
Abstract
Reduced insulin-like growth factor 2 (IGF2) levels in Alzheimer’s disease (AD) may be the mechanism relating age-related metabolic disorders to dementia. Since Igf2 is an imprinted gene, we examined age and sex differences in the relationship between amyloid-beta 1–42 (Aβ42) accumulation and epigenetic regulation of the Igf2/H19 gene cluster in cerebrum, liver, and plasma of young and old male and female 5xFAD mice, in frontal cortex of male and female AD and non-AD patients, and in HEK293 cell cultures. We show IGF2 levels, Igf2 expression, histone acetylation, and H19 ICR methylation are lower in females than males. However, elevated Aβ42 levels are associated with Aβ42 binding to Igf2 DMR2, increased DNA and histone methylation, and a reduction in Igf2 expression and IGF2 levels in 5xFAD mice and AD patients, independent of H19 ICR methylation. Cell culture results confirmed the binding of Aβ42 to Igf2 DMR2 increased DNA and histone methylation, and reduced Igf2 expression. These results indicate an age- and sex-related causal relationship among Aβ42 levels, epigenomic state, and Igf2 expression in AD and provide a potential mechanism for Igf2 regulation in normal and pathological conditions, suggesting IGF2 levels may be a useful diagnostic biomarker for Aβ42 targeted AD therapies.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is the leading cause of dementia, in which synaptic loss and cerebral atrophy are associated with progressive cognitive decline and behavioural deficits1. In AD, dysregulation of amyloid-beta (Aβ) production and clearance leads to the appearance of amyloid plaques that primarily consist of Aβ peptides 1–40 (Aβ40) and 1–42 (Aβ42). Deposition of Aβ42 in the brain is associated with increases in oxidative stress, neuroinflammation, tau hyperphosphorylation, and synaptic dysfunction, as functional biomarkers that correlate with AD onset and progression2,3,4. Aβ aggregates, especially Aβ42, can translocate to neuronal nuclei and interact with regulatory elements of candidate genes involved in Aβ accumulation, such as amyloid precursor protein (APP), β-secretase, and apolipoprotein E (ApoE)5,6,7,8. We have reported that Aβ42 association with the thioredoxin interacting protein (Txnip) gene promoter region results in enhanced Txnip expression and TXNIP levels, which inhibits thioredoxin (TRX), and increases oxidative stress signalling that promotes DNA damage and cell death9, further demonstrating that intranuclear Aβ42 species can regulate gene expression.
Many metabolic disorders (e.g., diabetes, hypertension, obesity, and dyslipidemia) are risk factors for AD, linking abnormal liver function with cognitive deficits10,11. For example, in diabetic patients, impaired growth factor signaling, energy metabolism, inflammation, and insulin resistance promote hepatic cell death and liver damage10. Blood plasma can carry circulating DNA fragments (cell-free DNA, cfDNA) originating from cell death and degradation12. The cfDNA population in the bloodstream is heterogeneous and differs between individuals in normal ageing and pathological conditions such as AD13, suggesting that the biological properties of cfDNA fragments (including DNA and histone modification) may be a useful diagnostic biomarker for Aβ42 pathologies and AD progression.
Mouse models have been developed to study the mechanistic role of Aβ in AD neuropathology14 and behavioural deficits15,16. The five times familial mouse model of AD (5xFAD) shows one of the most aggressive Aβ pathologies due to the five mutations it carries on the APP [V717I (London), I716V (Florida) and K670N/M671L (Swedish)] and presenilin 1 (M146L and L286V) genes17. The 5xFAD mice show cerebral Aβ42 accumulation as early as 2-months of age17, metabolic deficits and reduced body weight as early as 6-months of age18, followed by synaptic and neuronal loss as well as a decline in cognitive and motor performance by 9-months of age19,20,21,22.
Insulin-like growth factor 2 (IGF2) is one of the most abundant growth factors in the central nervous system (CNS)23 and is involved in the regulation of metabolism, tissue growth, and endocrine function24,25 in fetal growth and adulthood26,27. In patients with AD, reduced IGF2 levels have been reported in the hippocampus, cerebrospinal fluid, and blood plasma28. In mouse models of AD with APP mutations, increased IGF2 availability is associated with increased acetylcholine release and adult neurogenesis, and reduced amyloidosis and synaptic deficits29,30. Enhanced IGF2 availability also protects against hypoglycaemic damage in cultured hippocampal neurons31, enhances hippocampus-dependent memory consolidation32,33 and ameliorates age-related memory loss in rats34. Together, these findings suggest that IGF2 may be a useful biomarker for the metabolic deficits, pathological weight loss and cognitive decline of AD.
IGF2 protein is encoded by Igf2, which was one of the earliest imprinted genes to be identified35,36,37. Genomic imprinting is the epigenetic silencing of an allele through DNA methylation, independent of the sex of the offspring38,39, resulting in the expression of a single allele from one parent, causing post-Mendelian parent-of-origin traits40. To date, 260 imprinted genes have been identified in mice and about 230 of these are conserved in humans41. The murine H19/Igf2 gene locus is located on chromosome 7; H19 is expressed from the maternal allele, and Igf2 is expressed from the paternal allele42,43. H19/Igf2 loci regulation (see Fig. 1a) is controlled by the DNA methylation status at well-characterized differentially methylated regions (DMRs), namely the H19 imprinting control region (ICR), which is located upstream of the H19 promoter (position: chr7: 142653816-142655810; see Fig. 1a). The unmethylated H19 ICR on the maternally expressed mouse Igf2 functions as a transcriptional insulator; CTCF binding to the H19 ICR inhibits distal enhancers from binding to the Igf2 promoter, resulting in Igf2 silencing and expression of H19 from the maternal allele. On the other hand, methylation of the H19 ICR on the paternally expressed mouse Igf2 gene inhibits CTCF binding, resulting in H19 silencing and expression of Igf2 from the paternal allele43,44,45,46,47.
Three DMRs have been identified in the mouse Igf2 gene region. The maternally methylated DMR0 is in exon U1, whereas the paternally methylated DMR1 is positioned upstream of promoter 1, and DMR2 is in exon 648,49,50,51 (Fig. 1a). In humans, the H19/Igf2 locus is located on chromosome 11; and the regions homologous to mouse DMR0 and DMR2 are differentially methylated, whereas the region homologous to mouse DMR1 is unmethylated52. Igf2 DMR1 and DMR2 knockout mouse studies revealed that DMR1 functions as a silencer53 and DMR2 as an activator54 of Igf2 expression. Bi-allelic Igf2 expression and hypermethylation at the Igf2 DMR2 have been shown in patients with Beckwith–Wiedemann syndrome52, while loss of imprinting of Igf2 DMRs has been shown in patients with Wilms tumour and colorectal cancer55,56. Consequently, loss of Igf2 DMR imprinting (by DNA mutation or epimutation) and abnormalities in IGF2 function indicate the critical role of IGF2 in development and disease57. These findings suggest that Aβ42 and IGF2 potentially link peripheral biomarkers of liver functioning to central biomarkers related to AD progression, including Aβ aggregation, neural atrophy, and cognitive dysfunction.
Although genomic imprinting has been investigated in various brain regions of mouse models of AD58,59, it is unclear whether IGF2 expression is differently affected in symptomatic younger versus older transgenic AD mice. Moreover, it remains unclear how changes in Igf2 expression are influenced by the accumulation and intranuclear role of Aβ42 as a transcription factor. We therefore compared Aβ42 and IGF2 levels in cerebrum, liver, and blood plasma from 6- and 12-month-old, male and female 5xFAD mice with age- and sex-matched B6SJLF1/J wild-type (WT) mice. The 5xFAD mice show initial weight loss and cognitive deficits at 6-months of age18,60, with severe deficits and increased mortality by 12-months of age, especially in males61, providing a useful model to study age-related expression patterns of Igf2 and IGF2 in central, peripheral, and circulatory systems in response to Aβ accumulation. To determine whether the findings from the mouse model translated to human disease, we compared Igf2 epigenetic regulation in frontal cortex from aged humans diagnosed with AD and non-AD controls. To examine whether Aβ42 association with the Igf2 promoter region can directly mediate Igf2 transcription, we treated the human-derived cell line, HEK293, with Aβ42 oligomers and harvested the cells at various time points to evaluate temporal changes in Igf2 epigenetic status and expression.
Materials and methods
Mouse housing and breeding
All experimental procedures were performed in accordance with the guidelines of the Canadian Council on Animal Care and were approved by the Dalhousie University Committee on Laboratory Animals. Fifteen 5xFAD mice and 15 BSJLF1/J WT mice (6 males and 9 females of each genotype) were used in this study. All mice were bred in-house from pairs of male 5xFAD (B6SJLT-Tg (APPSwFlLon, PSEN1*M146L*L286V) 6799Vas/Mmjax; stock #034840) and female WT (C57BL/6JxSJL/J F1 mice; stock #100012) mice, originally purchased from the Jackson Laboratory (Bar Harbor, Maine). Pups were weaned at 21-days of age and housed in same sex, mixed genotype groups of 2–4 in transparent polyethylene cages (35 × 12 × 12 cm) with ad libitum food (Purina Rodent Chow, #5001) and tap water. Housing cages contained pine chip bedding and a polyvinyl chloride tube (5 cm diameter, 8 cm long) for enrichment and mice were provided with clean cages once a week. Mice were housed in a climate-controlled (20 ± 2 °C) vivarium under a 12:12 h reversed light/dark cycle with lights off between 09:30 and 21:30. Each mouse was individually marked with ear punches and genotyped for the APP and PS1 transgenes using polymerase chain reaction (PCR) and the tissue samples from ear-punches. The research described here was conducted in compliance with the ARRIVE 2.0 Guidelines for Reporting Animal Research62,63.
Tissue collection
At 5-weeks, 6-months, and 12-months of age mice were euthanized with sodium phenobarbital (200 mg/kg). After checking the toe-pinch reflex for signs of pain perception, blood was collected via cardiac puncture using 1 mL syringes washed with 10% ethylenediaminetetraacetic acid (EDTA). The blood was centrifuged at 1000g for 10 min at 4 °C and the plasma was collected. Following the cardiac puncture, mice were perfused with phosphate-buffered saline (PBS, 10%) solution for 2 min and livers and brains were harvested. Tissues and blood plasma were collected in 1.5-mL microcentrifuge tubes, frozen with dry ice, and stored at − 80 °C.
