein kinase inhibitor H7, suggesting a requirement for protein synthesis and phosphorylation (Tanimoto et al, 1999). Downregulation of Bcl-2 expression and dephosphorylation of protein kinase B and Bad were shown to be associated with PBR-ligand-induced apoptosis of hepatic stellate cells (Fischer et al, 2001). However, little is known about other transcriptional responses to treatment with PBR-specific ligands.
Cell cycle regulators are frequently disabled in human cancer. Hence, the modulation of cell cycle regulation is a feasible strategy for treating cancer (Shapiro and Harper, 1999). The induction of cell cycle arrest is a well-studied property of PBR-specific ligands. In oesophageal cancer cells, PBR-specific ligands induce a tumour-selective growth arrest at the G1/S checkpoint (Sutter et al, 2002). Likewise, a PBR-ligand-induced G1/S arrest was found in colorectal cancer cells (Maaser et al, 2001). Additionally, PBR ligands induce a cell cycle arrest at both major restriction points, the G1/S- and the G2/M junction (Carmel et al, 1999; Sa"nger et al, 2000) in breast cancer cells, whereas in lung and melanoma cells an accumulation is observed in the G2/M phase (Landau et al, 1998). However, the exact mechanism of PBR-ligand-mediated cell cycle arrest is not yet understood.
Employing cDNA arrays and RT–PCR, we assessed the PBR-ligand-induced downstream transcriptional profiles in oesophageal cancer cells. Here, we report on p38MAPK activation and gadd overexpression in response to PBR-specific ligands, leading to apoptosis and cell cycle arrest.
Material and methods
Cell lines and drugs
The human oesophageal squamous carcinoma cell line KYSE-140 (Shimada et al, 1992) and the human colorectal adenocarcinoma cell line HT-29 were grown in RPMI 1640 medium supplemented with 10% FBS. The human oesophageal adenocarcinoma cell line OE-33 (Rockett et al, 1997) was grown in RPMI 1640 medium supplemented with 10% FBS and 2 mM L-glutamine. Cell lines were cultured in a humidified atmosphere (5% CO2) at 37°C. To evaluate the effects of PBR-specific ligands, cells were incubated with either control medium or medium containing FGIN-1-27, PK 11195 (Tocris, Bristol, UK), clonazepam (Sigma, Deisenhofen, Germany), or FGIN-1-52 (Kozikowski et al, 1993). SB202190 (2–20 μ M, Calbiochem-Novabiochem, Bad Soden, Germany) was used for p38MAPK inhibition. DEVD-CHO (10 μg ml−1, Calbiochem-Novabiochem, Bad Soden, Germany) was used for caspase-3 inhibition.
RNA extraction and polyA+ mRNA preparation
Total RNA was extracted from cultured cell lines with RNAClean following the recommendation of the manufacturer (Hybaid, London, UK). Polyadenylated (polyA+) mRNAs were enriched using magnetic Dynabeads according to the instructions of the supplier (Dynal, Oslo, Norway). The quality of polyA+ and total RNA was controlled by agarose gel electrophoresis.
cDNA array
KYSE-140 and OE-33 cells were treated with FGIN-1-27 (50 μ M) for 24 h to determine PBR-ligand-induced differential gene expression. Untreated cells served as controls. We used an Atlas Human Apoptosis cDNA array with 205 human cDNAs spotted in duplicate on a nylon membrane (Clontech, Palo Alto, CA, USA). A complete list of the cDNAs and controls as well as their accession numbers is available on the web (http://atlasinfo.clontech.com/genelists/huApop.xls). The membranes were hybridised with labelled cDNA probes prepared by reverse transcription from 1 μg polyA+ mRNA using the respective protocol from Clontech. Probes used for hybridisation were equalised to a radioactivity of 2 × 106 cpm ml−1 hybridisation solution. Overnight incubation was followed by stringent washing as recommended by the manufacturer. The membranes were then exposed to X-ray film for quantification. The hybridisation signals were photometrically evaluated using TINA software (raytest Isotopenmessgeräte, Straubenhardt, Germany).
cDNA array data analysis
Alteration in the expression of a respective gene is given as fold increase/or decrease compared to the signal of the untreated control (Höpfner et al, 2002). For determining up- and downregulation, the mean optical density × mm−2 (OD × mm−2) of each gene was identified and normalised to the expression of different housekeeping genes (ubiquitin, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), tubulin α 1 subunit, HLA class I histocompatibility antigen C-4 α subunit (HLAC), cytoplasmic β-actin, 60S ribosomal protein L13A, 40S ribosomal protein S9). Then the ratio of gene expression in treated vs untreated cells was calculated. Data were used only when both signals were 50% or more above background, and if the deviations between duplicates did not exceed the difference between treated and untreated conditions. Each hybridisation experiment was repeated three times.
Semiquantitative RT–PCR
Semiquantitative analysis of mRNA expression of the genes coding for gadd45, gadd153, and for the housekeeping gene β-actin was carried out by RT–PCR with the number of cycles at which the band intensity increased linearly with the amount of mRNA used. For RT–PCR, 2 μg of total RNA was digested with 1 U DNAse I (Gibco, Karlsruhe, Germany) for 15 min at room temperature. Oligo-dT-primers and the SuperScript Preamplification-Kit (Gibco, Karlsruhe, Germany) were used for cDNA synthesis. PCR reactions were carried out in a total volume of 50 μl containing 400 nM of each primer, 200 μ M of each dNTP (Pharmacia, Uppsala, Sweden), 50 mM KCl, 1.5 mM MgCl2, 10 mM Tris, and 1 U Taq-Polymerase (Pharmacia, Uppsala, Sweden). PCR was performed in a Peltier thermocycler (PTC-200, MJ-Research, USA) with the primers and at the conditions indicated in Table 1 (Oh-Hashi et al, 2001).
