
Abstract
Endophytic fungi play an important role in the growth and development of traditional Chinese medicine plants. We isolated a strain of Acrocalymma vagum from the endophytic fungi of the traditional Chinese plants Paris. To accurately identify this endophytic fungal species of interest, we sequenced the mitochondrial genome of A. vagum, which is the first discovered mitochondrial genome in Massarineae. The A. vagum mitochondrial genome consists of a 35,079-bp closed circular DNA molecule containing 36 genes. Then, we compared the general sequence characteristics of A. vagum with those of Pleosporales, and the second structure of the 22 tRNAs was predicted. The phylogenetic relationship of A. vagum was constructed using two different data sets (protein-coding genes and amino acids). The phylogenetic tree shows that A. vagum is located at the root of Pleosporales. The analysis of introns shows that the number of introns increases with the increase in branch length. The results showed that monophyly was confirmed for all families in Pleosporales except for Pleosporaceae. A. vagum is an ancient species in the Pleosporales, and Pleosporaceae may require further revision. In Pleosporales, the number of introns is positively correlated with branch length, providing data for further study on the origin of introns.
Similar content being viewed by others


Introduction
Acrocalymma vagum belongs to the family Morosphaeriaceae, suborder Massarineae, and order Pleosporales1. The Pleosporales includes a wide range of complex organisms and is one of the largest orders of Dothideomycetes2,3. Most previous studies on Pleosporales relationships have focused on their morphological characteristics4. The different criteria employed by different fungal taxonomists to describe species result in often divergent morphological descriptions of the same fungus5. This has led to controversy over the taxonomic status of species in Pleosporales6. With the recent development of molecular sequencing technologies, molecular identification is commonly used for the identification of fungi7. The taxonomic status of many species in Pleosporales has been determined by molecular identification (internally transcribed spacer [ITS], 18S, and 28S)8,9,10. Differences in morphological and molecular identification result in frequent changes in the taxonomic status of many Pleosporales species11,12,13,14. For example, Rhizopycnis vagum was initially identified morphologically and then molecularly, leading to its transfer from R. vagum to Acrocalymma15.
In our previous study, we isolated an endophytic fungus from the roots of Paris polyphylla var. yunnanensis, which was morphologically and molecularly identified as belonging to A. vagum. Paris polyphylla var. yunnanensis is a valuable medicinal plant in traditional Chinese medicine. The market demand for Paris has increased with time, but its growth is slow and its natural reproduction rate is low. Endophytic fungi significantly promoted the growth of Paris polyphylla var. yunnanensis. Therefore, it is important to determine the distribution and characteristics of the fungal species in Paris polyphylla var. yunnanensis. In previous studies, the taxonomic status of A. vagum was generally determined by ITS sequencing. However, this has certain limitations due to its short molecular fragment. The mitochondrial genome is more suitable for the dating of evolutionary events, as it is believed to conform to the molecular clock hypothesis with a stable and constant mutation rate. The coding regions of the mitochondrial genome, characterized by increased length and a slower mutation rate, exhibit parallel mutations or decreased homology, which increases the reliability of phylogenetic estimates.
Owing to these advantages, mitochondrial genomes are widely used for fungal species classification. For example, Yildiz et al. identified the plant pathogenic fungus Monilinia laxa using a mitochondrial genome16; Jelen et al. used a mitochondrial genome to identify the fungus Verticillium-wilt, a plant Verticillium nonalfalfae pathogen17; and Kortsinoglou et al. used a mitochondrial genome to identify Metarhizium, a biocontrol agent component of entomopathogenic fungi18. Therefore, phylogenetic analysis of the mitochondrial genomes of A. vagum is necessary.
To date, Pleosporales has only 36 sequenced mitochondrial genomes, and there is one incomplete mitochondrial genome gene in Massarineae, with its sequence being too short to be representative. The genome’s status in the NCBI remains unverified. To fill this gap, we sequenced the complete mitochondrial genome of A. vagum. The complete mitochondrial genome of a species from Massarineae was obtained for annotation and phylogenetic analysis. The aim of this study was to address the following issues: (1) The general characterization of a mitochondrial genome from Massarineae; (2) Exploration of the interspecific developmental relationships among families in the order Pleosporlaes. (3) Exploration of the phylogenetic position of Massarineae in Pleosporales.
