
Abstract
Dental enamel is the highly mineralized tissue covering the tooth surface and is formed by ameloblasts. Ameloblasts have been known to be impossible to detect in adult tooth because they are shed by apoptosis during enamel maturation and tooth eruption. Owing to these, little was known about appropriate cell surface markers to isolate ameloblast-like cells in tissues. To overcome these problems, epithelial cells were selectively cultivated from the gingival tissues and used as a stem cell source for ameloblastic differentiation. When gingival epithelial cells were treated with a specified concentration of BMP2, BMP4, and TGFβ-1, the expression of ameloblast-specific markers was increased, and both the MAPK and Smad signaling pathways were activated. Gingival epithelial cells differentiated into ameloblast-like cells through epithelial-mesenchymal transition. By RNA-Seq analysis, we reported 20 ameloblast-specific genes associated with cell surface, cell adhesion, and extracellular matrix function. These cell surface markers might be useful for the detection and isolation of ameloblast-like cells from dental tissues.
Similar content being viewed by others


Introduction
Dentin, dental pulp, periodontal ligament, and dental enamel are developed by reciprocal interactions between dental epithelium and ectomesenchyme. Neural crest cell-derived ectomesenchyme differentiates into odontoblasts, periodontal ligament progenitors, cementoblasts, as well as various fibroblasts. On the other hand, enamel-forming ameloblasts differentiate from epithelial cells originating from oral ectoderm. In the process of enamel formation, the inner enamel epithelium differentiates into ameloblasts1. Ameloblastic differentiation possibly occurs after the initial dentin matrix protein secretion and deposition by odontoblasts2,3. The enamel matrix proteins (EMPs) are degraded by various proteinases secreted by ameloblasts and replaced by minerals during the maturation stage4. Hertwig’s epithelial root sheath/epithelial cell rests of Malassez (HERS/ERM) have been reported to be a unique epithelial cell source5,6. Bone marrow stromal cells, embryonic stem cells, and skin epithelial cells are alternative sources for the construction of ameloblasts7. Induction mechanism of various progenitors is strictly regulated by growth factors and cytokines, such as TGFs, FGFs, Wnts, and BMPs, as well as the extracellular matrix in the epithelium and mesenchyme8,9. In ameloblastic differentiation, BMP2 and BMP4 are secreted by ectomesenchymal odontoblasts and play important roles in the expression of EMPs and terminal differentiation of ameloblasts10,11. Ameloblast differentiation is prevented by follistatin by antagonizing the inductive effect of BMP4 from the odontoblasts. The expression of follistatin is shown to be induced by activin A from the overlying mesenchymal follicle cells. Thus, a balance between BMP4 and activin A, is required for proper ameloblast differentiation12. In addition, knockout of a BMP receptor, Bmpr1a/ALK3, causes defective enamel formation on tooth crowns13. Besides BMPs, TGFβ-1 stimulates the expression and secretion of EMPs in ameloblasts. The inhibition of the TGFβ-1 signaling pathway causes tooth and enamel malformations14,15. The Smad signaling is known as an intracellular canonical pathway activated by TGF-β superfamily members through a heteromeric receptor complex, comprised of type I and type II receptors16,17. According to the activation of receptors by TGFβ-1 and BMPs, Smad2/3 and Smad1/5/8, which are known as the regulatory Smads (R-Smads) are phosphorylated, respectively, and then, a complex of phospho-R-Smads and Smad4 regulates the expression of target genes in the nucleus18,19.
In this study, we isolated and characterized the epithelial cells from human gingival tissue, which is comparatively easy to obtain, and successfully induced differentiation into ameloblast-like cells through epithelial-mesenchymal transition. In addition, we revealed potential surface markers of ameloblast-like cells, which are categorized into those involved in cell adhesion and extracellular matrix functions.