Western blotting
Whole-cell extracts were prepared by harvesting and lysing the cells with lysis buffer (SDS 0.1%, sodium deoxycholic acid 0.5%, Nonidet P-40 1%, PMSF 0.1 mM, aprotinin 1 μg ml−1, pepstatin A 1 μg ml−1). The protein content of the lysate was determined using the BCA protein assay kit (Pierce, Rockford, IL, USA). The cell lysate was mixed with gel loading buffer (Tris-HCl 62.5 mM, glycerol 10%, SDS 1%, β-mercaptoethanol 2.5%). After boiling for 5 min, the lysates were subjected to SDS–polyacrylamide gel electrophoresis (20 μg of protein per lane; gel: polyacrylamide 12%, SDS 0.1%, Tris-HCl 25 mM; running buffer: Tris 25 mM, glycine 50 mM, 0.1% SDS). After electrophoresis, gels were equilibrated with transfer buffer (Tris 25 mM, glycine 50 mM, 20% methanol). Proteins were transferred to PVDF membranes by electroblotting. Blots were blocked in 1.5% BSA, and then incubated at 4°C overnight with anti-human p38MAPK or phospho-p38MAPK (1 : 500, Santa Cruz Biotechnology, CA, USA). After washing with PBS containing 0.1% Tween and incubation with horseradish peroxidase-coupled anti-IgG antibody (1 : 10000, Amersham, Uppsala, Sweden) at room temperature for 1 h, the blot was washed extensively and developed using enhanced chemiluminescent detection (Amersham, Uppsala, Sweden). Blots were exposed to Hyperfilm ECL film (Amersham, Uppsala, Sweden) for 1–30 min and analysed densitometrically using TINA software (raytest Isotopenmessgeräte, Straubenhardt, Germany).
Caspase-3 activity assay
To determine caspase-3 activity, cells were washed twice with PBS and stored at −80°C until use. Cells (106) were lysed with lysis buffer (Tris-HCl 10 mM, NaH2PO4/Na2HPO4 10 mM, NaCl 130 mM, Triton X-100 1%, NaPPi 10 mM, pH 7.5), and the total protein content was quantified using the BCA protein assay kit (Pierce, Rockford, IL, USA). The activity of caspase-3 was calculated from the cleavage of the fluorogenic substrate DEVD-AMC (Calbiochem-Novabiochem, Bad Soden, Germany). In brief, cell lysates were incubated with substrate solution (caspase-3 substrate AC-DEVD-AMC 20 μg ml−1, HEPES 20 mM, glycerol 10%, DTT 2 mM, pH 7.5) for 1 h at 37°C. The cleavage of DEVD-AMC was measured with a VersaFluor fluorometer (excitation: 360 nm emission: 460 nm) from Biorad, Munich, Germany (Maaser et al, 2002b).
DNA fragmentation
DNA fragmentation was determined by Cell Death Detection ELISA (Roche Molecular Biochemicals) according to the manufacturer's instructions (Höpfner et al, 2003). Briefly, after incubation with the indicated compounds, cells were lysed in incubation buffer. The cytoplasmic fractions were diluted to contain 2.5 × 103 cell equivalents per ml, and presence of mono- and oligonucleosomes was tested using antibodies directed against DNA and histones. DNA fragments were detected by a peroxidase system, with colour development analysed at 405 nm by an ELISA reader.
Cell cycle analysis
Cell cycle analysis was performed by the method of Vindelov and Christensen (1990). Cells were trypsinised, washed, and the nuclei were isolated using CycleTest PLUS DNA Reagent Kit (Becton Dickinson, Heidelberg, Germany). DNA was stained with propidium iodide according to the manufacturer's instructions. The DNA content of the nuclei was detected by flow cytometry and analysed using CellFit software (Becton Dickinson, Heidelberg, Germany).
Statistical analysis
Individual drug therapy was compared by the unpaired, two-tailed Mann–Whitney U-test. The unpaired Student's t-test was used for cell cycle analysis. P-values were considered to be significant at <0.05. If not stated otherwise, all experiments were performed in quadruplicate.
Results
Peripheral benzodiazepine receptor-ligand-induced differential gene expression
Peripheral benzodiazepine receptor-specific ligands have been shown to induce apoptosis and cell cycle arrest potently in human oesophageal cancer cells (Sutter et al, 2002). We used cDNA arrays to analyse changes in the expression of apoptosis- and cell cycle-regulating genes elicited by PBR-specific ligands (Höpfner et al, 2002). For cDNA array experiments, we chose a concentration of 50 μ M FGIN-1-27 and a 24-h incubation time, which is sufficient to induce apoptosis and cell cycle arrest (Maaser et al, 2001; Sutter et al, 2002).