Material and Methods
Sample collection and DNA extraction
The strains were collected from the Chinese herbal medicine plant Paris polyphylla var. yunnanensis, located in Anshun City, Guizhou Province, China (105° 58′ 13.22″ E and 26° 02′ 22.85″ N). After screening and purification, the mycelium of A. vagum was scraped off the potato dextrose agar solid medium and ground into a powder after being frozen in liquid nitrogen. Genomic DNA was extracted using the Fungal Genome Extraction Kit (Omega Bio-tek America). Genomic DNA integrity was determined using 1% agarose gel electrophoresis. The extracted DNA was stored at − 80 °C until use.
Mitogenome sequencing, assembly, and annotation
The whole genome of A. vagum was sequenced by Shanghai Sangon on the HiSeq 2500 platform (Illumina) with 150-bp paired-end reads. After quality evaluation and splicing, 6 GB of raw data was obtained. The sequences were assembled using Geneious Prime version 2019.219. The assembled gene sequences were compared with homologous sequences retrieved from GenBank and identified through BLAST searches in the NCBI database to confirm sequence accuracy20. We used the MITOS21 web server and BLAST searches in the NCBI database (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to annotate the assembled mitogenomes with mold protozoan mitochondrial genetic codes20 and the tRNAscan-SE 1.21 search server to identify the locations of the tRNA genes22,23. The 12S and 16S rRNA genes were identified based on the locations of the adjacent tRNA genes and compared with the sequences of other Pleosporales mitogenomes in the NCBI. ORF Finder in Geneious Prime was used to predict the protein-coding gene (PCG) locations in the mold protozoan mitochondria.
Evolutionary rates of the mitochondrial genes
The synonymous sites (Ks), nonsynonymous substitution sites (Ka), and their ratios (Ka/Ks) are often employed to measure evolutionary rates24. Therefore, we chose 13 PCGs (atp6, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4l, nad5, rps3, and nad6) of 18 Pleosporales to calculate the values of Ka, Ks, and Ka/Ks. These genes were aligned using MEGA 7.025 according to the codons (parameters: Gap opening penalty: 400; Gap extension penalty: 0.2; and Delay divergent cutoff: 30%), and the Ka/Ks ratio was calculated using DnaSP ver. 526.
Mitogenome annotation and sequence analysis
Mapping and comparative analysis of the mitochondrial genomes were performed on CGview (https://proksee.ca/). The codon usage frequencies of amino acids in the mitochondrial genome were analyzed using CodonW and MEGA 7.025. The relative synonymous codon usage value and codon usage frequency in the mitochondrial genome were analyzed with MEGA 7.025. Finally, the chain asymmetry was calculated using the following formula27:
Phylogenetic analysis
Eighteen Pleosporales [Astrosphaeriellaceae (1), Corynesporascaceae (1), Coniothyriaceae (1), Didymellaceae (3), Morosphaeriaceae (1), Phaeosphaeriaceae (1), Pleosporaceae (9), and Shiraiaceae (1)]28,29,30,31,32,33,34,35,36,37,38,39,40,41,42 were selected to construct the phylogenetic tree after the removal of unverified sequences, those that lacked an accurate scientific name, or were repetitive. Due to the loss of the rsp3 gene in some species during evolution, only 12 PCGs were used to construct the phylogenetic tree. Phylogenetic analysis was performed using the alignments of the 12 PCGs of complete or near-complete mitogenomes of the Pleosporales species. The three species of Zymoseptoria tritici, Pseudocercospora fijiensis, and Zasmidium cellare were used as outgroups43,44,45 (Table S1).