Results
Culture of the epithelial cells derived from human gingival tissue
To establish ameloblast-like cells from commonly available dental tissue, we at first attempted to isolate the epithelial cells from gingival tissue of ten donors (Fig. 1). Fibroblastic cells mostly grew out from gingival tissue under continuous culture in α-MEM/20% FBS. However, gingival epithelial cells were obtained within 1–2 weeks through selective transfer culture in a serum-free keratinocyte growth medium. During selective culture, residual fibroblastic cells were selectively eliminated by treatment with a low concentration of trypsin. The gingival fibroblasts exhibited bipolar fibroblastic shapes, whereas the gingival epithelial cells exhibited polygonal shapes that are a typical cellular morphology of epithelial cells (Fig. 2A). The expression of vimentin, a typical fibroblast marker, dramatically decreased in epithelial cells (Fig. 2B). Integrin α-6, EpCAM, and p75NTR have been used as epithelial stem cell markers in human HERS/ERM and ectomesenchymal stem cells20,21. The expressions of EpCAM, integrin α-6, and p75NTR were 8.9, 2.3, and 1.9 times greater in gingival epithelial cells than in gingival fibroblasts, respectively (Fig. 2C, a & b). On the other hand, the expressions of CD44, CD73, CD90, and CD146, which are known as mesenchymal stem cell markers22,23,24,25, in gingival epithelial cells were 5.5, 8.0, 16.7, and 3.9 times lower than those in fibroblasts, respectively (Fig. 2C, c & d). These results indicated that although most of the primary cells cultured from adult gingival tissue grew as fibroblasts, epithelial cells present in small amounts can be grown using selective trypsinization and specific medium.

Schematic strategy of the epithelial cell culture derived from human gingival tissue. The detailed scheme of the selective cell culture process was described in Materials and Methods.

Characterization of the gingival epithelial cells cultured from human gingival tissue. (A) Microscopic observation of cellular morphology of the gingival fibroblasts (GF) (a) and the gingival epithelial cells (GE) (b). (B) Expression of endogenous vimentin, a fibroblastic cell marker in GF and GE. Total cell extracts were analyzed on SDS-PAGE, and endogenous vimentin was detected by western blot analysis with anti-vimentin antibody (α-vimentin). For normalization of protein amounts used, actin was detected by anti-actin antibody (α-actin). (C) Immunophenotyping of cells by using epithelial and mesenchymal cell markers. Intact cells harvested by non-enzymatic method were incubated with the primary antibodies, and expression of the cell surface antigens was analyzed by FACS as described in Materials and Methods. The mean fluorescence values from the FACS data were estimated from peak data using Cell Quest software and the WinMDI 2.9 program. a & b, FACS histogram and mean fluorescence value on the expression of epithelial cell markers. b & d, FACS histogram and mean fluorescence value on the expression of mesenchymal cell markers. The data were originated from a cell line of 4 individual cultures of gingival fibroblast (A, B, a & c in C), and from average value of 4 individual cultures (b & d in C). *P < 0.1, **P < 0.01, or ***P < 0.001 was determined by using Student t-test.
Co-treatment with BMPs and TGFβ-1 induces ameloblastic differentiation of gingival epithelial cells
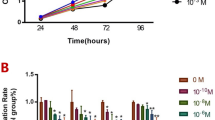
When gingival epithelial cells were treated with 100 ng/ml BMP4 or 100 ng/ml BMP4 and BMP2 for 7 days, morphological phenotype did not change much from the initial epithelial phenotype (Fig. 3A, panels a–c). However, bipolar fibroblastic phenotypes appeared when gingival epithelial cells were treated with TGFβ-1 alone (Fig. 3A, panel d) and treated with BMP4, BMP2, and TGFβ-1 simultaneously (Fig. 3A, panel e). Under these conditions, the expressions of ameloblast-specific markers such as amelogenin, enamelin, and ameloblastin was notably increased in cells co-treated with BMP4, BMP2, and TGFβ-1 by 1.7, 4.7, and 15.9 times, respectively (Fig. 3B, bar 5 in a–c) compared with those in cells treated individually or with BMPs only (Fig. 3B, bars 2–4 in a–c). Although we also analyzed the mRNA expressions of KLK4 and MMP20, which are known as the enamel specific markers, these gene expressions were much increased by co-treatment with BMP2, 4, and TGFβ-1 (Fig. 3B, g & h). As is the case in ameloblast-specific markers, the expressions of osteogenic markers were also highly increased in cells co-treated with BMP4, BMP2, and TGFβ-1 (Fig. 3B, bar 5 in d–f) than in cells treated individually or with BMPs only (Fig. 3B, bars 2–4 in d–f). Because tooth enamel is a highly mineralized tissue, we analyzed the ALP activity and mineralization efficiency to prove the efficiency of enamel formation in ameloblast-like cells. After treatment with BMPs and/or TGFβ-1 for 7 days, cells were cultured in a medium containing ascorbic acid, β-glycerophosphate, and dexamethasone for the induction of mineralization. Although the ALP activity was not much increased in epithelial cells treated with BMPs or TGFβ-1 only (Fig. 3C, bars 2 & 3), it was highly increased by 2.8 times on co-treatment (Fig. 3C, bars 1 & 4). In addition, when cells were stained by alizarin red for analyzing mineralization efficiency, the intensity was highly increased in cells co-treated with BMPs and TGFβ-1 (Figs 3D, 4 in a & b). These results suggested that gingival epithelial cells are efficiently cytodifferentiated into ameloblast-like progenitors by co-treatment with BMPs and TGFβ-1 in a synergistic manner.