We found over 45 genes that were differentially expressed in both KYSE-140 and OE-33 cells. FGIN-1-27 treatment resulted in an asymmetric distribution of overexpressed vs suppressed genes. Moreover, a comparison of the up- and downregulated genes revealed discrepancies in the FGIN-1-27-induced regulation between the two cell lines: in KYSE-140 (12 overexpressed, 35 suppressed), 25.5% of the regulated genes were overexpressed and 74.5% were suppressed. In contrast, 65.2% of the genes were overexpressed and 34.8% were suppressed in OE-33 cells (30 overexpressed, 16 suppressed). Tables 2 and 3 show the genes regulated by FGIN-1-27 in KYSE-140 and OE-33 cells, respectively.
Tanimoto et al (1999) have demonstrated that PBR-ligand-induced apoptosis required protein de novo synthesis. Therefore, we focused on the genes being overexpressed in both the cancer cell lines. In KYSE-140, five genes were induced by FGIN-1-27 to a level exceeding the expression ratio of 2.0. Three of these genes (gadd153, gadd45, and factor associated with neutral sphingomyelinase activation) were also induced in OE-33 cells, suggesting that these genes are involved in a common signalling pathway. gadd45 and gadd153, both of which are activated by p38MAPK (Kultz et al, 1998), have been associated with apoptosis and growth arrest induced by various extracellular stimuli (Kultz et al, 1998; Maytin et al, 2001; Oh-Hashi et al, 2001). These two genes were selected for further analysis.
Peripheral benzodiazepine receptor-specific ligands induce gadd45 and gadd153 mRNA expression
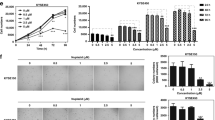
Semiquantitative RT–PCR analysis was performed to confirm the overexpression of gadd45 and gadd153 observed by cDNA array analysis and to monitor their temporal induction. FGIN-1-27 and PK 11195 induced gadd45 and gadd153 rapidly after 2–4 h of treatment, with maximal expression of both transcripts occurring after 8–24 h for FGIN-1-27 and after 4–6 h for PK 11195. The kinetics of induction, however, differed for the two ligands: during treatment with PK 11195, both transcripts returned to basal level after 8 h, but remained elevated up to 24 h after treatment with FGIN-1-27 (Figure 1A, B). To study if gadd overexpression commonly occurred in response to PBR activation, we also analysed gadd expression in FGIN-1-27- or PK 11195-treated HT-29 colorectal cancer cells. HT-29 cells have previously been characterised regarding PBR expression and PBR-ligand-induced apoptosis (Maaser et al, 2001). Both FGIN-1-27 and PK 11195 induced a transient overexpression of gadd153, reaching a maximal induction after 24 h (50 μ M FGIN-1-27, ratio=7.0±3.2) or 6 h (50 μ M PK 11195, ratio=3.3±0.4). Similar to the findings in oesophageal cancer cells, gadd45 was also overexpressed in HT-29 cells after a 24-h incubation with PBR ligands (data not shown).

mRNA expression of gadd45 and gadd153 in response to PBR-ligands: involvement of the p38MAPK signalling pathway. mRNA expression of gadd45 and gadd153 in KYSE-140 cells (A) or OE-33 cells (B) was detected after incubation with FGIN-1-27 or PK 11195. (C) mRNA expression of gadd45 and gadd153 in KYSE-140 cells treated with FGIN-1-27 for 8 h in the presence or absence of SB202190. Pretreatment with SB202190 markedly reduced gadd induction elicited by FGIN-1-27.
p38MAPK activation contributes to gadd45 and gadd153 induction
Mitogen-activated protein (MAP) kinases represent one of the most important signalling cascades in response to extracellular stimuli (Chan-Hui and Weaver, 1998). To gain an insight into the PBR-ligand-mediated signal transduction pathways responsible for gadd45 and gadd153 induction, we determined the influence of the p38MAPK (stress-activated protein kinase 2) cascade. We used the potent p38MAPK inhibitor SB202190 (Herlaar and Brown, 1999; Lee et al, 2000; Mayr et al, 2002) to determine whether p38MAPK activation is directly associated with the induction of gadd messages in oesophageal cancer cells. SB202190 belongs to a family of pyridinyl imidazole compounds that have been shown to inhibit specifically p38MAPkinase activity at the concentrations used, but do not exhibit any significant effect upon a variety of other kinases such as JNK, ERK-1, and MAPKAP kinase 2 (Lee et al, 1994; Cuenda et al, 1995). The FGIN-1-27-mediated induction of gadd45 and gadd153 transcripts was markedly decreased after preincubating the cells with SB202190 for 1 h (Figure 1C). SB202190 alone had no effect on gadd expression (data not shown). These data suggest that p38MAPK activation contributes to the induction of gadd45 and gadd153 by the PBR-specific ligand FGIN-1-27.
p38MAPK activation by PBR-specific ligands
Phosphorylation-mediated activation of the p38MAPK by PBR-specific ligands was determined by Western blotting. Both PBR-specific ligands, FGIN-1-27 and PK 11195, induced a time- and dose-dependent phosphorylation of p38MAPK, thereby showing high correlation with the induction of gadd transcripts (Figure 2A, B). The maximum of p38MAPK activation was observed after 4 h (FGIN-1-27) or 1–8 h (PK 11195) of treatment. After 4 h, we observed an about 1.7-fold activation of p38MAPK in response to 10 μ M of the respective PBR ligand and an about 3.1-fold activation in response to 50 μ M of either ligand. At 100 μ M, we detected an up to 4.1-fold increase of activated p38MAPK. As activation was pronounced at 50 μ M of either ligand already, we chose this concentration for further experiments. PBR-specific ligands did not affect the expression of either p38MAPK protein (Figure 2) or mRNA (data not shown) analysed within a period of 24 h.