The 12 PCGs were aligned using the TranslatorX online tool, employing MAFFT to perform the protein alignment34,46,47,48. The 12 resulting alignments were assessed and manually corrected using MEGA version 7.0 program25. For the phylogenetic analyses, the maximum likelihood (ML) and Bayesian inference (BI) methods were employed to construct the ML and BI trees based on two datasets (PCG, the 12 PCGs; AA and amino acid sequences of the 12 PCGs). ML analysis was performed with 1000 rapid bootstrapping replicates using iqtree49, whereas the BI analysis was performed in MrBayes 3.2.7a with four chains and sampling of the chains every 1000 generations50. Two independent runs of 10 million generations were performed. After the average standard deviation of the split frequencies decreased to < 0.001, the initial 25% of the samples were discarded as the burn-in and the remaining trees were used to generate a consensus tree and calculate the posterior probabilities51. The BI and ML analyses were performed on the CIPRES Science Gateway (https://www.phylo.org) website, and the phylogenetic trees were visualized using FigTree 1.4.252.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Results
Genome organization
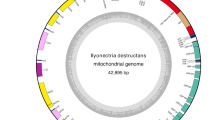
The mitochondrial genome of the fungal strain is a closed circular DNA molecule with a length of 35,079 bp. It contains 36 genes, including 12 PCGs, 2 rRNA genes, and 22 tRNA genes (Fig. 1). The 12 PCGs ranged in length from 270 bp (nad4l) to 2886 bp (nad4). The AT content of these 12 PCGs ranged from 67.3 to 78.2%, which was consistent with the previously reported genomes of mitotic Pleosporales species28,42. The AT skew of these 12 PCGs was − 0.167–0.167, and the GC skew was − 0.089–0.205. The 12 PCGs contained a subunit in the F0 region of the ATP synthase complex (atp6), three cytochrome oxidases (cox1, cox2, and cox3), seven subunits of the electron transport chain complex I (nad1, nad2, nad3, nad4, nad4l, nad5, and nad6), and a subunit (cob) of complex III. The strand where most genes are located is referred to as the J-strand (majority strand), and the strand where few genes are located is referred to as the N-strand (minority strand). It has five PCGs, which are encoded by the N-strand (cob, nad1, nad4, nad4l, and nad5), and the remaining seven PCGs are encoded by the J-strand (atp6, nad2, nad3, nad6, cox1, cox2, and cox3) (Table S2).

Circular map of the Acrocalymma vagum mitochondrial genome.
PCGs with codon usage and nucleotide composition
To understand the bias in codon usage of the mitchondrial genes in A. vagum, the codon usage in the PCGs was determined. A statistical analysis of codon usage in the PCGs region of A. vagum was conducted (Fig. 2A). Among the A. vagum, the most frequently used codon was UUA (for leucine; Leu) and the most frequently used amino acids were 58.5 leucine (Leu), 44.4 isoleucine (Ile), 36.2 serine (Ser), and 33.5 phenylalanine (Phe). In addition, CGC, GGC, CGG, CUC, and CGG were the five codons with the lowest usage rates (Fig. 2B).

Basic characteristics of codons in the Acrocalymma vagum mitochondrial genome. (A) Codon distribution in the Acrocalymma vagum mitogenome. Numbers on the y-axis refer to the total number of codons. Codon families are shown on the x-axis. (B) Codon usage in the mitochondrial genome in Acrocalymma vagum. Different colored boxes represent codon usage.
To understand the differences in mitochondrial genome composition between two species, the mitochondrial genomes of A. vagum and outgroup species (Z. cellare and A. vagum relatives from close to distant Shiraia bambusicola, Edenia gomezpompae, Phoma sp., and Bipolaris oryzae) were linearized and plotted (Fig. 3). The figure shows that both A. vagum and Phoma sp. lack the rsp3 gene. Therefore, we further performed the evolutionary analysis of rsp3.

The order of the species in this figure, from top to bottom, is determined by their relatedness to Acrocalymma vagum, ranging from closely related to distant. Additionally, positioned at the top are the outgroup species Z. cellare. Comparative analysis of the nucleotide sequences for each protein-coding gene and two ribosomal DNA genes among A. vagum, S. bambusicola, E. gomezpompae, Phoma sp., and B. oryzae. The same genes are marked with the same color, and the black color in a gene represents an intron in that gene.
Evolutionary rates of mitochondrial genes
To detect the nature of the evolutionary selection pressure in A. vagum, we selected 13 mitochondrial genes to calculate Ka, Ks, and Ka/Ks values. The results (Fig. 4) showed that the Ka/Ks ratio ranged from 0.06 to 1.02 for the mitochondrial genes, suggesting that these genes underwent negative selection pressure in the evolution process. The mean Ka/Ks ratio of these mitochondrial genes was 0.407. The mitochondrial genes with the highest Ka/Ks ratio were rps3 (1.02), nad6 (0.69), and nad4 (0.62), with rps3 undergoing positive selection. This may explain the phenomenon of the rps3 gene absence in A. vagum, while other genes underwent purifying selection during their evolutionary process.