Post-mortem human brain samples
All methods were carried out in accordance with the Tri-Council Policy Statement on Ethical Conduct for Research Involving Humans (Government of Canada). Informed, written consent forms were obtained for all subjects. Ethical approval was obtained from the Health Sciences Research Ethics Board (Halifax, Nova Scotia, Canada). Human brain samples and corresponding clinical and neuropathological diagnoses were provided by the Maritime Brain Tissue Bank (Halifax, Nova Scotia, Canada). A total of 12 frontal-cortex (orbitofrontal cortex, Brodmann’s area 11) samples from 6 AD and 6 non-AD patients (3 of each sex) were used in this study. The mean age of donors at the time of death was 76 years, which did not differ between the disease conditions or sexes (FDisease1, 8 = 2.32, p = 0.163; FSex1, 8 = 0.41, p = 0.537). The clinical and neuropathological characteristics summarized in Table 1 show that cases 1–6 had no neuropathological hallmarks of AD sufficient for a clinical diagnosis, while cases 7–12 had progressive dementing illness and fulfilled the neuropathological criteria for AD64. During autopsy, the brains were removed and bisected through the midline. Half of the brain was fixed in formalin and used for neuropathologic diagnosis. The remaining (non-fixed) half was cut in 1–2 cm slabs, vacuum-sealed, frozen on dry ice and stored at − 80 °C until used in this study.
Cell culture, Aβ42 peptide preparation, and treatment
Human embryonic kidney (HEK) 293 cells were grown as adherent cultures in Dulbecco’s Modified Eagle Medium (DMEM; Corning, VWR International Co., cat# 17-207-CV) supplemented with fetal bovine serum (FBS, 10%; Corning, VWR International Co., cat# 35-077-CV), 1 mM Sodium Pyruvate, 4 mM L-Glutamine, 25 mM d-Glucose, 100 units/mL penicillin, and 100 mg/mL streptomycin (VWR International Co., cat# K952-100ML) and maintained in a humidified incubator (5% CO2, 37 °C). Cultured cells were then counted using a haemocytometer and cell passaging was performed by washing residual media from cells using PBS (–Ca/–Mg; Gibco, Thermo Fisher Scientific Inc., cat# 14190144) and dissociating cells from the plate using Trypsin–EDTA (0.05%; Gibco, Thermo Fisher Scientific Inc., cat# 15400054). In each experiment, cultured cells were seeded into 6-well plates with 1.0 × 106 cells per well. Human Aβ42 ‘click peptide’ (GenScript Biotech Inc., cat# RP10017-1) was dissolved in deionized H2O to make a 500 μM stock solution, which was further diluted to 5 μM with Dulbecco's PBS and incubated on a shaker (1.5 h, 300 rpm, 21 °C), as previously described65. Upon reaching 70% confluency, the HEK293 cultures received a single treatment of Aβ42 oligomer solution (500 nM) or saline vehicle control. The medium was replaced every 3 days and the cultures were harvested after 3-, 6-, and 9-days of incubation, the cell lysates were then collected for in vitro analysis. Treatments were repeated a minimum of three times using different cultures of HEK293 cells.
Enzyme-linked immunosorbent assay (ELISA)
Protein isolation and ELISAs were performed as previously described9,66. ELISA was used to detect mouse IGF2 [detection range (DR), 15.6–1000 pg/mL; Cloud-Clone Corp; cat#: SEA051Mu], mouse and human reactive Aβ42 [DR, 7.4 to 250 pg/mL; BioLegend, San Diego, CA; cat#: 842401], human IGF2 [DR, 0.625–40 ng/mL; Cloud-Clone Corp; Cat#: SEA051HU, and human Aβ40 and Aβ42 [DR, 15.63 to 1000 pg/mL; Novus Biologicals Canada; cat# NBP2-69909 and NBP2-69913, respectively] according to the manufacturer’s protocols. The Novus Biologicals Canada's ELISA for beta-amyloid detection is specific for either human Aβ40 or human Aβ42 with minimal or no cross reactivity with other isoforms. Samples were loaded in triplicate alongside an eight-point standard curve in duplicate (with controls) in the 96-well microtiter plates supplied by each manufacturer. Detection was performed with horseradish peroxidase-labelled, Fc-specific IgG and read in the microplate reader at a wavelength of 620 nm (BioLegend mouse and human reactive Aβ42 kit) or 450 nm. The blank corrected levels of each target protein are reported in pg/mL.
Real-time quantitative PCR (RT-qPCR)
The RNeasy Mini (QIAGEN Group, cat# 74004) and iScript cDNA Synthesis (Bio-Rad Laboratories Inc., cat# 1708891) kits were used to purify RNA and generate cDNA. Amplification reactions, containing SsoFast EvaGreen Supermix (Bio-Rad Laboratories Inc., cat# 1725201) with mouse or human Igf2 and rpl13a primers (listed in Table 2), were run on the CFX Connect Real-Time PCR System (Bio-Rad Laboratories Inc.) as previously reported9. The 2−∆∆CT method was used to analyze the relative changes in gene expression67.
Restriction enzyme-based DNA methylation analysis (GlucMS-qPCR)
The DNA methylation (5mC) and DNA hydroxymethylation (5hmC) levels in the mouse H19 ICR were measured by restriction enzyme-based assay (EpiMark kit; New England Biolabs Inc., cat# E3317S) using the differential susceptibility of methylated and hydroxy-methylated DNA to cleavage by HpaII and MspI, as previously described9. Genomic DNA was extracted from cerebrum and liver tissues using the DNeasy Mini Kit (QIAGEN Group, cat# 69504), while circulating cfDNA was prepared from blood plasma using the QIAamp MinElute cfDNA Kit (QIAGEN Group, cat# 55284), according to the manufacturer’s protocols. The isolated DNA was treated with or without T4 Phage β-glucosyltransferase (T4-BGT; New England Biolabs Inc., cat#M0357S) to glucosylate 5hmC residues, rendering existing MspI sites non-cleavable. HpaII cleavage is prevented by 5mC and 5hmC. Glucosylated genomic DNA (100 ng) was digested with 10 U of either HpaII, MspI or no enzyme (8 h, 37 °C), followed by inactivation (20 min, at 80 °C). The HpaII- and MspI-resistant fraction was quantified by qPCR using mouse or human primers (listed in Table 2) designed around six HpaII/MspI sites (Fig. S1a and S2a), normalizing to the mock digestion control and two regions lacking HpaII/MspI sites. Resistance to MspI directly translates into percentage of 5hmC. 5mC levels were obtained by subtracting the 5hmC contribution from the total HpaII resistance.
Chromatin immunoprecipitation assays (ChIP-qPCR)
ChIP-qPCR assays were performed as previously described9,68. Tissues were fixed with formaldehyde (1%) and the resulting crosslinked protein–DNA complexes were sonicated using the Q800R2 Sonicator system (Qsonica LLC) into 150–250 bp length fragments confirmed using the QIAxcel Advanced System (QIAGEN Group). Immunoprecipitation was then performed using rabbit polyclonal IgG antibodies against 5mC (AnaSpec Inc., cat# BI-MECY-0500), Aβ42 (MilliporeSigma Co., cat# AB5078P), CTCF (MilliporeSigma Co., cat# 07-729), H3K9Ac (Diagenode, cat# C15410004), H3K9me3 (Diagenode, cat# C15410193), as well as the control normal rabbit IgG (Santa Cruz Biotechnology Inc, cat# sc-2027). Post immunoprecipitation, the precipitated DNA–protein complexes were dissociated from the protein A conjugated Dynabeads (Invitrogen, Thermo Fisher Scientific Inc., cat# 10001D) and isolated using a Purelink PCR purification kit (Invitrogen, Thermo Fisher Inc. Scientific, cat# K310001) along with their respective total input controls (pre-immunoprecipitation DNA). RT-qPCR analyses were then performed using mouse and human primers (listed in Table 2) targeting the CTCF and AβID RE sites spanning the H19 ICR (Fig. S1a and S2a) and Igf2 DMR2 (Figs. 3a and 5a). Results are expressed as the fold change of the enrichment of the DNA detected under the treatment conditions against the DNA detected under the no treatment conditions. This was determined by dividing the signals obtained from the ChIP by the signals obtained from the total input control sample and normalizing for the DNA detected by the non-immune IgG (negative control). For sequential ChIP–reChIP experiments, the protein bound to the beads with the first antibody was incubated (30 min, 37 °C) twice with DTT (20 mM) and the combined elutes were suspended in ChIP dilution buffer, which was then immunoprecipitated (14 h, 4 °C) with the second antibody.
Experimental design and statistical analyses
Each mouse was ear-punched and given a unique number. During the experiments, the investigators (E.F., G.M.H., I.C.G.W.) were blinded to sample genotype, sex, age, diagnosis, and treatment. The R Project Statistical Computing version 4.0.0 (2020-04-24)—"Arbor Day" was used for all statistical analyses, the graphs were generated in GraphPad Prism 7.0a for Mac OS X, and the cartoon figures were created with BioRender.com. Data from the mouse experiments were analysed within each tissue type (cerebrum, liver, plasma) between the genotypes (WT, 5xFAD), sexes (male, female) and ages (6-, 12-months). Human brain data were analysed between the disease status (non-AD, AD) and sexes (male, female). Data from the cell culture studies were analysed between the treatment conditions (vehicle treatment, Aβ42 treatment) and time points (3-, 6-, 9-days). All the analyses mentioned above were done using ANOVAs with Type 2 sums of squares and effect sizes were reported as partial eta squared (ηp2). Post-hoc group differences were determined by 95% confidence intervals (CI)69,70. Data for Aβ42 association with DNA were analyzed with Welch Two Sample t-tests and effect sizes were reported as Cohen’s d (d). Sample sizes were confirmed to be appropriate to achieve a type 2 error rate smaller than 0.05 for the majority of the measures by using the G*Power software71.
Results
Introductory overview
Because changes in IGF2 levels in the CNS and peripheral tissues have been associated with age-related cognitive decline and AD pathology in humans and mouse models72, we first measured IGF2 levels, Igf2 expression, H19 ICR methylation, CTCF binding, and histone modification in the cerebrum, liver, and blood plasma of 6- and 12-month-old, male and female 5xFAD and WT mice. The 5xFAD mice carry five mutations on two transgenes, which collectively increases Aβ production and the Aβ42 to Aβ40 ratio in the brain17. We therefore measured Aβ42 levels in the same three tissues to examine the age-related pattern of Aβ42 accumulation between 6- and 12-months of age in 5xFAD and WT mice. The next question concerned the temporal mechanism that potentially mediates changes in Igf2 expression in the 5xFAD mice independent of H19 ICR DNA methylation. In 5xFAD mice, the earliest timepoint Aβ42 accumulation has been reported in the brain is between 1.5- and 2-month of age17,73. We therefore used 5-week-old 5xFAD and WT mice to characterize Igf2 epigenetic regulation and IGF2 levels prior to Aβ40 and Aβ42 accumulation. The results from the 5xFAD mice raised the question of whether changes in Igf2 expression in AD patients are associated with changes in levels of Aβ42 binding to AβID regions on the Igf2 promoter. We therefore characterized Aβ40 and Aβ42 accumulation, Igf2/H19 epigenetic regulation, and IGF2 levels in the frontal cortex from aged individuals diagnosed with AD and non-AD patients. To further characterise the temporal epigenetic marks associated with Aβ42 binding to AβID regions on the Igf2 promoter, we treated HEK293 cell cultures with a single dose of either Aβ42 oligomers or saline vehicle and then harvested the cultures at various time points for analysis of Aβ42 levels, Igf2 epigenetic regulation, and IGF2 levels. The full results of statistical tests (including DoF, F- and p-values and CI) are given in the supplemental material.