Activation of p38MAPK by PBR-specific ligands: involvement of caspase-3. Posphorylation of p38MAPK was analysed by Western blotting using antibodies against the active form (P-p38MAPK) and nonphosphorylated p38MAPK. p38MAPK was transiently phosphorylated in KYSE-140 (A) and OE-33 cells (B) by FGIN-1-27 or PK 11195. (C) p38MAPK phosphorylation in KYSE-140 cells treated with either vehicle, FGIN-1-27 (4 h), or DEVD-CHO for 1 h followed by FGIN-1-27 for 4 h. Pretreatment with DEVD-CHO attenuated FGIN-1-27-induced p38MAPK phosphorylation.
FGIN-1-27-induced caspase-3 activation contributes to activation of p38MAPK
Peripheral benzodiazepine receptor-ligand-mediated apoptosis involves caspase-3 activation, leading to DNA fragmentation and cell death (Sutter et al, 2002). The PBR-specific ligand FGIN-1-27 induced a dose- (Sutter et al, 2002) and time-dependent increase in caspase-3 activity in both KYSE-140 (Figure 3A) and OE-33 (Figure 3B) cells. The time course of PBR-ligand-induced caspase-3 activation correlates with p38MAPK phosphorylation, suggesting a link between caspase-3 and p38MAPK activation. Thus, a possible regulatory relationship between caspase-3 and p38MAPK during PBR-ligand-mediated apoptosis was investigated. KYSE-140 cells were treated with FGIN-1-27 (50 μ M) for 4 h in the presence or absence of the caspase-3 inhibitor DEVD-CHO (10 μg ml−1). This concentration was previously shown to be sufficient to inhibit caspase-3 activation and DNA fragmentation (Sutter et al, 2002). Cells pretreated with DEVD-CHO displayed a markedly (−42%) reduced activation of p38MAPK (Figure 2C), indicating that in FGIN-1-27-induced apoptosis of KYSE-140 cells, caspase-3 activation contributes to p38MAPK activation. On the other hand, pretreatment of KYSE-140 cells with SB202190 did not prevent caspase-3 activation by FGIN-1-27, even at the highest concentration of the p38MAPK inhibitor, demonstrating that p38MAPK activation is not required for caspase-3 activation (data not shown).

PBR-ligand-induced caspase-3 activation. KYSE-140 (A) or OE-33 cells (B) were treated with FGIN-1-27 (50 μ M). Caspase-3 activity was measured fluorometrically by the cleavage of DEVD-AMC. Data are given as percentage of untreated control (means±s.e.m. of four independent experiments). * Statistical significance (P<0.05) compared to untreated control.
FGIN-1-27-induced p38MAPK activation contributes to DNA fragmentation
Finally, we investigated if p38MAPK activation contributes to FGIN-1-27-induced DNA fragmentation. Thus, KYSE-140 cells were treated with FGIN-1-27 (50 μ M) either in the presence or in the absence of SB202190. Cells pretreated with SB202190 for 1 h displayed a reduced DNA fragmentation, providing evidence that p38MAPK activation is involved in DNA fragmentation (Figure 4A).

p38MAPK activation is required for FGIN-1-27-mediated DNA fragmentation and G1/S arrest. (A) KYSE-140 cells, incubated with 50 μ M FGIN-1-27 alone or in combination with SB202190, were analysed for DNA fragmentation. Cells were preincubated for 1 h with SB202190 prior to FGIN-1-27 addition and subsequent incubation for 14 h. Untreated cells (ctrl), and cells treated only with SB202190, were also run as controls. SB202190 strongly diminished FGIN-1-27-induced DNA fragmentation. (B) KYSE-140 cells were treated with either vehicle, FGIN-1-27 for 24 h, SB202190 for 25 h, or SB202190 for 1 h followed by FGIN-1-27 plus SB202190 for 24 h. Treatment with FGIN-1-27 increased the proportion of cells in the G0/G1 phase (black columns), whereas the proportion of cells in the S (hatched columns) and G2/M phases (white columns) decreased. Pretreatment with SB202190 abolished FGIN-1-27-mediated G1/S arrest. Means of three independent experiments±s.e.m are shown. *P<0.05, NS=not significant.
Activation of the p38MAPK pathway is responsible for FGIN-1-27-mediated cell cycle arrest
Peripheral benzodiazepine receptor ligands have been shown to arrest the cell cycle of human oesophageal cancer cells in the G0/G1 phase, thereby causing a G1/S arrest (Sutter et al, 2002). Thus, we analysed whether p38MAPK activation was involved in PBR-ligand-mediated cell cycle arrest. Preincubating KYSE-140 cells with SB202190 completely abolished FGIN-1-27-induced G1/S arrest in oesophageal cancer cells (Figure 4B), suggesting that p38MAPK activation is required for the cell cycle arrest observed. SB202190 alone did not have any significant impact on the cell cycle.