The Ka (nonsynonymous) and Ks (synonymous) values, as well as their Ka/Ks ratios appear as red, green, and blue box plots, respectively. The bar represents the standard deviation, median, and the values between the first and third quartiles of Ka, Ks, and Ka/Ks.
tRNAs and rRNAs
Twenty-two tRNA genes were located in the A. vagum mitochondrial genome, Arg, Ser, and Leu were encoded by two tRNA genes with different anticodons; Val and Met were encoded by three tRNA genes with the same anticodon; and the tRNA arms of the same AA codon were different. These tRNAs have a typical highly conserved clover secondary structure (Fig. 5), and 55 G-U mismatches were found, most of which were distributed around rrnL. These tRNAs ranged in length from 71 to 85 bp, with a total length of 2003 bp, representing 5.71% of the mitochondrial genome length. The AT content of the tRNAs was 60.2%, displaying an AT skew of − 0.167–0.190 and a GC skew of − 0.22–0.375. The mitochondrial genome of A. vagum, as with other eukaryotes, had two rRNA genes (16S and 12S), namely the small subunit rRNA (rrnS) and large subunit rRNA (rrnL). The lengths of the rrnL and rrnS genes were 1792 and 3493 bp, respectively.

Predicted secondary structures of the 22 tRNAs of the Acrocalymma vagum mitochondrial genome.
Phylogenetic relationships
To obtain more evidence to conduct the classification and understand the evolutionary history of the mitochondrial genome, we used the identical and well-supported tree topology based on two datasets, PCG and AA, using BI and ML methods. The branching nodes of all trees showed a high degree of node support (bootstrap support BS > 90) and BI (posterior probabilities PP = 1.00). In the four phylogenetic trees of BI-AA, ML-AA, BI-PCG, and ML-PCG, A. vagum has the smallest evolutionary distance. The main difference between the AA and PCG analyses was that the species closest to A. vagum in the phylogenetic relationship were Ascochyta rabiei and S. bambusicola, respectively (Fig. 6, Figs. S1–S3). The BI-PCG and ML-PCG analyses align more closely with previous phylogenetic studies41,53.

Phylogenetic relationships of Pleosporales inferred by MrBayes 3.2.6 based on the nucleotides of the first and second codons of the 12 protein-coding genes (11,688 bp) and two rRNAs. Numbers on nodes are the percentage frequency with which a cluster appears in a bootstrap test of 1000 runs (≥ 95%). The number on the branch represents the length of the branch.
At the family level, the results of phylogenetic relationships indicate the well-verified monophyletic groups of Astrosphaeriellaceae, Orynesporascaceae, Coniothyriaceae, Didymellaceae, Morosphaeriaceae, Phaeosphaeriaceae, and Shiraiaceae, while the monophyletic group of Pleosporaceae is questioned. At the genus level, the monophylies of all genera, except for Bipolaris, were well verified in Pleosporaceae, and B. oryzae was distantly related to the other two species.
Our results (Fig. 6) show that A. vagum belongs to the family Morosphaeriaceae and is located at the base of the phylogenetic tree of Pleosporales, which is an ancient species under the Pleosporales. Morosphaeriaceae is most closely related to Shiraiaceae based on phylogenetic distance, exhibiting a greater genetic distance from species within Pleosporaceae. The results showed that the combined mitochondrial gene set was suitable as a reliable molecular marker for the analysis of the phylogenetic relationships among the Pleosporales species.