Age-related changes in IGF2 levels, Igf2 expression and H19 ICR methylation in 5xFAD and WT mice
Based on the results of ELISA assays, the levels of IGF2 in the cerebrum were significantly lower in 5xFAD than WT mice (Fig. 1b), significantly lower in females than males, and lower in 12-month than 6-month-old mice. While this age effect was significant for 5xFAD mice it was not significant for WT mice. In the liver, there was a genotype by sex by age interaction for IGF2 levels (Fig. 1c); all mice showed a decrease in IGF2 levels at 12-months of age except WT males which had significantly higher levels of IGF2 than all other 12-month-old mice. Circulating IGF2 levels in the blood plasma were significantly lower in 5xFAD than WT mice (Fig. 1d), significantly lower in 12-month than 6-month-old mice, and significantly lower in females than males. Together, these results show that genotype-, sex-, and age-related differences in IGF2 levels occur in the liver and blood plasma as well as in the cerebrum of 5xFAD and WT mice, indicating that IGF2 levels in peripheral tissues and the circulatory system are concomitant with those in the brain.

IGF2 levels, Igf2 expression, and H19 ICR methylation in the cerebrum, liver, and plasma from 6- and 12-month-old male and female WT and 5xFAD mice. (a) Schematic representation of the Igf2/H19 gene cluster (see text for details). Parental-specific DNA methylation of the H19 ICR defines the imprinted status of both H19 and Igf2. Igf2 expression on the paternal allele (upper row) is associated with H19 ICR DNA methylation, H19 silencing, and an unmethylated Igf2 DMR2. Conversely, Igf2 silencing on the maternal allele (lower row) is associated with an unmethylated H19 ICR, transcription factor (CTCF) binding, H19 expression, and Igf2 DMR2 DNA methylation. (b–d) ELISA levels of IGF2, (e–f) RT-qPCR levels of Igf2 mRNA, and (g–i) GlucMS-qPCR levels of H19 ICR DNA methylation (5mC) in the cerebrum, liver, and plasma of female and male 5xFAD and WT mice at 6- and 12-months of age. Data are expressed as means + / − SEM. IGF2/Igf2, insulin-like growth factor 2; DMR2, differentially methylated region 2; ICR, imprinting control region; Me, DNA methylation; CTCF, CCCTC-binding factor; rpl13a, ribosomal protein L13a.; HpaII/Msp1, DNA restriction enzymes. See supplemental materials for results of statistical analyses.
To determine whether these differences in IGF2 levels were associated with differences in Igf2 expression, we performed RT-qPCR analyses in cerebrum and liver tissue. Igf2 mRNA transcript levels in the cerebrum were significantly lower in 5xFAD than WT mice (Fig. 1e), significantly lower in 12-month than 6-month-old mice, and significantly lower in females than males. The Igf2 mRNA levels in the liver were significantly lower in 5xFAD than WT mice and lower in females than males. There was also a significant genotype by sex by age interaction (Fig. 1f) as the Igf2 mRNA levels were lower for all mice at 12-months than at 6-months of age, except for the WT males.
In the H19/Igf2 locus, an ICR associated with a DMR2 sequence upstream of H19 regulates the reciprocal expression of Igf2 and H19 (see Fig. 1a). Hypomethylation of the DMR2 on the maternal allele blocks enhancer driven Igf2 transcription. To determine whether the differences in Igf2 expression were associated with alterations in DNA methylation or DNA hydroxymethylation, we analysed 5mC and 5hmC levels in six distinct loci that are known to be differentially methylated in this region48,49,50,51. In both 6- and 12-month-old mice, the levels of H19 ICR methylation (5mC) in the cerebrum were significantly lower in females than males (Fig. 1g), which reflects the reduced Igf2 expression (Fig. 1e) and lower IGF2 levels (Fig. 1b). There was no significant effect of genotype or age on H19 ICR 5mC levels in the cerebrum, suggesting that mechanisms independent of H19 ICR methylation status regulate Igf2 expression in aging 5xFAD mice. The H19 ICR 5mC levels in the liver were significantly lower in 5xFAD than WT mice (Fig. 1h), which reflects the reduced Igf2 expression levels (Fig. 1f) and lower IGF2 levels (Fig. 1c). Likewise, levels of H19 ICR 5mC were significantly lower in the liver of females than males and significantly lower in the liver from 12-month than 6-month-old mice. The levels of DNA methylation (5mC) on the H19 ICR of cfDNA in the blood plasma were significantly lower in 5xFAD than in WT mice, lower in females than males, and lower in 12-month than 6-month-old mice (Fig. 1i). The levels H19-ICR 5hmC levels did not differ between groups in the cerebrum, liver, and blood plasma (all p > 0.05; Table S1).
5xFAD-associated tissue-specific variation in H19 ICR methylation, CTCF binding and histone modification
Since hypermethylation of CpG dinucleotides of the H19 ICR is associated with increased Igf2 expression, while hypomethylation of CpG dinucleotides of the H19 ICR is associated with CTCF binding and Igf2 silencing, we tested the hypothesis that sex- and age-related differences in IGF2 levels, Igf2 expression, and H19 promoter hypomethylation in 5xFAD mice are associated with increased CTCF binding to the H19 ICR (Fig. S1a). The results showed sex differences in DNA methylation (5mC) and in the levels of CTCF association with the H19 ICR with females having lower 5mC and higher CTCF binding than males, and no genotype difference (Fig. S1b–d). Females also had higher levels of histone acetylation (H3K9Ac) following CTCF binding to the H19 ICR than males (Fig. S1e–g), while males had higher levels of histone methylation (H3K9me3) following CTCF binding to the H19 ICR than females (Fig. S1h–j).
Overall, these findings show that IGF2 levels are associated with sex-differences in H19 ICR DNA and histone modifications in the cerebrum, liver, and plasma, and that these enduring epigenetic marks potentially program chromatin accessibility and Igf2 expression through CTCF association with the H19 ICR in the cerebrum and liver. The lack of effect of genotype on group differences in CTCF association with the H19 ICR methylation, CTCF binding, and histone modification in the cerebrum suggests that the changes in Igf2 expression in 5xFAD mice may be caused by temporal mechanisms independent of H19 ICR DNA methylation. Given the bidirectional communication between the nervous and liver systems (i.e., the liver-brain axis)74 and the potential role of Aβ42 as a transcription factor, this raised the question of whether changes in Igf2 expression are linked to changes in Aβ42 deposition in the cerebrum and liver of 5xFAD mice.
5xFAD-associated tissue-specific variation in Aβ42 levels
The results of ELISA analyses show that the levels of Aβ42 were higher in the cerebrum of 5xFAD than WT mice and increased with age in 5xFAD but not in WT mice, resulting in an age by genotype interaction, with no sex difference (Fig. 2a). Levels of Aβ42 were higher in the liver of 5xFAD than WT mice and increased significantly with age in 5xFAD mice but not in WT mice resulting in an age by genotype interaction. There was also a sex by age interaction, as levels of Aβ42 in the liver increased more in males from 6- and 12-months of age than females (Fig. 2b). The levels of Aβ42 were higher in the blood plasma of 5xFAD than WT mice, and decreased with age in 5xFAD mice, but not in WT mice. There was no sex difference in levels of Aβ42 in the blood plasma (Fig. 2c). These results confirm the earlier findings17 of elevated Aβ42 levels in the 5xFAD mice in an age-dependent manner and show that this increase occurs in the liver as well as the brain.

ELISA levels of Aβ42 in the cerebrum, liver, and plasma of male and female, 5xFAD and WT mice at 6- and 12-months of age. Data are expressed as means + / − SEM. Aβ42, amyloid beta 1–42.
5xFAD-associated tissue-specific variation in Igf2 DMR2 methylation, Aβ 42 binding and histone modification
Results from neural (SH-SY5Y) cell cultures75 and the triple transgenic mouse model of AD9 provide evidence that Aβ peptides, especially Aβ42, can translocate to neuronal nuclei and bind to the Aβ interacting domain (AβID) within gene regulatory and promoter elements to alter gene expression. This raises the question of whether changes in Igf2 expression in 5xFAD mice are associated with changes in levels of Aβ42 binding to AβID regions on the Igf2 promoter. Three DMRs have been identified in the mouse Igf2 promoter region (Fig. 3a). The paternally methylated DMR2 functions as a transcriptional activator of Igf2 expression when hypomethylated and contains AβID response elements (REs)54. We first performed ChIP-qPCR analyses with an antibody toward Aβ42 to test the specificity of the association of Aβ42 with the potential AβID region of Igf2 DMR2 in cerebrum from 12-month-old mice and found that Aβ42 association with Igf2 DMR2 was significantly higher than with the H19 ICR that did not include the potential AβID region in 5xFAD mice (Fig. 3b). This raises the question of whether the decrease in IGF2 levels and Igf2 expression in the 5xFAD mice is associated with increased Aβ42 binding with Igf2 DMR2. To test this, we performed ChIP analysis with antibodies toward 5mC and Aβ42 with the AβID region of Igf2 DMR2 in cerebrum, liver, and blood plasma from 6- and 12-month-old male and female 5xFAD and WT mice.

Epigenetic marks associated with Aβ42 binding to Igf2 DMR2 in mice. (a) Schematic representation of the mouse Igf2 promoter region (also see Fig. 1a). Mouse Igf2 has four promoters (P, 0–3) and three DMRs (0–2). Beneath is shown the Igf2 DMR2 DNA sequence we analyzed, with the location of seven CpG sites (bold) relative to the predicted Aβ42 interacting domain (AβID; boxed area). (b) ChIP-qPCR analyses of Aβ42 association with H19 ICR and Igf2 DMR2 in cerebrum from 12-month-old 5xFAD mice. ChIP- and double ChIP-qPCR analyses of DNA methylation (5mC) and Aβ42 association with Igf2 DMR2 in (c) cerebrum, (d) liver, and (e) plasma of male and female, 5xFAD and WT, 6- and 12-month-old mice. ChIP-qPCR analyses of H3K9Ac (f–h) and H3K9me3 (i–k) association with Igf2 DMR2 in cerebrum, liver, and plasma of male and female, 5xFAD and WT, 6- and 12-month-old mice. Data are expressed as means − / + SEM. Igf2, insulin-like growth factor 2; DMR2, differentially methylated region 2; ICR, imprinting control region; 5mC, 5-methylcytosine; Aβ42, amyloid beta 1–42; H3K9Ac, histone 3 lysine-9 acetylation; H3K9me3, histone 3 lysine-9 tri-methylation.