Co-treatment of BMP2, BMP4, and TGFβ-1 induces ameloblastic cytodifferentiation in the gingival epithelial cells. (A) Microscopic observation of cellular morphology of cells treated with cytokines for 7 days. a, cells without treatment; b, cells treated with BMP4; c, cells co-treated with BMP4 and BMP2; d, cells treated with TGFβ-1; e, cells co-treated with BMP4, BMP2, and TGFβ-1. (B) Relative mRNA expressions of amelogenic and osteogenic markers in cells treated with cytokines. mRNA expression was analyzed by qPCR as described in the Materials and Methods. See also Table S1. a, expression of amelogenin; b, expression of enamelin; c, expression of ameloblastin; d, expression of bone sialoprotein; e, expression of osteocalcin; f, expression of osteopontin; g, expression of KLK4; h, expression of MMP20. 1, control without treatment; 2, treatment with BMP2; 3, treatment with BMP4; 4, co-treatment with BMP4 and BMP2; 5, treatment with TGFβ-1; 6, co-treatment with BMP4, BMP2, and TGFβ-1. The data were originated from average value of 3 individual cultures. Statistical significance of *P < 0.1, **P < 0.01, or ***P < 0.001 was determined by using Student t-test. (C,D) Estimation of alkaline phosphatase activity and mineralization efficiency. Epithelial cells were incubated in KGM containing 50 μg/ml ascorbic acid, 10 mM β-glycerophosphate, and 5 μM dexamethasone for 7~14 days after ameloblastic cytodifferentiation. The data were originated from average value of 2 individual cultures. Statistical significance of *P < 0.1, **P < 0.01, or ***P < 0.001 was determined by using Student t-test. 1, without treatment; 2, co-treatment with BMP4 and BMP2; 3, treatment with TGFβ-1; 4, co-treatment with BMP4, BMP2 & TGFβ-1; 5, control of buffer only.

Epithelial-mesenchymal transition is occurred in the gingival epithelial cells during ameloblastic cytodifferentiation. (A) Expression of vimentin, a mesenchymal fibroblast marker in cells treated with cytokines for 7 days. Endogenous vimentin (α-vimentin) and actin (α-actin) were detected by western blot analysis. 1, control without treatment; 2, co-treatment with BMP4 & BMP2; 3, treatment with TGFβ-1; 4, co-treatment with BMP4, BMP2, & TGFβ-1. (B) Immunophenotyping of the gingival epithelial cells treated with cytokines. Expression of the mesenchymal cell surface was analyzed by FACS analysis after incubation with anti-CD44, anti-CD73, anti-CD90, and anti-CD146 antibodies. a, FACS histograms; b, mean fluorescence calculated from FACS data. This FACS histogram was representative of one of the three experiments using three individual cultures of gingival epithelial cells.