Discussion
The basic information regarding the mitochondrial genome reveals a significantly higher AT than GC content in A. vagum, which is attributed to the fact that the four most frequently used codons (UUA, AUA, UUU, and AAU) in the mitochondrial genome of A. vagum are all composed of A and T. This is the same reason for the high AT content in Pleosporales in general42,54. Mitochondrial genome length varied from 30,836 bp (Phoma sp.) to 13,600 bp (B. sorokiniana) in 18 Pleosporales. There is a large gap in the mitochondrial genome size among species, and this is related to the number of introns. As the number of introns increases, so does the length of the genome, which is supported by previous studies on fungi55,56. It was observed that the number of introns increased with the evolutionary distance. In the outer group species Zasmudium cellare and the species with the smallest evolutionary distance A. vagum, no introns were present. Among the three species closely related to A. vagum, namely S. bambusicola, E. gomezpompae, and Phoma sp., the number of introns was 1, 0, and 2, respectively. The phylogenetic results showed that the number of introns increased with the evolutionary distance of the species; e.g., species in the clade containing B. oryzae have higher numbers of introns than species more closely related to A. vagum. There are two generally accepted theories regarding the origin of introns. The first theory suggests that introns were abundant in the ancestral mitochondrial genome but were subsequently lost in most lineages57,58. The second theory supports intron mobility and expansion within genes due to events of horizontal transfer, even between distant phylogenetically related species59,60. The trend of the number of introns gradually increasing with evolutionary distance is consistent with the second conjecture. With the gradual evolution of the species, the horizontal and vertical transfer of genes in the mitochondrial genome leads to an increased number of introns.
The results of the phylogenetic relationships show that Pleosporaceaeas is a paraphyletic group. Among them, E. gomezpompae is genetically distant from other species of Pleosporaceae but closely related to Shiraiaceae. This result differs from the report by Cui et al., who proposed placing Edenia in Pleosporaceae based on the phylogenetic analysis of ITS rDNA61. However, our results are consistent with the phylogenetic tree of the mitochondrial genome reported by Huang et al.41. There are two reasons for this taxonomic difference: (1) There are limitations in constructing phylogenetic trees from ITS rDNA sequences alone, and species closer to Edenia may not have been included in the phylogenetic tree construction. (2) Morphological characteristics were not compared between species in the family Pleosporaceae. The phylogenetic tree constructed in this study included more species of the Pleosporales order. Therefore, we suggest that Edenia be merged into Shiraiaceae after being removed from Pleosporaceae. To further confirm the relationship between the genus Edenia and the family Shiraiaceae, more morphological features of other species of Edenia and Shiraiaceae need to be compared. We observed that B. cooke and B. sorokiniana in the genus Bipolaris are in the same clade. Another species in the genus Bipolaris, B. oryzae, was more closely related to the genus Stemphylium. Therefore, we question the monophyly of Bipolaris. However, establishing the relationship between B. oryzae and Stemphylium requires additional morphological and molecular data to prove this relationship.
Line plots of the mitochondrial genome show that although the PCGs and rRNAs were arranged in vastly different orders in the mitochondria, they still showed considerable homology. Our results support the widespread distribution of the nad2–nad3, nad4l–nad5, and cox1–cox2 pairs in Pleosporlaes, and tRNA was mostly distributed on both sides of the rrnl42. A. vagum and the other four strains showed significant differences in the mitochondrial gene arrangement. However, there were four genes in the same position, maintaining a certain degree of homology. This indicates that numerous rearrangement events occurred in A. vagum and its close relatives during its evolution. The mitochondrial gene arrangements of S. bambusicola and E. gomezpompae are highly similar. This provides further evidence that Edenia should be incorporated into the family Shiraiaceae.
Conclusion
In this study, we sequenced and annotated the complete genome of A. vagum, an endophytic fungus of the highly valued Chinese herbal medicine Paris polyphylla var. yunnanensis. Then, the mitochondrial genome sequence was uploaded to NCBI (GenBank accession number: OQ509476). The mitochondrial characteristics (gene content, size, and order, base composition, PCG codon usage, and tRNA secondary structure) were placed into the order Pleosporlaes for their comparison. It was proven that A. vagum belongs to the family Massarineae and is an ancient species of the order Pleosporales. This study is the first to report the complete mitochondrial genome obtained from the family Massarineae. In the mitochondrial genome signature, it was found that the mitochondrial introns gradually increased with species evolution. Mitochondria within Massarineae and closely related species have a lower intron count than distant relatives, as observed in the species Bipolaris. In addition, we performed a phylogenetic analysis of Pleosporlaes, which was used to determine the phylogenetic relationships between the species in Pleosporlaes. Based on the phylogenetic tree, Edenia should be placed in the family Shiraiaceae, and Morosphaeriaceae and Shiraiaceae are sister groups with high homology ratings. This study not only improved our phylogenetic understanding of Pleosporlaes but also provided a new method to identify the distribution of endophytic fungi in Chinese herbal medicine.