Levels of DNA methylation on the AβID region of Igf2 DMR2 in the cerebrum showed no genotype difference but were significantly higher in 12-month than 6-month-old mice, and there was a sex by genotype interaction. While female WT mice had higher levels of Igf2 DMR2 methylation in the cerebrum than male WT mice, there was no sex difference in 5xFAD mice (Fig. 3c). However, levels of Aβ42 association with the AβID region of Igf2 DMR2 in the cerebrum were significantly higher in 5xFAD than WT mice, with no effect of sex or age (all p > 0.05; Fig. 3c). Levels of DNA methylation on the AβID region of Igf2 DMR2 in the liver were significantly higher in 5xFAD than WT mice, and were significantly higher in females than males, with no effect of age (Fig. 3d). Levels of Aβ42 association with the AβID region of Igf2 DMR2 in the liver were significantly higher in 5xFAD than WT mice, with no effect of sex or age (all p > 0.05; Fig. 3d). There was no effect of genotype on levels of Igf2 DMR2 methylation of cfDNA fragments from the plasma, but there was a sex by age interaction: females had lower levels at 12-months than 6-months of age, while males had higher levels at 12-months than 6-months of age (Fig. 3e). The 5xFAD mice showed significantly higher Aβ42 binding in the plasma than WT mice, and there were genotype by age and genotype by sex interactions: Aβ42 association with the Igf2 promoter increased in 5xFAD mice as they aged, and the increase was greater in 5xFAD females than males (Fig. 3e), suggesting that cfDNA epigenetic marks at transcription factor binding sites in plasma also reflect age-related AD pathologies. The double ChIP analyses showed low levels Igf2 DMR2 enrichment in lanes labelled 5mC/Aβ42 and Aβ42/5mC, with no significant genotype, sex, or age differences in cerebrum, liver, or blood plasma (all p values > 0.05; Fig. 3c–e).
Aβ42 binding at the Igf2 DMR2 is associated with Igf2 silencing, suggesting that an association of Aβ42 with specific changes in chromatin through histone modification could also influence Igf2 DMR2 activity. To test this hypothesis, we performed ChIP-qPCR analyses measuring histone acetylation (H3K9Ac) and histone methylation (H3K9me3) within the AβID region in 5xFAD and WT mice. Levels of H3K9Ac association with the AβID region of Igf2 DMR2 in the cerebrum were significantly lower in 5xFAD than WT mice, lower in females than males, and lower in 12-month than 6-month-old mice (Fig. 3f). While there was no genotype effect, there was a significant genotype by sex interaction for H3K9Ac association with the AβID region of Igf2 DMR2 in the liver: while males had overall higher levels than females, the sex difference was only significant within the WT mice and not the 5xFAD mice. Moreover, the levels of H3K9Ac association were lower in 12-month than 6-month-old mice (Fig. 3g). Levels of H3K9Ac association with the AβID region of Igf2 DMR2 in the blood plasma were also significantly lower in 5xFAD than WT mice, lower in females than males, and lower in 12-month than 6-month-old mice (Fig. 3h).
The overall levels of H3K9me3 association with the AβID region of Igf2 DMR2 in the cerebrum were higher in 5xFAD than WT mice, higher in 12-month than 6-month-old mice, and higher in females than males, but there was a genotype by sex by age interaction as 5xFAD males had a decrease with age whereas all other groups showed an increase with age (Fig. 3i). In the liver, there was no significant genotype difference for H3K9me3 association, but there was a sex by age interaction; females showed an increase from 6- to 12-months of age but males did not (Fig. 3j). Levels of H3K9me3 association with the AβID region of Igf2 DMR2 in the blood plasma were significantly higher in females than males, and higher in 12-month than 6-month-old mice, with no effect of genotype (Fig. 3k). These results suggest that Aβ42 association with the Igf2 DMR can alter the chromatin structure and decrease Igf2 expression by changing both histone acetylation and methylation in a sex, age, and tissue specific manner.
IGF2 levels, Igf2 expression, and epigenetic status of the H19/Igf2 locus in 5-week-old 5xFAD and WT mice
There was no difference in Aβ40 or Aβ42 accumulation in 5-week-old 5xFAD compared to WT mice, and no significant genotype differences in IGF2 levels, Igf2 mRNA levels, or epigenetic status of the H19/Igf2 locus (Table S2). These findings suggest that Aβ40 and Aβ42 accumulation either precede, or occur concomitantly with, the tissue-specific alterations in Igf2 epigenetic regulation and IGF2 levels observed in 6- and 12-month-old 5xFAD mice.
Frontal cortex IGF2 and Igf2 mRNA reduction and H19 ICR methylation in AD patients
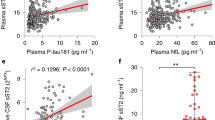
To determine whether the findings from the 5xFAD mouse model translated to humans, we compared Igf2 epigenetic regulation and IGF2 levels in the frontal cortex from aged humans diagnosed with AD and age-matched non-AD controls. As in 5xFAD mice, IGF2 levels in the frontal cortex were lower in AD than non-AD patients and lower in females than males (Fig. 4a; Table S3). In agreement with this, Igf2 mRNA levels in the frontal cortex were lower in AD than non-AD patients, and lower in females than males (Fig. 4b; Table S3).

IGF2 levels, Igf2 expression, H19 ICR methylation, and Aβ40 and Aβ42 levels in the frontal cortex of male and female AD and non-AD patients. (a) ELISA levels of IGF2, (b) RT-qPCR levels of Igf2 mRNA, (c) GlucMS-qPCR levels of H19 ICR DNA methylation (5mC), and ELISA levels of (d) Aβ40, (e) Aβ42, and (f) the Aβ42/Aβ40 ratio in the frontal cortex of male and female, AD and non-AD patients. Data are expressed as means + / − SEM. IGF2/Igf2, insulin-like growth factor 2; rpl13a, ribosomal protein L13a; HpaII/Msp1, DNA restriction enzymes; Aβ40, amyloid beta 1–40; Aβ42, amyloid beta 1–42.
To determine whether the differences in Igf2 expression were associated with alterations in DNA methylation and hydroxymethylation we analysed 5mC and 5hmC levels in six distinct loci that are known to be differentially methylated in the H19 ICR (Fig. S2a). The levels of H19 ICR 5mC in the frontal cortex were significantly lower in females than males (Fig. 4c) but did not differ between AD and non-AD patients. Levels of H19 ICR 5hmC did not differ between the disease conditions or sexes (all p > 0.05; Table S3). Results of ELISA analyses showed that AD patients had higher levels of Aβ40 (Fig. 4d), Aβ42 (Fig. 4e), and a higher Aβ42/Aβ40 ratio (Fig. 4f) than non-AD patients. There were no sex differences in any of these measures (all p > 0.05; Table S3). Together these results demonstrate that Aβ42 accumulation in the frontal cortex is associated with Igf2 expression in AD pathology in human patients.
H19 ICR DNA methylation, CTCF binding, and histone modifications in AD patients
We tested the hypothesis that the variations in frontal cortex levels of IGF2 and Igf2 mRNA transcripts, and H19 promoter hypomethylation in AD are associated with altered CTCF association with the H19 ICR (Fig. S2a). Overall, these findings (see Fig. S2 and Table S3 for statistical analyses) show that males had higher levels of H19 ICR DNA methylation than females, with no effect of AD, and that CTCF association was higher in non-AD females than non-AD males (Fig. S2b), with no AD- or sex-differences in H3K9ac or H3K9me3 (Fig. S2c,d).
AD status- and sex-specific variation in Igf2 DMR2 methylation, Aβ 42 binding, and histone modification in humans
Two DMRs have been identified in the human Igf2 promoter region (Fig. 5a). Using ChIP-qPCR analyses with an antibody toward Aβ42 we found that there was significantly greater Aβ42 association with the Igf2 DMR2 sequence containing the potential AβID region in frontal cortex of AD patients than there was with the H19 ICR that did not include this AβID region (Fig. 5b; Table S3). Using ChIP-qPCR analyses with antibodies toward 5mC and Aβ42 we found that levels of Igf2 DMR2 methylation (5mC) in the frontal cortex were not significantly different between AD and non-AD patients, but were significantly higher in females than males (Fig. 5c). Conversely, Aβ42 association with Igf2 DMR2 was significantly higher in the frontal cortex from AD compared to non-AD patients, but there was no sex difference (Fig. 5c). Similar to the results in mice, the double ChIP assays demonstrated that Aβ42 did not strongly bind to methylated DNA (evidenced by low levels Igf2 DMR2 enrichment in lanes labelled 5mC/Aβ42 and Aβ42/5mC; Fig. 5c). The ChIP-qPCR analyses measuring H3K9Ac and H3K9me3 within the AβID region found that the levels H3K9Ac association with Igf2 DMR2 in the frontal cortex were higher in female than male non-AD patients but not in AD patients (Fig. 5d). Conversely, H3K9me3 association with Igf2 DMR2 in the frontal cortex was significantly higher in AD than non-AD patients, with no significant sex difference (Fig. 5e).

Epigenetic marks associated with Aβ42 binding to Igf2 DMR2 in human frontal cortex. (a) Schematic representation of the human Igf2 promoter region (also see Fig. 1a). Human Igf2 has five promoters (human P1, HuP1; and P, 0–3) and two DMRs (0 and 2). There is no mouse homologue for HuP1; however, P0–3, DMR0 and DMR2 are homologous in human and mouse. Beneath is shown the Igf2 DMR2 DNA sequence we analyzed, with the location of twelve CpG sites (bold) relative to the predicted Aβ42 interacting domain (AβID; boxed area). (b) ChIP-qPCR analyses of Aβ42 association with H19 ICR and Igf2 DMR2 in AD and non-AD patients. ChIP- and double ChIP-qPCR analyses of DNA methylation (5mC) and Aβ42 association with the Igf2 DMR2 in male and female, AD and non-AD patients. ChIP-qPCR analyses of H3K9Ac (d) and H3K9me3 (e) association with Igf2 DMR2 in male and female, AD and non-AD patients. Data are expressed as means + / − SEM.
Effects Aβ 42 exposure on Igf2 promoter regulation in HEK293 cell culture
Results of the ELISA analyses found that levels of Aβ42 in the cell nuclear fractions were higher in the Aβ42-treated than the vehicle-treated HEK293 cells as a function of the number of days the cells spent in culture following Aβ42 treatment (Fig. 6a; Table S4). Three-days following treatment, the magnitude of the difference in Aβ42 levels between Aβ42- and vehicle-treated cultures was greatest. While still significant, 6-days after treatment the magnitude of the difference in Aβ42 levels between Aβ42- and vehicle-treated cultures was reduced and was not significant 9-days following Aβ42 treatment (Fig. 6a). These results suggest that exogenous Aβ42 can enter the cell nucleus, followed by temporal removal (clearing). The ChIP-qPCR analyses with an antibody toward Aβ42 showed that in the Aβ42 treated cultures, there was significantly greater Aβ42 binding with Igf2 DMR2 than there was with H19 ICR, while there was no difference in the vehicle-treated cultures (Fig. 6b). The levels of Aβ42 binding to Igf2 DMR2 were significantly higher in Aβ42-treated cultures than control cultures at all time points (Fig. 6c), as were the levels of Igf2 DMR2 DNA methylation (Fig. 6d).