Discussion
In this study we provide an insight into the signal transduction pathway by which PBR-specific ligands induce cell cycle arrest and apoptosis in oesophageal cancer cells. Peripheral benzodiazepine receptor-specific ligands activate the p38MAPK signalling pathway, leading to overexpression of gadd45 and gadd153 and cell cycle arrest. Furthermore, we show that PBR-ligand-induced caspase-3 activation contributes to p38MAPK activation, resulting in DNA fragmentation. This suggests an involvement of p38MAPK in PBR-mediated apoptosis.
The p38MAPK pathway is known to be activated by a variety of stimuli including UV irradiation, hydrogen peroxide, DNA damage, heat, and hyperosmotic shock. Activation of the p38MAPK pathway results in growth arrest and apoptosis (Kultz et al, 1998; Ichijo, 1999). We used the pyridinyl imidazole inhibitor SB202190 to demonstrate the involvement of p38MAPK in PBR-ligand-mediated apoptosis and cell cycle arrest. SB202190 has been shown to inhibit p38MAPK α and β at the concentrations applied, whereas it shows no effect against a large panel of other related protein kinases tested (Davies et al, 2000). SB202190 has widely been used to study the involvement of p38MAPK in proliferation, apoptosis, and differentiation (Kultz et al, 1998; Oh-Hashi et al, 2001). In this study, SB202190 prevented PBR-ligand-induced apoptosis and G1/S arrest, suggesting an involvement of the p38MAPK pathway.
Mitogen-activated protein kinase (MAPK) signal transduction pathways are known to regulate the expression of the gadd genes (Kultz et al, 1998; Oh-Hashi et al, 2001). Thus, our next goal was to elucidate if p38MAPK is involved in PBR-ligand-mediated gadd overexpression. The expression of the gadd45 gene has been correlated with the presence of strong growth arrest (Zhan et al, 1994a), and it has been shown to associate with proliferating cell nuclear antigen (PCNA), where it may play a role in DNA repair (Smith et al, 1994). Overexpression of each gadd gene causes growth inhibition and/or apoptosis, and combined overexpression of the gadd genes leads to a synergistic suppression of cell growth (Zhan et al, 1994b). In this study, inhibition of p38MAPK activity by SB202190 suppressed the expression of gadd genes induced by PBR-specific ligands. These results confirm earlier findings that gadd induction occurs as a direct consequence of p38MAPK activation (Oh-Hashi et al, 2001). The potency of SB202190 to inhibit gadd45 and gadd153 overexpression correlated well with its ability to decrease apoptosis and cell cycle arrest, suggesting an involvement of gadd genes in apoptosis and G1/S arrest. In accordance with our findings, it has been reported that G1/S arrest is a result of gadd induction by p38MAPK (Smith et al, 1994). However, as even the highest concentration of SB202190 only partially prevented the increase in gadd45 and gadd153 expression, other still unidentified, p38MAPK-independent pathways (Maytin et al, 2001) may contribute to PBR-ligand-mediated gadd induction: the generation of reactive oxygen species, the activation of the p53 pathway or the JNK pathway are well known to induce gadd genes, too (Guyton et al, 1996; Sheikh et al, 2000). In our previous report on PBR-ligand-mediated apoptosis of oesophageal cancer cells, we showed that disruption of the mitochondrial membrane potential was required for caspase-3 activation (Sutter et al, 2002). Using the caspase-3-specific inhibitor DEVD-CHO, we now demonstrate that caspase activation is upstream of p38MAPK activation, supporting previous findings in Jurkat T lymphocytes (Juo et al, 1997,1998; Matsuda et al, 1999). Interestingly, caspase-3 activation occurs independently of p38MAPK, as the p38MAPK inhibitor SB202190 failed to prevent the activation of caspase-3. Furthermore, FGIN-1-27-induced DNA fragmentation is decreased by pretreatment with SB202190, suggesting an involvement of p38MAPK in PBR-ligand-mediated formation of oligonucleosomes.
As p38MAPK activation also proved to be a prerequisite for PBR-ligand-mediated cell cycle arrest, we provide a link between PBR-ligand-mediated induction of apoptosis and G1/S arrest. Furthermore, we extend our current model on PBR-ligand-mediated apoptosis. The signalling cascade comprises mitochondrial membrane permeabilisation leading to caspase-3 activation, followed by p38MAPK activation and finally DNA fragmentation.
To clarify the PBR specificity of the effects observed, we applied FGIN-1-52, a structural analogue of FGIN-1-27, and the benzodiazepine clonazepam; neither substance binding to PBR (Wang et al, 1984b; Kozikowski et al, 1993). Clonazepam or FGIN-1-52 did not affect p38MAPK phosphorylation, gadd expression (data not shown), apoptosis, or the cell cycle (Maaser et al, 2001; Sutter et al, 2002). This indicates that the dose- and time-dependent induction of the p38MAPK signalling pathway by FGIN-1-27 and PK 11195 is PBR specific.