Relevant legislations, permitting and consent
The collection of plant material must comply with relevant institutional, national, and international guidelines and legislation as well as IUCN Policy Statement on Research Involving Species at Risk of Extinction and Convention on the Trade in Endangered Species of Wild Fauna and Flora.
Data availability
The data that support the findings of this study will be available in GenBank at https://www.ncbi.nlm.nih.gov/, with accession number OQ509476.
References
Crous, P. W. et al. Fungal Planet description sheets: 214–280. Persoonia 32, 184–306 (2014).
Zhang, Y. et al. Multi-locus phylogeny of Pleosporales: A taxonomic, ecological and evolutionary re-evaluation. Stud. Mycol. 64, 85–105 (2009).
Schoch, C. L. et al. A class-wide phylogenetic assessment of Dothideomycetes. Stud. Mycol. 64, 1–15 (2009).
Zhang, Y., Crous, P. W., Schoch, C. L. & Hyde, K. D. Pleosporales. Fungal Divers 53, 1–221 (2012).
Xu, J. Fungal DNA barcoding. Genome 59, 913–932 (2016).
Mugambi, G. K. & Huhndorf, S. M. Molecular phylogenetics of Pleosporales: Melanommataceae and Lophiostomataceae re-circumscribed (Pleosporomycetidae, Dothideomycetes, Ascomycota). Stud. Mycol. 64, 103–121 (2009).
Guo, L. D. et al. Molecular identification of white morphotype strains of endophytic fungi from Pinus tabulaeformis. Mycol. Res. 107, 680–688 (2003).
Teimoori-Boghsani, Y. et al. Endophytic fungi of native salvia abrotanoides plants reveal high taxonomic diversity and unique profiles of secondary metabolites. Front. Microbiol. 10, 3013 (2019).
Ameen, F., Stephenson, S. L., AlNadhari, S. & Yassin, M. A. Isolation, identification and bioactivity analysis of an endophytic fungus isolated from Aloe vera collected from Asir desert, Saudi Arabia. Bioprocess Biosyst. Eng. 44, 1063–1070 (2021).
Xiao, J. L. et al. Isolation and screening of stress-resistant endophytic fungus strains from wild and cultivated soybeans in cold region of China. Appl. Microbiol. Biotechnol. 105, 755–768 (2021).
Manamgoda, D. S. et al. The genus Bipolaris. Stud. Mycol. 79, 221–288 (2014).
Tanaka, K. et al. Revision of the Massarineae (Pleosporales, Dothideomycetes). Stud. Mycol. 82, 75–136 (2015).
Ahmed, S. A. et al. Revision of agents of black-grain eumycetoma in the order Pleosporales. Persoonia 33, 141–154 (2014).
Wanasinghe, D. N. et al. Phylogenetic revision of Camarosporium (Pleosporineae, Dothideomycetes) and allied genera. Stud. Mycol. 87, 207–256 (2017).
Trakunyingcharoen, T. et al. Mycoparasitic species of Sphaerellopsis, and allied lichenicolous and other genera. IMA Fungus 5, 391–414 (2014).
Yildiz, G. & Ozkilinc, H. First characterization of the complete mitochondrial genome of fungal plant-pathogen Monilinia laxa which represents the mobile intron rich structure. Sci. Rep. 10, 13644 (2020).
Jelen, V., de Jonge, R., Van de Peer, Y., Javornik, B. & Jakše, J. Complete mitochondrial genome of the Verticillium-wilt causing plant pathogen Verticillium nonalfalfae. PLoS One 11, e0148525 (2016).
Kortsinoglou, A. M., Saud, Z., Eastwood, D. C., Butt, T. M. & Kouvelis, V. N. The mitochondrial genome contribution to the phylogeny and identification of Metarhizium species and strains. Fungal Biol. 124, 845–853 (2020).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res, 25, 3389–3402 (1997).
Bernt, M. et al. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 69, 313–319 (2013).
Laslett, D. & Canbäck, B. ARWEN: A program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175 (2008).
Schattner, P., Brooks, A. N. & Lowe, T. M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686-689 (2005).
Kryazhimskiy, S. & Plotkin, J. B. The population genetics of dN/dS. PLoS Genet. 4, e1000304 (2008).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Perna, N. T. & Kocher, T. D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 41, 353–358 (1995).