Effects of Aβ42 on Igf2 regulation and IGF2 levels in HEK239 cells. Cell cultures received a single treatment of Aβ42 oligomer solution or saline vehicle, which were replaced every 3 days (removing Aβ42 from the media), and the cells were harvested after 3-, 6-, or 9-days for in vitro assays. (a) ELISA levels of Aβ42 from nuclear extracts in Aβ42-treated cultures. (b) ChIP-qPCR analyses of Aβ42 association with H19 ICR and Igf2 DMR2 (containing the AβID) 6-days following Aβ42-treatment. ChIP-qPCR analyses of (c) Aβ42, (d) DNA methylation (5mC), (e) H3K9Ac, and (f) H3K9me3 association with Igf2 DMR2 in Aβ42-treated cultures. (g) RT-qPCR analyses of Igf2 mRNA levels and (h) ELISA analysis of IGF2 levels in Aβ42-treated cultures. Data are expressed as means + / − SEM (*p < 0.05; **p < 0.01; ***p < 0.001).
Levels of Igf2 DMR2 histone acetylation were decreased in the Aβ42-treated cultures compared to control cultures after 6 and 9 days (Fig. 6e), while levels of Igf2 DMR2 histone methylation increased as a function of the number of days the cells spent in culture following Aβ42 treatment (Fig. 6f). The levels of Igf2 DMR2 histone methylation in Aβ42-treated and vehicle-treated cultures were not significantly different after 3-days of Aβ42 treatment, while after 6- and 9-days they were significantly higher in Aβ42-treated than control cultures. Levels of Igf2 mRNA were significantly lower in Aβ42-treated cultures at 6- and 9-days, but not at 3-days following Aβ42 treatment (Fig. 6g). Likewise, levels of IGF2 were significantly lower in Aβ42-treated than control cultures at 6- and 9-days, but not at 3-days after Aβ42 treatment (Fig. 6h). Together, these results suggest a causal relationship between Aβ42 accumulation, Aβ42 binding and DNA methylation on the Igf2 DMR2, combined with histone modifications (deacetylation and methylation), leading to a stable reduction of Igf2 expression and IGF2 levels (Table S4). Stable association of Aβ42 with the Igf2 DMR2 provides a potential mechanism underlying the temporal stability of altered Igf2 expression patterns in 5xFAD mice and humans diagnosed with AD.
Discussion
Our findings demonstrate that IGF2 levels and Igf2 expression in WT mice is associated with increased histone acetylation, transcription factor (CTCF) binding and H19 ICR hypomethylation, whereas the reduced IGF2 levels and Igf2 expression in the cerebrum, liver, and blood plasma of 5xFAD mice is associated with increased histone methylation, transcription factor (Aβ42) binding and Igf2 DMR2 hypermethylation (Table S1). The results from 1.5-month-old 5xFAD mouse pups (Table S2), which lack Aβ42 accumulation and do not differ from WT mice in IGF2 levels suggest that Aβ42 accumulation precedes or is associated with the genotype- and tissue-specific alterations in the epigenetic regulation of Igf2 expression and IGF2 levels observed in symptomatic 5xFAD mice. Results from humans diagnosed with AD (Table S3) suggest a similar causal relationship among Aβ42 levels, epigenomic state, and Igf2 expression in the frontal cortex, reflecting a conserved mechanism for Igf2 gene regulation in AD (Fig. 7a). In addition, Aβ42 treatment of human derived HEK239 cells (Table S4) induced DNA methylation, histone deacetylation and histone methylation, and a reduction in Igf2 expression through stable binding of Aβ42 to the Igf2 DMR2 promoter region (Fig. 7b). Thus, Aβ42 association with the Igf2 DMR on the Igf2 promoter provides a nonconical mechanism for reduced IGF2 levels in 5xFAD mice and AD patients, independent of H19 ICR DNA methylation. These findings suggest a causal relationship among epigenomic state, Igf2 expression, and IGF2 levels in a mouse model with age-related changes in metabolism, reduced weight gain, tissue degradation, and cognitive/motor decline and provide a potential mechanism for mouse and human Igf2 gene regulation in normal and pathological conditions.

Dynamics of histone modification, CpG methylation, and transcription factor binding that control Igf2 expression associated with Aβ42 accumulation in AD. (a) In Non-AD (WT mice): enhanced Igf2 expression on the paternal allele (upper row) is associated with histone hypermethylation (H3K9me3), DNA hypermethylation (5mC), and reduced transcription factor (CTCF) binding to the H19 ICR, with concomitant histone hyperacetylation (H3K9Ac) and DNA hypomethylation of Igf2 DMR2. Conversely, attenuated Igf2 expression on the maternal allele (lower row) is associated with histone hyperacetylation (H3K9Ac), DNA hypomethylation, and increased transcription factor (CTCF) binding to the H19 ICR, with concomitant histone hypermethylation (H3K9me3), and DNA hypermethylation (5mC) of Igf2 DMR2. In AD (5xFAD mice): as in WT mice, attenuated Igf2 expression on the paternal allele (upper row) is associated with histone hypermethylation (H3K9me3), and DNA hypermethylation (5mC); however, enhanced transcription factor (Aβ42) binding to Igf2 DMR2 is associated with histone hypoacetylation (H3K9Ac), histone hypermethylation (H3K9me3), DNA hypermethylation (5mC) and attenuation of Igf2 expression. As in WT mice, attenuated Igf2 expression on the maternal allele (lower row) is associated with histone hyperacetylation (H3K9Ac), DNA hypomethylation (5mC), and increased transcription factor (CTCF) binding to the H19 ICR; however, enhanced transcription factor (Aβ42) binding to Igf2 DMR2 is associated with histone hypoacetylation (H3K9Ac), histone hypermethylation (H3K9me3), DNA hypermethylation (5mC) and further attenuation of Igf2 expression. In this context, the regions of DNA that bind Aβ42 (i.e., AβID, Aβ interacting domains) have repressive chromatin marks and increased Aβ42 binding association. (b) Temporal effects of Aβ42 on Igf2 regulation. In cultured cells: Increased Aβ42 levels are associated with increased binding of Aβ42 to, and DNA hypermethylation of, the Igf2 differentially methylated region 2 (DMR2) (3-Days). Persistent Igf2 DMR2 Aβ42 binding and DNA hypermethylation combined with histone modifications (deacetylation and methylation) are associated with a stable reduction of Igf2 expression and IGF2 levels (6-Days, 9-Days). IGF2/Igf2, insulin growth factor 2; DMR2, differentially methylated region 2; ICR, imprinting control region; CTCF, CCCTC-binding factor, Aβ42, amyloid beta 1–42.
In agreement with previous reports68, our results show reduced IGF2 levels in the CNS of young 5xFAD mice, which further decline with age and lower IGF2 levels in females than males at both 6- and 12-months of age (Fig. 1b). To determine whether attenuated IGF2 levels were due to tissue-specific epigenetic regulation, we measured H19 ICR methylation, Igf2 expression and IGF2 levels and found sex- and age-dependent changes in IGF2 protein, as well as epigenetic regulation of the Igf2/H19 locus in the cerebrum, liver, and plasma of young and aged 5xFAD mice (Fig. 1b–i). To our knowledge, this is the first study to investigate H19 ICR promoter methylation, Igf2 expression and IGF2 levels in neural and peripheral tissues of young and aged 5xFAD mice. Overall, the Igf2 mRNA and IGF2 protein levels were lower in 1) 5xFAD mice than WT mice, 2) females than males, and 3) older mice than younger mice.
These results suggest that it might be important to determine the nature of brain-liver activities and how such interactions result in sustained changes in gene expression and function over the lifespan and in AD. As in the cerebrum, IGF2 levels were significantly lower in the liver of 5xFAD mice compared to the WT mice (Fig. 1c). The qPCR analysis showed decreased Igf2 mRNA transcript levels in the liver of 5xFAD mice (Fig. 1f), suggesting that IGF2 is endogenously produced in the liver rather than carried in from other tissues. Reduced IGF2 levels in the liver of 5xFAD mice suggests reduced metabolic activity in the liver, as occurs in the mouse76 (and human77) brain. IGF2 depletion accelerates the onset of metabolic deficits, reduced cell survival, and cognitive impairments by altering growth hormone-related gene expression. Indeed, memory and synaptic deficits in mouse models of AD can be reversed by treatment with IGF2 analogs26,30. Likewise, liver Igf2 expression and plasma IGF2 levels respond to metabolic status and promote tissue survival and proliferation, along with adipocyte signaling78,79. Both liver volume and blood flow decrease with age in mice80 (and humans81). Together with our findings of Aβ42-related reduction of Igf2 expression, this may help explain the pathophysiology of peripheral metabolic deficits observed in AD mouse models18 and humans diagnosed with AD82.
There are several CTCF binding sites on the mouse H19 promoter (Fig. 1a and S1a). We therefore examined the potential mechanism for Igf2 regulation by CTCF. The 5xFAD mice had lower levels of Igf2 mRNA and IGF2 in the cerebrum and liver (Fig. 1e,f). However, we did not find any genotype differences in H19 ICR methylation or CTCF binding (Fig. S1b–d), suggesting that the reduced Igf2 expression observed in the cerebrum and liver of 5xFAD mice is independent of canonical H19 imprinting.
A switch from monoallelic to biallelic human Igf2 promoter methylation during aging has been documented83, and this may be partly responsible for the reduced levels seen in rodents and humans34,84,85. This is further supported by our results; while there were no age differences in H19 ICR methylation or CTCF in the cerebrum, IGF2 promoter methylation was higher in the 12-month-old than the 6-month-old mice (Fig. S1b). On the other hand, there was a direct relationship between H19 and Igf2 regulation for the sex differences observed in this study: the female mice had lower IGF2 levels than males, accompanied by lower H19 ICR methylation and higher CTCF binding. In support of this, lower expression levels of IGF2 and H19, along with other imprinted genes such as Peg3 and Zim1 have previously been reported in female mice, measurable from later stages of pregnancy onwards86.