Protein de novo synthesis was shown to be required for PBR-ligand-mediated induction of apoptosis (Tanimoto et al, 1999). Therefore, we analysed the transcriptional changes in response to treatment with FGIN-1-27. Using an apoptosis- and cell-cycle-specific cDNA array spotted with 205 genes related to proliferation, apoptosis and cell cycle, we have identified 78 genes, about 45 genes in either cell line, responsive to FGIN-1-27 treatment. The fact that PBR-specific ligands exert antiproliferative effects in different tumours (Wang et al, 1984a; Maaser et al, 2001) suggests that PBR ligands interfere with a common signalling pathway. The expression patterns elicited by FGIN-1-27 partially overlapped between KYSE-140 and OE-33 cells, suggesting that those genes commonly regulated in both cell lines are of general importance for apoptosis and cell cycle arrest. Out of the 205 genes, only three of those exceeding an expression ratio of 2.0 in KYSE-140 cells were also overexpressed in OE-33 cells. Two of them were gadd45 and gadd153, both of which are known to be transcriptionally regulated (Kultz et al, 1998). Our functional data indicate that gadd45 and gadd153 overexpression plays an important role in PBR-ligand-mediated apoptosis and cell cycle arrest. Many genes were regulated by FGIN-1-27 treatment in only one of the two cell lines. Apparently, cell-type-specific differences occur in the signalling pathways involved in the effects of FGIN-1-27. Moreover, differences between the two cell lines may also reflect differences in the cellular stress response to the initial stimulus. For example, in both the cell lines we found an overexpression of glutathione transferases in response to treatment with FGIN-1-27. However, different isoforms of the antioxidant enzyme were induced. In spite of the differences, the expression patterns of both cell lines after treatment with FGIN-1-27 reflect the apoptotic and growth-arrested phenotype of oesophageal cancer cells. In OE-33 cells, FGIN-1-27 treatment strongly decreased the expression of survivin, which is an antiapoptotic protein with prognostic relevance in oesophageal cancer (Grabowski et al, 2003). Furthermore, programmed cell death 2 protein (PDCD2), a gene associated with apoptosis of thymocytes (Kawakami et al, 1995), is induced by FGIN-1-27. The growth arrest is reflected in the overexpression of cdki2A and PCNA, the interaction partner of gadd45, and the downregulation of cdc2. In KYSE-140 cells, we observed a downregulation of cyclin C, cyclin D1 and cdc25A, all of which are associated with G1/S transition. However, the functional involvement of each protein has to be evaluated.
In summary, p38MAPK is dose- and time-dependently activated by PBR-ligands. Furthermore, gadd genes are overexpressed and apoptosis and cell cycle arrest are induced, all of which are known consequences of p38MAPK activation. Intriguingly, all effects can be antagonised by SB202190, which is described as a potent p38MAPK inhibitor. Thus, our data suggest that activating the p38MAPK pathway is a necessary step for inducing apoptosis and cell cycle arrest by PBR-specific ligands. Understanding the mechanisms of action will facilitate the design of combination chemotherapies that act additively or synergistically. Furthermore, some of the molecular targets like gadd153 and gadd45 might be used as surrogate biomarkers for future PBR-ligand intervention trials. Interestingly, using gadd153 induction as a predictor of clinical response has already been evaluated for paclitaxel treatment of cancer patients (Las Alas et al, 2000). Hence, our data on the pathways responding to PBR-specific ligands, in combination with the knowledge that signalling pathways may be defective in tumours, will be helpful in predicting the responsiveness of tumours to PBR ligands in the future.
Change history
16 November 2011
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Beinlich A, Strohmeier R, Kaufmann M, Kuhl H (1999) Specific binding of benzodiazepines to human breast cancer cell lines. Life Sci 65: 2099–2108
Beurdeley-Thomas A, Miccoli L, Oudard S, Dutrillaux B, Poupon MF (2000) The peripheral benzodiazepine receptors: a review. J Neurooncol 46: 45–56
Braestrup C, Squires RF (1977) Specific benzodiazepine receptors in rat brain characterized by high-affinity (3 H)diazepam binding. Proc Natl Acad Sci USA 74: 3805–3809
Carmel I, Fares FA, Leschiner S, Scherübl H, Weisinger G, Gavish M (1999) Peripheral-type benzodiazepine receptors in the regulation of proliferation of MCF-7 human breast carcinoma cell line. Biochem Pharmacol 58: 273–278
Chan-Hui PY, Weaver R (1998) Human mitogen-activated protein kinase kinase kinase mediates the stress-induced activation of mitogen-activated protein kinase cascades. Biochem J 336: 599–609
Cornu P, Benavides J, Scatton B, Hauw JJ, Philippon J (1992) Increase in omega 3 (peripheral-type benzodiazepine) binding site densities in different types of human brain tumours. A quantitative autoradiography study. Acta Neurochir 119: 146–152
Cuenda A, Rouse J, Doza YN, Meier R, Cohen P, Gallagher TF, Young PR, Lee JC (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 364: 229–233
Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105
Fischer R, Schmitt M, Bode JG, Häussinger D (2001) Expression of the peripheral-type benzodiazepine receptor and apoptosis induction in hepatic stellate cells. Gastroenterology 120: 1212–1226
Garnier M, Boujrad N, Oke BO, Brown AS, Riond J, Ferrara P, Shoyab M, Suarez-Quian CA, Papadopoulos V (1993) Diazepam binding inhibitor is a paracrine/autocrine regulator of Leydig cell proliferation and steroidogenesis: action via peripheral-type benzo-diazepine receptor and independent mechanisms. Endocrinology 132: 444–458
Grabowski P, Kühnel T, Mühr-Wilkenshoff F, Heine B, Stein H, Höpfner M, Germer CT, Zeitz M, Scherübl H (2003) Prognostic value of nuclear survivin expression in oesophageal squamous cell carcinoma. Br J Cancer 88: 115–119
Guyton KZ, Xu Q, Holbrook NJ (1996) Induction of the mammalian stress response gene GADD153 by oxidative stress: role of AP-1 element. Biochem J 314: 547–554
Hardwick M, Fertikh D, Culty M, Li H, Vidic B, Papadopoulos V (1999) Peripheral-type benzodiazepine receptor (PBR) in human breast cancer: correlation of breast cancer cell aggressive phenotype with PBR expression, nuclear localization, and PBR-mediated cell proliferation and nuclear transport of cholesterol. Cancer Res 59: 831–842
Herlaar E, Brown Z (1999) p38 MAPK signalling cascades in inflammatory disease. Mol Med Today 5: 439–447
Höpfner M, Maaser K, Theiss A, Lenz M, Sutter AP, Riecken EO, Zeitz M, Scherübl H (2003) Apoptotic and antiproliferative effects of photoactivated hypericin on esophageal cancer cells. Int J Colorectal Dis 18: 239–247
Höpfner M, Sutter AP, Beck N, Barthel B, Maaser K, Jockers-Scherübl M, Zeitz M, Scherübl H (2002) Meta-iodobenzylguanidine induces growth inhibition and apoptosis of neuroendocrine gastrointestinal tumor cells. Int J Cancer 101: 210–216
Ichijo H (1999) From receptors to stress-activated MAP kinases. Oncogene 18: 6087–6093
Juo P, Kuo CJ, Reynolds SE, Konz RF, Raingeaud J, Davis RJ, Biemann HP, Blenis J (1997) Fas activation of the p38 mitogen-activated protein kinase signalling pathway requires ICE/CED-3 family proteases. Mol Cell Biol 17: 24–35
Juo P, Kuo CJ, Yuan J, Blenis J (1998) Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr Biol 8: 1001–1008
Katz Y, Ben Baruch G, Kloog Y, Menczer J, Gavish M (1990a) Increased density of peripheral benzodiazepine-binding sites in ovarian carcinomas as compared with benign ovarian tumours and normal ovaries. Clin Sci 78: 155–158
Katz Y, Eitan A, Gavish M (1990b) Increase in peripheral benzodiazepine binding sites in colonic adenocarcinoma. Oncology 47: 139–142
Kawakami T, Furukawa Y, Sudo K, Saito H, Takami S, Takahashi E, Nakamura Y (1995) Isolation and mapping of a human gene (PDCD2) that is highly homologous to Rp8, a rat gene associated with programmed cell death. Cytogenet Cell Genet 71: 41–43
Kozikowski AP, Ma D, Brewer J, Sun S, Costa E, Romeo E, Guidotti A (1993) Chemistry, binding affinities, and behavioral properties of a new class of “antineophobic” mitochondrial DBI receptor complex (mDRC) ligands. J Med Chem 36: 2908–2920
Kultz D, Madhany S, Burg MB (1998) Hyperosmolality causes growth arrest of murine kidney cells. Induction of GADD and (GADD153 by osmosensing via stress-activated protein kinase 2. J Biol Chem 273: 13645–13651
Landau M, Weizman A, Zoref-Shani E, Beery E, Wasseman L, Landau O, Gavish M, Brenner S, Nordenberg J (1998) Antiproliferative and differentiating effects of benzodiazepine receptor ligands on B16 melanoma cells. Biochem Pharmacol 56: 1029–1034
Las Alas MM, Christen RD, Gately DP, Weiner DE, Benbatoul K, Kirmani S, D'Agostino HR, Plaxe SC, Darrah D, McClay EF, Aebi S, Howell SB, Los G (2000) Increase in tumor GADD153 mRNA level following treatment correlates with response to paclitaxel. Cancer Chemother Pharmacol 45: 381–388
Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, Adams JL (2000) Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology 47: 185–201
Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler SE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR, (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739–746
Maaser K, Grabowski P, Sutter AP, Höpfner M, Foss HD, Stein H, Berger G, Gavish M, Zeitz M, Scherübl H (2002a) Overexpression of the peripheral benzodiazepine receptor is a relevant prognostic factor in stage III colorectal cancer. Clin Cancer Res 8: 3205–3209
Maaser K, Höpfner M, Jansen A, Weisinger G, Gavish M, Kozikowski AP, Weizman A, Carayon P, Riecken EO, Zeitz M, Scherübl H (2001) Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human colorectal cancer cells. Br J Cancer 85: 1771–1780
Maaser K, Höpfner M, Kap H, Sutter AP, Barthel B, von Lampe B, Zeitz M, Scherübl H (2002b) Extracellular nucleotides inhibit growth of human esophageal cancer cells via P2Y(2)-receptors. Br J Cancer 86: 636–644
Matsuda S, Minowa A, Suzuki S, Koyasu S (1999) Differential activation of c-Jun NH2-terminal kinase and p38 pathways during FTY720-induced apoptosis of T lymphocytes that is suppressed by the extracellular signal-regulated kinase pathway. J Immunol 162: 3321–3326