Zaccaron, A. Z. & Bluhm, B. H. The genome sequence of Bipolaris cookei reveals mechanisms of pathogenesis underlying target leaf spot of sorghum. Sci. Rep. 7, 17217 (2017).
Song, N., Geng, Y. & Li, X. The mitochondrial genome of the phytopathogenic fungus Bipolaris sorokiniana and the utility of mitochondrial genome to infer phylogeny of Dothideomycetes. Front. Microbiol. 11, 863 (2020).
Ma, Q. et al. Comparative mitochondrial genome analyses reveal conserved gene arrangement but massive expansion/contraction in two closely related Exserohilum pathogens. Comput. Struct. Biotechnol. J. 20, 1456–1469 (2022).
Liao, M., Chen, C. & Li, Q. The complete mitochondrial genome of Alternaria alternata (Hypocreales: Nectriaceae). Mitochondrial. DNA B Resour. 2, 587–588 (2017).
Franco, M. E. E. et al. The mitochondrial genome of the plant-pathogenic fungus Stemphylium lycopersici uncovers a dynamic structure due to repetitive and mobile elements. PLoS One 12, e0185545 (2017).
Yuan, X. L. et al. Characterization of nuclear and mitochondrial genomes of two tobacco endophytic fungi Leptosphaerulina chartarum and Curvularia trifolii and their contributions to phylogenetic implications in the Pleosporales. Int J Mol Sci 21, 2641 (2020).
Deng, G. et al. The complete mitochondrial genome of Cochliobolus miyabeanus (Dothideomycetes, Pleosporaceae) causing brown spot disease of rice. Mitochondrial. DNA B Resour. 4, 2832–2833 (2019).
Chen, C. et al. Characterization of the complete mitochondrial genome of Corynespora cassiicola (Pleosporales: Dothideomycetes), with its phylogenetic analysis. Mitochondrial. DNA B Resour. 4, 2938–2939 (2019).
Shah, R. M. et al. Reference Genome Assembly for Australian Ascochyta rabiei Isolate ArME14. G3 10, 2131–2140 (2020).
Panteleev, S., Mozharovskaya, L., Kiryanov, P., Kagan, D. & Baranov, O. Y. Structural and functional organisation of the phytopathogenic fungi Phoma sp. 1 mitochondrial genome. Proc. Natl. Acad. Sci. Belarus Biol. Ser. 67, 359–357 (2022).
Hane, J. K. et al. Dothideomycete plant interactions illuminated by genome sequencing and EST analysis of the wheat pathogen Stagonospora nodorum. Plant Cell 19, 3347–3368 (2007).
Stone, C. L. et al. Annotation and analysis of the mitochondrial genome of Coniothyrium glycines, causal agent of red leaf blotch of soybean, reveals an abundance of homing endonucleases. PLoS One 13, e0207062 (2018).
Wu, D., Zhou, L., Xue, J., Xia, Q. & Meng, L. Characterization of two new Apodemus mitogenomes (Rodentia: Muridae) and mitochondrial phylogeny of Muridae. Diversity 14, 1089 (2022).
Huang, L., Mu, Y., Zhang, X., Chang, K. & Zhang, J. The mitochondrial genome of the endophyte Edenia gomezpompae CRI Eg3 isolated from sweet potato. Mitochondrial. DNA B Resour. 7, 454–455 (2022).
Shen, X. Y. et al. Characterization and phylogenetic analysis of the mitochondrial genome of Shiraia bambusicola reveals special features in the order of pleosporales. PLoS One 10, e0116466 (2015).
Torriani, S. F., Brunner, P. C. & McDonald, B. A. Evolutionary history of the mitochondrial genome in Mycosphaerella populations infecting bread wheat, durum wheat and wild grasses. Mol. Phylogenet. Evol. 58, 192–197 (2011).
Arcila-Galvis, J. E., Arango, R. E., Torres-Bonilla, J. M. & Arias, T. The mitochondrial genome of a plant fungal pathogen Pseudocercospora fijiensis (Mycosphaerellaceae), comparative analysis and diversification times of the sigatoka disease complex using fossil calibrated phylogenies. Life 11, 215 (2021).