To determine whether attenuated IGF2 levels were associated with AD neuropathology, we measured Aβ42 levels in the cerebrum, liver, and blood plasma of young and aged 5xFAD mice (Fig. 2a–c). To our knowledge, this is the first study to show the presence of Aβ, as well as decreased IFG2, in the liver of 5xFAD mice (Fig. 2b). Although the exact source needs to be determined, the Aβ42 found in the hepatic tissue may have come from the circulating Aβ that we detected in the plasma (Fig. 2c). Our results show that: 1) Aβ42 levels were higher in the cerebrum and liver of 5xFAD mice than WT mice; 2) the accumulation of Aβ42 begins before 6-months of age; 3) Aβ42 levels increase as the mice continue to age; and that 4) IGF2 levels decrease as Aβ42 levels increase (indicated by reduced IGF2 levels in older 5xFAD mice). These results enhance the validity of the 5xFAD mice as a model for AD, as enriched Aβ in peripheral tissues in humans carrying the Swedish mutation has been reported87, and shown to slow down Aβ clearance in the brain88. Indeed, increased peripheral tissue damage (including the liver) in mild AD and cognitive impairment has been linked to ApoE-ε4 allele carriers89.
We found an Aβ42 binding (AβID) region on the mouse Igf2 promoter (Fig. 3a), implying that the Igf2 gene may be co-regulated by Aβ42, which has been shown to act as a transcription factor for the APP, β-secretase, and ApoE genes, as well as Txnip5,9. In support of this theory, Aβ42 selectively associated with genomic DNA sequences that contained the potential AβID region, in cerebrum of 12-month-old mice and human frontal cortex as well as ex vivo in human-derived HEK293 cells (compare Figs. 3b, 5b and 6b). We therefore examined the potential mechanism for Igf2 gene regulation by CTCF and Aβ42. ChIP- and double-ChIP-qPCR assays measuring chromatin modifications showed lower histone acetylation and higher DNA methylation on the AβID response element region of the 5xFAD mice, suggesting that cerebrum and liver Igf2 expression is silenced both by Aβ42 and CTCF (Fig. 3c–k). Our results show that Aβ42 can interact with gene regulatory elements and recruit heterochromatin marks associated with gene silencing (i.e., decreased H3K9Ac and increased H3K9me3) through specific binding to the AβID sequence. While consistent with other reports90, this further demonstrates that the effect of Aβ42 on transcription (either increasing or decreasing mRNA expression) is gene-specific5,75. These results were supported by double ChIP-qPCR assays to study DNA methylation with Aβ42 and CTCF binding (Fig. 3c–e and S1b–d). WT mice had hypomethylated AβID and hypermethylated CTCF regions accompanied by low Aβ42 and CTCF binding. Conversely, 5xFAD mice had hypermethylated AβID and hypomethylated CTCF regions accompanied by higher Aβ42 and CTCF binding, which explains the low amounts of IGF2 in the cerebrum, liver, and plasma of the 5xFAD mice.
IGF2 reduction contributes to AD pathogenesis due to its role in Aβ42 clearance91. Reduced IGF2 levels in the liver could accelerate the disease process. In hepatic cells, during normal aging, IGF2 directly interacts with growth hormone to increase its transcriptional activity for promotion of somatic growth and the regulation of metabolism92. The reduction of IGF2 levels in the cerebrum and liver may help to explain the pathological weight loss and cognitive deficits seen in the 5xFAD mice18,60. Higher IGF2 levels in the young WT mice may be a part of normal development, protecting developing blood cells against oxidative damage. Since the levels of IGF2 are much lower in the 5xFAD mice than WT mice, this may contribute to blood-related abnormalities, such as higher oxygen consumption93 or elevated oxidative damage of DNA by reactive oxygen species, as shown by our findings in 3xTg-AD mice9. We found higher levels of Aβ42 binding to a potential AβID region of Igf2 DMR2 (Fig. 3e) along with reduced IGF2 levels (Fig. 1d) in plasma samples of the 12-month-old 5xFAD mice compared to the 6-month-old mice, even though the levels of Aβ42 were lower in the older mice (Fig. 2c). Reductions in plasma Aβ42 levels occur in later stages of AD94, and may result in reduced clearance of Aβ42 from the CNS due to formation of large Aβ aggregates and plaques or clearance of other toxic species that contribute to AD progression. Higher binding to the Igf2 promoter, even at lower concentrations, suggest that various Aβ species are present in the 5xFAD mice in an age dependent manner and more toxic species may be forming as the mice age. While our ELISA method was not capable of differentiating between these species, which is a limitation of this study, the role of different sizes and shapes of Aβ oligomers on different AD mechanisms has been reported95.
The canonically imprinted Igf2/H19 locus is conserved between mice and humans in both DNA sequence and epigenetic regulation52. To determine whether the findings from the 5xFAD mouse model supported relevance of mechanism to human disease, we compared Igf2 epigenetic regulation in frontal cortex from aged humans diagnosed with AD and non-AD patients. Our findings show that the levels of IGF2 and Igf2 mRNA expression were lower in the frontal cortex of AD than non-AD patients and lower in females than males (Fig. 4a,b), with the sex differences driven by H19 ICR DNA methylation (Fig. 4c with S2b), whereas genotype differences were driven by Aβ42 bound to the Igf2 DMR2 (Fig. 5c), independent of the levels of H19 ICR DNA methylation (compare Figs. 5c with 4c and S2b). These results, together with the Aβ42 association with the Igf2 AβID region (Fig. 5c), provide evidence for conserved mechanisms for Igf2 regulation in 5xFAD mice and AD patients. To the best of our knowledge, this the first time Aβ42 has been shown to have a role as a transcription factor and epigenetic regulator in the human brain, altering transcription of an imprinted gene independently of canonical imprinting.
Exogenous Aβ40 and Aβ42 oligomers have been shown to translocate to the cell nucleus in culture8, where Aβ42 can bind to genomic DNA and alter gene expression5,9. To further characterise the temporal order of events that lead to a decrease in IGF2 and to examine whether Aβ42 bound to the Igf2 promoter region can directly mediate Igf2 transcription, HEK293 cultures were treated with Aβ42 oligomers and then harvested at various time points to evaluate temporal changes in Igf2 epigenetic status and expression.
Our results show exogenous Aβ42 was detectable in the cell nucleus 3-days following Aβ42 treatment, but levels decreased 6- and 9-days following Aβ42 treatment, suggesting leakage or an active clearance mechanism (Fig. 6a). While the endocytosis and nuclear transport mechanisms of Aβ42 have not been fully characterised, these processes have been shown to involve the alpha-7 nicotinic96 and toll-like receptors97,98. Interestingly, while the nuclear Aβ42 levels decreased over time (Fig. 6a), the levels of Aβ42 bound to DNA did not change (Fig. 6c), suggesting that only the Aβ42 unbound to DNA is cleared from the cell nucleus (compare Fig. 6a,c). In addition, Igf2 expression and IGF2 levels remained low following Aβ42 treatment (Fig. 6g,h). These results suggest stable association of Aβ42 with the Igf2 DMR2 may provide a potential mechanism underlying the temporal stability of altered Igf2 expression patterns in 5xFAD mice and AD.
These results suggest a causal relationship between Aβ42 accumulation, Aβ42 binding and DNA methylation on the Igf2 DMR2, combined with histone deacetylation and methylation, leading to a stable reduction of Igf2 expression and IGF2 levels. Similar to our previous findings in 3xTg-AD mice9, these results support of the role of Aβ42 as a transcription factor. However, binding of Aβ42 with the Igf2 DMR AβID region results in enhanced histone methylation and Igf2 silencing (rather than enhanced histone acetylation and activation as seen with the TXNIP AβID region).
Limitations
While the sample size used in the mouse studies was justified by power analysis, 3 mice per group is still at the lower end. However, the high agreement between the mouse, human, and cell culture results indicates a relatively conserved biological mechanism in mammalian cells and that the findings are biologically valid. Further studies are required to determine the temporal time point(s) of the functional changes in cerebrum and liver of the 5xFAD mice, and how the transgenes in the 5xFAD mice or their derivatives alter brain-liver metabolism, tissue growth, liver function, and the epigenetic status of the Igf2 gene promoter. In addition, the exact causal relationship between DNA methylation and altered histone acetylation and transcription factor (Aβ42, CTCF) binding remains to be defined, as well as the process by which Aβ42 enters the cell, translocates to the nucleus, and is then cleared. It also remains unclear whether a similar mechanism occurs at AβID elements within other imprinted genes or whether neurons and different types of glial cells show the same response. Genetically modified cell lines and animal models with induced disruption to the Igf2 DMR AβID region would permit us to validate the DNA sequence specificity of Aβ42 binding and monitor the dynamics of epigenetic regulation in parallel with behavioral assessments. Future work should also consider the use of allelic-specific primers to examine the possibility of tissue-specific variation in maternal and paternal allele expression, while longitudinal sampling would allow us to determine if these changes are the result of gradual interactions between Aβ42 (and other Aβ species) with AβID regions in neural and non-neuronal cell types.
Conclusions
This study systematically examined molecular and functional deficits in the cerebrum, liver, and plasma of young and aged 5xFAD mice and showed the transcriptional relationship between Aβ42 and IGF2 in the cerebrum and liver. This transcriptional relationship between Aβ42 and IGF2 was shown to be conserved in the human frontal cortex of individuals diagnosed with AD and was modeled in Aβ42-treated human derived HEK293 cell cultures. Consistently Aβ42 was shown to override gene imprinting and control gene expression. Alterations in histone structure, both in terms of methylation99 and acetylation100 have been shown in the brains of AD patients, however these changes have been related to tau pathology101,102. Thus, our results contribute to the already known role of Aβ42 as a transcription factor, and for the first time confirm it in the human brain. Overall, these findings provide a potential mechanism for the enduring effect of pathological inflammation on brain function in a common chronic progressive neurodegenerative disorder and suggest IGF2 levels could potentially be a useful predictive or diagnostic biomarker for Aβ42-targeted AD therapies.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Scheltens, P. et al. Alzheimer’s disease. Lancet 397, 1577–1590. https://doi.org/10.1016/S0140-6736(20)32205-4 (2021).
Hampel, H. et al. The amyloid-beta pathway in Alzheimer’s disease. Mol. Psychiatry https://doi.org/10.1038/s41380-021-01249-0 (2021).
Hardy, J. & Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388 (1991).
Selkoe, D. J. & Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 8, 595–608. https://doi.org/10.15252/emmm.201606210 (2016).
Maloney, B. & Lahiri, D. K. The Alzheimer’s amyloid beta-peptide (Abeta) binds a specific DNA Abeta-interacting domain (AbetaID) in the APP, BACE1, and APOE promoters in a sequence-specific manner: characterizing a new regulatory motif. Gene 488, 1–12. https://doi.org/10.1016/j.gene.2011.06.004 (2011).
Huang, Y. A., Zhou, B., Nabet, A. M., Wernig, M. & Sudhof, T. C. Differential signaling mediated by ApoE2, ApoE3, and ApoE4 in human neurons parallels Alzheimer’s disease risk. J. Neurosci. 39, 7408–7427. https://doi.org/10.1523/JNEUROSCI.2994-18.2019 (2019).