Mayr M, Hu Y, Hainaut H, Xu Q (2002) Mechanical stress-induced DNA damage and rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J 16: 1423–1425
Maytin EV, Ubeda M, Lin JC, Habener JF (2001) Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res 267: 193–204
McEnery MW, Snowman AM, Trifiletti RR, Snyder SH (1992) Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci 89: 3170–3174
Neary JT, Jorgensen SL, Oracion AM, Bruce JH, Norenberg MD (1995) Inhibition of growth factor-induced DNA synthesis in astrocytes by ligands of peripheral-type benzodiazepine receptors. Brain Res 675: 27–30
Oh-Hashi K, Maruyama W, Isobe K (2001) Peroxynitrite induces GADD34, 45, and 153 via p38 MAPK in human neuroblastoma SH-SY5Y cells. Free Radic Biol Med 30: 213–221
Papadopoulos V, Amri H, Boujrad N, Cascio C, Culty M, Garnier M, Hardwick M, Li H, Vidic B, Brown AS, Reversa JL, Bernassau JM, Drieu K (1997) Peripheral benzodiazepine receptor in cholesterol transport and steroidogenesis. Steroids 62: 21–28
Rockett JC, Larkin K, Darnton SJ, Morris AG, Matthews HR (1997) Five newly established oesophageal carcinoma cell lines: phenotypic and immunological characterization. Br J Cancer 75: 258–263
Sänger N, Strohmeier R, Kaufmann M, Kuhl H (2000) Cell cycle-related expression and ligand binding of peripheral benzodiazepine receptor in human breast cancer cell lines. Eur J Cancer 36: 2157–2163
Shapiro GI, Harper JW (1999) Anticancer drug targets: cell cycle and checkpoint control. J Clin Invest 104: 1645–1653
Sheikh MS, Hollander MC, Fornace Jr AJ (2000) Role of Gadd45 in apoptosis. Biochem Pharmacol 59: 43–45
Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T (1992) Characterization of 21 newly established esophageal cancer cell lines. Cancer 69: 277–284
Smith ML, Chen IT, Zhan Q, Bae I, Chen CY, Gilmer TM, Kastan MB, O'Connor PM, Fornace Jr AJ (1994) Interaction of the p53-regulated protein Gadd45 with proliferating cell nuclear antigen. Science 266: 1376–1380
Sutter AP, Maaser K, Höpfner M, Barthel B, Grabowski P, Faiss S, Carayon P, Zeitz M, Scherübl H (2002) Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human esophageal cancer cells. Int J Cancer 102: 318–327
Tanimoto Y, Onishi Y, Sato Y, Kizaki H (1999) Benzodiazepine receptor agonists modulate thymocyte apoptosis through reduction of the mitochondrial transmembrane potential. Jpn J Pharmacol 79: 177–183
Venturini I, Zeneroli ML, Corsi L, Avallone R, Farina F, Alho H, Baraldi C, Ferrarese C, Pecora N, Frigo M, Ardizzone G, Arrigo A, Pellicci R, Baraldi M (1998) Up-regulation of peripheral benzodiazepine receptor system in hepatocellular carcinoma. Life Sci 63: 1269–1280
Vindelov L, Christensen IJ (1990) An integrated set of methods for routine flow cytometric DNA analysis. Methods Cell Biol 33: 127–137
Wang JK, Morgan JI, Spector S (1984a) Benzodiazepines that bind at peripheral sites inhibit cell proliferation. Proc Natl Acad Sci 81: 753–756
Wang JK, Taniguchi T, Spector S (1984b) Structural requirements for the binding of benzodiazepines to their peripheral-type sites. Mol Pharmacol 25: 349–351
Zhan Q, Bae I, Kastan MB, Fornace Jr AJ (1994a) The p53-dependent gamma-ray response of GADD45. Cancer Res 54: 2755–2760
Zhan Q, Lord KA, Alamo Jr I, Hollander MC, Carrier F, Ron D, Kohn KW, Hoffman B, Liebermann DA, Fornace Jr AJ (1994b) The gadd and MyD genes define a novel set of mammalian genes encoding acidic proteins that synergistically suppress cell growth. Mol Cell Biol 14: 2361–2371
Acknowledgements
This study was supported by grants of the Deutsche Krebshilfe, Wilhelm-Sander Stiftung, and Berliner Krebsgesellschaft. Andreas P Sutter was supported by a scholarship from the DFG, Graduiertenkolleg 276/2, ‘signal transduction and recognition’. We thank Dr Alan P Kozikowski for generously providing us with FGIN-1-52, Mr Nikolai I Beck for excellent technical assistance, and Dr Michael Höpfner for careful revision of the manuscript and helpful discussions. We are indebted to the Institute of Physiology, Free University Berlin, Germany, for laboratory facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Sutter, A., Maaser, K., Barthel, B. et al. Ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in oesophageal cancer cells: involvement of the p38MAPK signalling pathway. Br J Cancer 89, 564–572 (2003). https://doi.org/10.1038/sj.bjc.6601125
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6601125
Keywords
This article is cited by
-
TSPO ligand residence time influences human glioblastoma multiforme cell death/life balance
Apoptosis (2015)
-
In vitro effect of FGIN-1-27, a ligand to 18 kDa mitochondrial translocator protein, in human osteoblast-like cells
Journal of Bioenergetics and Biomembranes (2014)
-
Identification, characterization and potent antitumor activity of ECO-4601, a novel peripheral benzodiazepine receptor ligand
Cancer Chemotherapy and Pharmacology (2008)
-
PK11195 potently sensitizes to apoptosis induction independently from the peripheral benzodiazepin receptor
Oncogene (2005)
-
Evidence in favour of a role for peripheral-type benzodiazepine receptor ligands in amplification of neuronal apoptosis
Apoptosis (2005)