Goodwin, S. B. et al. The mitochondrial genome of the ethanol-metabolizing, wine cellar mold Zasmidium cellare is the smallest for a filamentous ascomycete. Fungal Biol. 120, 961–974 (2016).
Abascal, F., Zardoya, R. & Telford, M. J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, W7-13 (2010).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166 (2019).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32, 268–274 (2015).
Du, Y., Zhang, C., Dietrich, C. H., Zhang, Y. & Dai, W. Characterization of the complete mitochondrial genomes of Maiestas dorsalis and Japananus hyalinus (Hemiptera: Cicadellidae) and comparison with other Membracoidea. Sci. Rep. 7, 14197 (2017).
Nobile, P. M. et al. Identification, classification and transcriptional profiles of dirigent domain-containing proteins in sugarcane. Mol. Genet. Genom. 292, 1323–1340 (2017).
Tao, L. et al. Plant growth-promoting activities of bacterial endophytes isolated from the medicinal plant Pairs polyphylla var. yunnanensis. World J. Microbiol. Biotechnol. 38, 15 (2021).
Oh, J., Kong, W. S. & Sung, G. H. Complete mitochondrial genome of the entomopathogenic fungus Beauveria pseudobassiana (Ascomycota, Cordycipitaceae). Mitochondrial DNA 26, 777–778 (2015).
Torriani, S. F. et al. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet. Biol. 62, 34–42 (2014).
Li, Q. et al. The complete mitochondrial genomes of two model ectomycorrhizal fungi (Laccaria): Features, intron dynamics and phylogenetic implications. Int. J. Biol. Macromol. 145, 974–984 (2020).
Goddard, M. R. & Burt, A. Recurrent invasion and extinction of a selfish gene. Proc. Natl. Acad. Sci. U. S. A. 96, 13880–13885 (1999).
Gonzalez, P., Barroso, G. & Labarère, J. Molecular gene organisation and secondary structure of the mitochondrial large subunit ribosomal RNA from the cultivated Basidiomycota Agrocybe aegerita: A 13 kb gene possessing six unusual nucleotide extensions and eight introns. Nucleic Acids Res. 27, 1754–1761 (1999).
Vaughn, J. C., Mason, M. T., Sper-Whitis, G. L., Kuhlman, P. & Palmer, J. D. Fungal origin by horizontal transfer of a plant mitochondrial group I intron in the chimeric CoxI gene of Peperomia. J. Mol. Evol. 41, 563–572 (1995).
Gonzalez, P., Barroso, G. & Labarère, J. Molecular analysis of the split cox1 gene from the Basidiomycota Agrocybe aegerita: Relationship of its introns with homologous Ascomycota introns and divergence levels from common ancestral copies. Gene 220, 45–53 (1998).
Cui, Y. et al. Characterization of Edenia gomezpompae isolated from a patient with keratitis. Mycopathologia 176, 75–81 (2013).
Funding
This work was supported by grants from the National Natural Science Foundation of China (32160668), the Science and Technology Program of Guizhou Province (2017)2833, the Academic New Seedling Cultivation and Innovation Exploration Project [Guizhou Science and Technology Platform Talents Fund (2018)5779-53, the Science and Technology Foundation of Health Commission of Guizhou Province (gzwkj2021-510), the National Natural Science Foundation of Guizhou Medical University (20NSP055), the Science and Technology Program of Guizhou Province (Qian Ke He Foundation-ZK [2022] General 364), and the project of State Key Laboratory of Functions and Applications of Medicinal Plants, Guizhou Medical University (FAMP2021Z1-1), special funds from the central finance to support the development of local universities (Qian Jiao Ji No [2023]036).
Author information
Authors and Affiliations
Contributions
G.W. contributed to the design and implementation of the study as well as the preparation and drafting of the manuscript. G.Z., X.L., Y.W., and Y.L. coordinated and participated in the data analysis and contributed to the discussion of the manuscript. X.W. and H.L. contributed to the design and implementation of the research work and revised the manuscript. All authors analyzed, discussed the data, reviewed, and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, G., Zhang, G., lv, X. et al. First complete mitogenome of Massarineae and its contribution to phylogenetic implications in Pleosporales. Sci Rep 13, 22431 (2023). https://doi.org/10.1038/s41598-023-49822-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-49822-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