Huang, Y. A., Zhou, B., Wernig, M. & Sudhof, T. C. ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Abeta secretion. Cell 168, 427–441e421. https://doi.org/10.1016/j.cell.2016.12.044 (2017).
Barucker, C. et al. Nuclear translocation uncovers the amyloid peptide Abeta42 as a regulator of gene transcription. J. Biol. Chem. 289, 20182–20191. https://doi.org/10.1074/jbc.M114.564690 (2014).
Fertan, E. et al. Cognitive decline, cerebral-spleen tryptophan metabolism, oxidative stress, cytokine production, and regulation of the txnip gene in a triple transgenic mouse model of Alzheimer disease. Am. J. Pathol. 189, 1435–1450. https://doi.org/10.1016/j.ajpath.2019.03.006 (2019).
Ahmadieh, H. & Azar, S. T. Liver disease and diabetes: Association, pathophysiology, and management. Diabetes Res. Clin. Pract. 104, 53–62. https://doi.org/10.1016/j.diabres.2014.01.003 (2014).
Teo, E. et al. Metabolic stress is a primary pathogenic event in transgenic Caenorhabditis elegans expressing pan-neuronal human amyloid beta. Elife https://doi.org/10.7554/eLife.50069 (2019).
Grabuschnig, S. et al. Putative origins of cell-free DNA in humans: A review of active and passive nucleic acid release mechanisms. Int. J. Mol. Sci. 21, 66. https://doi.org/10.3390/ijms21218062 (2020).
Ranucci, R. Cell-free DNA: Applications in different diseases. Methods Mol. Biol. 1909, 3–12. https://doi.org/10.1007/978-1-4939-8973-7_1 (2019).
Esquerda-Canals, G., Montoliu-Gaya, L., Guell-Bosch, J. & Villegas, S. Mouse models of Alzheimer’s disease. J. Alzheimers Dis. 57, 1171–1183. https://doi.org/10.3233/JAD-170045 (2017).
Tai, L. M., Maldonado Weng, J., LaDu, M. J. & Brady, S. T. Relevance of transgenic mouse models for Alzheimer’s disease. Prog. Mol. Biol. Transl. Sci. 177, 1–48. https://doi.org/10.1016/bs.pmbts.2020.07.007 (2021).
Watamura, N., Sato, K. & Saido, T. C. Mouse models of Alzheimer’s disease for preclinical research. Neurochem. Int. 158, 105–361. https://doi.org/10.1016/j.neuint.2022.105361 (2022).
Oakley, H. et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140. https://doi.org/10.1523/JNEUROSCI.1202-06.2006 (2006).
Gendron, W. H. et al. Age related weight loss in female 5xFAD mice from 3 to 12 months of age. Behav. Brain Res. 406, 113214. https://doi.org/10.1016/j.bbr.2021.113214 (2021).
Fertan, E. & Brown, R. E. Age-related deficits in working memory in 5xFAD mice in the Hebb–Williams maze. Behav. Brain Res. 424, 113806. https://doi.org/10.1016/j.bbr.2022.113806 (2022).
Kimura, R. & Ohno, M. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol. Dis. 33, 229–235. https://doi.org/10.1016/j.nbd.2008.10.006 (2009).
Jawhar, S., Trawicka, A., Jenneckens, C., Bayer, T. A. & Wirths, O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer’s disease. Neurobiol. Aging 33(196), e129-140. https://doi.org/10.1016/j.neurobiolaging.2010.05.027 (2012).
O’Leary, T. P., Mantolino, H. M., Stover, K. R. & Brown, R. E. Age-related deterioration of motor function in male and female 5xFAD mice from 3 to 16 months of age. Genes Brain Behav. 19, e12538. https://doi.org/10.1111/gbb.12538 (2020).
Troncoso-Escudero, P. & Vidal, R. L. Insulin-like growth factor 2: Beyond its role in hippocampal-dependent memory. J. Cell Immunol. 3, 46–52 (2021).
Cianfarani, S. Insulin-like growth factor-II: New roles for an old actor. Front Endocrinol. 3, 118. https://doi.org/10.3389/fendo.2012.00118 (2012).
Livingstone, C. & Borai, A. Insulin-like growth factor-II: Its role in metabolic and endocrine disease. Clin. Endocrinol. 80, 773–781. https://doi.org/10.1111/cen.12446 (2014).
Holly, J. M. P., Biernacka, K. & Perks, C. M. The neglected insulin: IGF-II, a metabolic regulator with implications for diabetes, obesity, and cancer. Cells https://doi.org/10.3390/cells8101207 (2019).
White, V. et al. IGF2 stimulates fetal growth in a sex- and organ-dependent manner. Pediatr. Res. 83, 183–189. https://doi.org/10.1038/pr.2017.221 (2018).
Hertze, J., Nagga, K., Minthon, L. & Hansson, O. Changes in cerebrospinal fluid and blood plasma levels of IGF-II and its binding proteins in Alzheimer’s disease: An observational study. BMC Neurol. 14, 64. https://doi.org/10.1186/1471-2377-14-64 (2014).
Mellott, T. J., Pender, S. M., Burke, R. M., Langley, E. A. & Blusztajn, J. K. IGF2 ameliorates amyloidosis, increases cholinergic marker expression and raises BMP9 and neurotrophin levels in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE 9, e94287. https://doi.org/10.1371/journal.pone.0094287 (2014).
Pascual-Lucas, M. et al. Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol. Med. 6, 1246–1262. https://doi.org/10.15252/emmm.201404228 (2014).
Cheng, B. & Mattson, M. P. IGF-I and IGF-II protect cultured hippocampal and septal neurons against calcium-mediated hypoglycemic damage. J. Neurosci. 12, 1558–1566 (1992).
Chen, D. Y. et al. A critical role for IGF-II in memory consolidation and enhancement. Nature 469, 491–497. https://doi.org/10.1038/nature09667 (2011).
Stern, S. A., Chen, D. Y. & Alberini, C. M. The effect of insulin and insulin-like growth factors on hippocampus- and amygdala-dependent long-term memory formation. Learn. Mem. 21, 556–563. https://doi.org/10.1101/lm.029348.112 (2014).
Steinmetz, A. B., Johnson, S. A., Iannitelli, D. E., Pollonini, G. & Alberini, C. M. Insulin-like growth factor 2 rescues aging-related memory loss in rats. Neurobiol. Aging 44, 9–21. https://doi.org/10.1016/j.neurobiolaging.2016.04.006 (2016).
Bartolomei, M. S., Zemel, S. & Tilghman, S. M. Parental imprinting of the mouse H19 gene. Nature 351, 153–155. https://doi.org/10.1038/351153a0 (1991).
DeChiara, T. M., Robertson, E. J. & Efstratiadis, A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64, 849–859. https://doi.org/10.1016/0092-8674(91)90513-x (1991).
Ferguson-Smith, A. C., Cattanach, B. M., Barton, S. C., Beechey, C. V. & Surani, M. A. Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nature 351, 667–670. https://doi.org/10.1038/351667a0 (1991).
Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 15, 517–530. https://doi.org/10.1038/nrg3766 (2014).
Ferguson-Smith, A. C. & Bourc’his, D. The discovery and importance of genomic imprinting. Elife https://doi.org/10.7554/eLife.42368 (2018).
Lawson, H. A., Cheverud, J. M. & Wolf, J. B. Genomic imprinting and parent-of-origin effects on complex traits. Nat. Rev. Genet. 14, 609–617. https://doi.org/10.1038/nrg3543 (2013).
Tucci, V., Isles, A. R., Kelsey, G., Ferguson-Smith, A. C. & Erice Imprinting, G. Genomic imprinting and physiological processes in mammals. Cell 176, 952–965. https://doi.org/10.1016/j.cell.2019.01.043 (2019).
Zemel, S., Bartolomei, M. S. & Tilghman, S. M. Physical linkage of two mammalian imprinted genes, H19 and insulin-like growth factor 2. Nat. Genet. 2, 61–65. https://doi.org/10.1038/ng0992-61 (1992).
Kaffer, C. R., Grinberg, A. & Pfeifer, K. Regulatory mechanisms at the mouse Igf2/H19 locus. Mol. Cell Biol. 21, 8189–8196. https://doi.org/10.1128/MCB.21.23.8189-8196.2001 (2001).
Thorvaldsen, J. L., Duran, K. L. & Bartolomei, M. S. Deletion of the H19 differentially methylated domain results in loss of imprinted expression of H19 and Igf2. Genes Dev. 12, 3693–3702. https://doi.org/10.1101/gad.12.23.3693 (1998).
Kaffer, C. R. et al. A transcriptional insulator at the imprinted H19/Igf2 locus. Genes Dev. 14, 1908–1919 (2000).
Srivastava, M. et al. H19 and Igf2 monoallelic expression is regulated in two distinct ways by a shared cis acting regulatory region upstream of H19. Genes Dev. 14, 1186–1195 (2000).
Nordin, M., Bergman, D., Halje, M., Engstrom, W. & Ward, A. Epigenetic regulation of the Igf2/H19 gene cluster. Cell Prolif. 47, 189–199. https://doi.org/10.1111/cpr.12106 (2014).
Sasaki, H. et al. Parental imprinting: potentially active chromatin of the repressed maternal allele of the mouse insulin-like growth factor II (Igf2) gene. Genes Dev. 6, 1843–1856. https://doi.org/10.1101/gad.6.10.1843 (1992).
Ariel, M. et al. Allele-specific structures in the mouse Igf2-H19 domain. Cold Spring Harb. Symp. Quant. Biol. 58, 307–313 (1993).
Feil, R., Walter, J., Allen, N. D. & Reik, W. Developmental control of allelic methylation in the imprinted mouse Igf2 and H19 genes. Development 120, 2933–2943 (1994).
Moore, T. et al. Multiple imprinted sense and antisense transcripts, differential methylation and tandem repeats in a putative imprinting control region upstream of mouse Igf2. Proc. Natl. Acad. Sci. USA 94, 12509–12514. https://doi.org/10.1073/pnas.94.23.12509 (1997).
Reik, W. et al. Imprinting mutations in the Beckwith-Wiedemann syndrome suggested by altered imprinting pattern in the IGF2-H19 domain. Hum. Mol. Genet. 4, 2379–2385. https://doi.org/10.1093/hmg/4.12.2379 (1995).
Constancia, M. et al. Deletion of a silencer element in Igf2 results in loss of imprinting independent of H19. Nat. Genet. 26, 203–206. https://doi.org/10.1038/79930 (2000).
Murrell, A. et al. An intragenic methylated region in the imprinted Igf2 gene augments transcription. EMBO Rep. 2, 1101–1106. https://doi.org/10.1093/embo-reports/kve248 (2001).
Sullivan, M. J., Taniguchi, T., Jhee, A., Kerr, N. & Reeve, A. E. Relaxation of IGF2 imprinting in Wilms tumours associated with specific changes in IGF2 methylation. Oncogene 18, 7527–7534. https://doi.org/10.1038/sj.onc.1203096 (1999).
Cui, H. et al. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 62, 6442–6446 (2002).
Selenou, C., Brioude, F., Giabicani, E., Sobrier, M. L. & Netchine, I. IGF2: Development, genetic and epigenetic abnormalities. Cells 11, 66. https://doi.org/10.3390/cells11121886 (2022).
Gregg, C. et al. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329, 643–648. https://doi.org/10.1126/science.1190830 (2010).
Zamarbide, M. et al. Maternal imprinting on cognition markers of wild type and transgenic Alzheimer’s disease model mice. Sci. Rep. 8, 6434. https://doi.org/10.1038/s41598-018-24710-7 (2018).
O’Leary, T. P. & Brown, R. E. Visuo-spatial learning and memory impairments in the 5xFAD mouse model of Alzheimer’s disease: Effects of age, sex, albinism, and motor impairments. Genes Brain Behav. 21, e12794. https://doi.org/10.1111/gbb.12794 (2022).
Rae, E. A. & Brown, R. E. The problem of genotype and sex differences in life expectancy in transgenic AD mice. Neurosci. Biobehav. Rev. 57, 238–251. https://doi.org/10.1016/j.neubiorev.2015.09.002 (2015).
Kilkenny, C., Browne, W. J., Cuthill, I. C., Emerson, M. & Altman, D. G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8, e1000412. https://doi.org/10.1371/journal.pbio.1000412 (2010).
Percie du Sert, N. et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biol. 18, e3000410. https://doi.org/10.1371/journal.pbio.3000410 (2020).
Khachaturian, Z. S. Diagnosis of Alzheimer’s disease. Arch. Neurol. 42, 1097–1105. https://doi.org/10.1001/archneur.1985.04060100083029 (1985).
Balducci, C. et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 107, 2295–2300. https://doi.org/10.1073/pnas.0911829107 (2010).
Fertan, E., Wong, A. A., Purdon, M. K., Weaver, I. C. G. & Brown, R. E. The effect of background strain on the behavioral phenotypes of the MDGA2(+/−) mouse model of autism spectrum disorder. Genes Brain Behav. 20, e12696. https://doi.org/10.1111/gbb.12696 (2021).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 (2001).
Kennedy, B. E., Hundert, A. S., Goguen, D., Weaver, I. C. G. & Karten, B. Presymptomatic alterations in amino acid metabolism and DNA methylation in the cerebellum of a murine model of Niemann–Pick type C disease. Am. J. Pathol. 186, 1582–1597. https://doi.org/10.1016/j.ajpath.2016.02.012 (2016).
du Prel, J. B., Hommel, G., Rohrig, B. & Blettner, M. Confidence interval or p-value? Part 4 of a series on evaluation of scientific publications. Dtsch. Arztebl. Int. 106, 335–339. https://doi.org/10.3238/arztebl.2009.0335 (2009).
O’Brien, S. F. & Yi, Q. L. How do I interpret a confidence interval?. Transfusion 56, 1680–1683. https://doi.org/10.1111/trf.13635 (2016).
Faul, F., Erdfelder, E., Lang, A. G. & Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191. https://doi.org/10.3758/bf03193146 (2007).
Bonham, L. W. et al. Insulin-like growth factor binding protein 2 is associated with biomarkers of Alzheimer’s disease pathology and shows differential expression in transgenic mice. Front. Neurosci. 12, 476. https://doi.org/10.3389/fnins.2018.00476 (2018).
Eimer, W. A. & Vassar, R. Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegener. 8, 2. https://doi.org/10.1186/1750-1326-8-2 (2013).
D’Mello, C. & Swain, M. G. Liver-brain inflammation axis. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G749-761. https://doi.org/10.1152/ajpgi.00184.2011 (2011).
Barucker, C. et al. Alzheimer amyloid peptide abeta42 regulates gene expression of transcription and growth factors. J. Alzheimers Dis. 44, 613–624. https://doi.org/10.3233/JAD-141902 (2015).
Andersen, J. V. et al. Hippocampal disruptions of synaptic and astrocyte metabolism are primary events of early amyloid pathology in the 5xFAD mouse model of Alzheimer’s disease. Cell Death Dis. 12, 954. https://doi.org/10.1038/s41419-021-04237-y (2021).
Liang, W. S. et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 105, 4441–4446. https://doi.org/10.1073/pnas.0709259105 (2008).
Wang, C. et al. Insulin-like growth factor 2 regulates the proliferation and differentiation of rat adipose-derived stromal cells via IGF-1R and IR. Cytotherapy 21, 619–630. https://doi.org/10.1016/j.jcyt.2018.11.010 (2019).
Alfares, M. N., Perks, C. M., Hamilton-Shield, J. P. & Holly, J. M. P. Insulin-like growth factor-II in adipocyte regulation: depot-specific actions suggest a potential role limiting excess visceral adiposity. Am. J. Physiol. Endocrinol. Metab. 315, E1098–E1107. https://doi.org/10.1152/ajpendo.00409.2017 (2018).
Ito, Y. et al. Age-related changes in the hepatic microcirculation in mice. Exp. Gerontol. 42, 789–797. https://doi.org/10.1016/j.exger.2007.04.008 (2007).
Zoli, M. et al. Total and functional hepatic blood flow decrease in parallel with ageing. Age Ageing 28, 29–33. https://doi.org/10.1093/ageing/28.1.29 (1999).
Cai, H. et al. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 9, 5–17. https://doi.org/10.2174/156720512799015064 (2012).
Issa, J. P., Vertino, P. M., Boehm, C. D., Newsham, I. F. & Baylin, S. B. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc. Natl. Acad. Sci. USA 93, 11757–11762. https://doi.org/10.1073/pnas.93.21.11757 (1996).
Kelijman, M. Age-related alterations of the growth hormone/insulin-like-growth-factor I axis. J. Am. Geriatr. Soc. 39, 295–307. https://doi.org/10.1111/j.1532-5415.1991.tb01654.x (1991).
Bartke, A. et al. Insulin-like growth factor 1 (IGF-1) and aging: Controversies and new insights. Biogerontology 4, 1–8. https://doi.org/10.1023/a:1022448532248 (2003).
Faisal, M., Kim, H. & Kim, J. Sexual differences of imprinted genes’ expression levels. Gene 533, 434–438. https://doi.org/10.1016/j.gene.2013.10.006 (2014).
Citron, M. et al. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl. Acad. Sci. USA 91, 11993–11997 (1994).
Marques, M. A. et al. Peripheral amyloid-beta levels regulate amyloid-beta clearance from the central nervous system. J. Alzheimers Dis. 16, 325–329. https://doi.org/10.3233/JAD-2009-0964 (2009).
Baldeiras, I. et al. Peripheral oxidative damage in mild cognitive impairment and mild Alzheimer’s disease. J. Alzheimers Dis. 15, 117–128 (2008).
Hong, W. K. et al. Amyloid-beta-peptide reduces the expression level of mitochondrial cytochrome oxidase subunits. Neurochem. Res. 32, 1483–1488. https://doi.org/10.1007/s11064-007-9336-7 (2007).
Garcia-Huerta, P. et al. Insulin-like growth factor 2 (IGF2) protects against Huntington’s disease through the extracellular disposal of protein aggregates. Acta Neuropathol. 140, 737–764. https://doi.org/10.1007/s00401-020-02183-1 (2020).
LeRoith, D., Holly, J. M. P. & Forbes, B. E. Insulin-like growth factors: Ligands, binding proteins, and receptors. Mol. Metab. 52, 101245 (2021).
Knight, E. M., Verkhratsky, A., Luckman, S. M., Allan, S. M. & Lawrence, C. B. Hypermetabolism in a triple-transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 33, 187–193. https://doi.org/10.1016/j.neurobiolaging.2010.02.003 (2012).
Giedraitis, V. et al. The normal equilibrium between CSF and plasma amyloid beta levels is disrupted in Alzheimer’s disease. Neurosci. Lett. 427, 127–131. https://doi.org/10.1016/j.neulet.2007.09.023 (2007).
De, S. et al. Different soluble aggregates of Abeta42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 10, 1541. https://doi.org/10.1038/s41467-019-09477-3 (2019).
Nagele, R. G., D’Andrea, M. R., Anderson, W. J. & Wang, H. Y. Intracellular accumulation of beta-amyloid(1–42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110, 199–211. https://doi.org/10.1016/s0306-4522(01)00460-2 (2002).
Latty, S. L. et al. Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub. Elife https://doi.org/10.7554/eLife.31377 (2018).
Hughes, C. et al. Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun. Biol. 3, 79. https://doi.org/10.1038/s42003-020-0792-9 (2020).
Smith, A. R. et al. The histone modification H3K4me3 is altered at the ANK1 locus in Alzheimer’s disease brain. Future Sci. OA 7, FSO665. https://doi.org/10.2144/fsoa-2020-0161 (2021).
Lu, X., Wang, L., Yu, C., Yu, D. & Yu, G. Histone acetylation modifiers in the pathogenesis of Alzheimer’s disease. Front. Cell Neurosci. 9, 226. https://doi.org/10.3389/fncel.2015.00226 (2015).
Ding, H., Dolan, P. J. & Johnson, G. V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 106, 2119–2130. https://doi.org/10.1111/j.1471-4159.2008.05564.x (2008).
Klein, H. U. et al. Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nat. Neurosci. 22, 37–46. https://doi.org/10.1038/s41593-018-0291-1 (2019).
Acknowledgements
We would like to thank Dr. Chris Sinal (Department of Pharmacology, Dalhousie University) and his lab for their assistance with animal genotyping, Dr. Tamara Franklin (Department of Psychology, Dalhousie University) and her lab for their assistance with animal generation, and Mr. Andrew Reid (Department of Medical Neuroscience, Dalhousie University) for their assistance at the Maritime Brain Tissue Bank.
Funding
This work was supported by Discovery Grants from the Natural Sciences and Engineering Research Council of Canada to I.C.G.W. (Grant RGPIN-2022-05208), R.E.B. (Grant RG7441) as well as Research Funding from Department of Psychiatry to I.C.G.W., and a Multi-Investigator Research Initiative from Brain Canada to R.E.B.
Author information
Authors and Affiliations
Contributions
E.F. and I.C.G.W.: Conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript. W.H.G., A.A.W., and G.M.H.: collection and/or assembly of data. R.E.B.: Conception and design, manuscript writing, final approval of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fertan, E., Gendron, W.H., Wong, A.A. et al. Noncanonical regulation of imprinted gene Igf2 by amyloid-beta 1–42 in Alzheimer’s disease. Sci Rep 13, 2043 (2023). https://doi.org/10.1038/s41598-023-29248-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-29248-x
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