As shown in Fig. 2B, when gingival epithelial cells were treated with TGFβ-1 alone or BMPs and TGFβ-1, the polygonal phenotype was changed into a fibroblastic shape, and the expression of vimentin, a fibroblastic marker, was increased (Fig. 4A, lanes 3 & 4 in upper panel). As expected, the expressions of mesenchymal cell markers, CD44, CD73, CD90, and CD146, were notably increased in cells treated with TGFβ-1 alone or with BMPs and TGFβ-1 (Fig. 4B, a & b). However, the expressions of epithelial stem cell markers, EpCAM, p75NTR, and integrin α-6, were unchanged by treatment with TGFβ-1 alone or with BMPs and TGFβ-1 (data not shown). These findings suggested that gingival epithelial cells are transformed into fibroblastic cells by TGFβ-1 or BMPs and TGFβ-1. Although treatment with TGFβ-1 alone seemed to be sufficient to induce the mesenchymal transition of epithelial cells, the expression of ameloblast-specific genes was efficiently stimulated by co-treatment with BMPs and TGFβ-1 (Fig. 3B). During ameloblastic induction, the activations of Smad and p38 mitogen-activated protein kinase (MAPK) were examined. Both Smad1/5/8 and Smad3 and p38 were highly phosphorylated in cells treated with BMPs alone or BMPs and TGFβ-1 (Supplementary Fig. 1), indicated that the non-canonical MAPK pathway as well as the canonical pathway might be stimulated by BMPs and TGFβ-1 during the epithelial-mesenchymal transition for the ameloblastic cytodifferentiation of epithelial cells.
Characterization of ameloblast-specific cell surface markers through RNA-Seq based transcriptome analysis
To perform a comparative analysis of the gene expression profiles during ameloblastic differentiation, four different states of cells were used: gingival fibroblasts (GF), gingival fibroblasts treated with BMPs/TGFβ-1 (dGF), gingival epithelial cells (GE), and gingival epithelial cells treated with BMPs/TGFβ-1 (dGE/amelo). For verification of the cell states before analysis, ameloblast-specific markers were validated by qPCR. As expected, the expressions of amelogenin, ameloblastin, and OPN in GF were lower than those in GE without treatment with cytokines (Fig. 5A, bars 1 & 3 in a–c). In contrast, treatment with BMPs/TGFβ-1 did not induce gene expression in GF (Fig. 5A, bars 1 & 2 in a–c), while the expressions of amelogenin, ameloblastin, and OPN were increased in GE treated with BMPs/TGFβ-1 (Fig. 5A, bars 3 & 4 in a–c). On the basis of the above results, GE treated with BMPs/TGFβ-1 was considered as ameloblast-like cells. RNA-Seq was performed to compare the gene expression profiling of the four different states of cells. According to Ensembl gene annotation, the expression of 18,313 genes in the four different cell types were identified, and the genes with log2FPKM ≥ 1 which are mainly expressed in four different cell states were used for hierarchical clustering analysis (Supplementary Table S2 and Supplementary Fig. S1A). The levels of gene expression showed a uniform distribution in all cell types, indicating the reliability of our data (Supplementary Fig. S1B). We established three pairwise comparisons as follows: dGE/amelo vs. GF, dGE/amelo vs. dGF, and dGE/amelo vs. GE. Totals of 1531 genes (700 up- and 831 down-regulated) and 1402 genes (816 up- and 586 down-regulated) were differentially expressed in dGE/amelo in comparison with GF and dGF, respectively (Fig. 5B and Supplementary Fig. S2A,B). In the dGE/amelo vs. GE comparison, we observed 495 DEGs (250 up- and 245 down-regulated) (Fig. 5B and Supplementary Fig. S2C). Five hundred thirty-seven of 1195 genes and 470 of 1148 genes showed higher and lower expression, respectively, in dGE/amelo rather than in the other cells (Fig. 5B). Based on the gene expression data, we performed hierarchical clustering analysis using a total of 1007 DEGs in dGE/amelo (Fig. 5C). Particularly, 34 up- and 44 down-regulated genes were shared by all comparison groups (Fig. 5B and Supplementary Fig. S3). The GO classification for 470 down-regulated genes is related to the processes of “cell cycle”, “DNA replication”, “nuclear division”, and “spindle organization” (Supplementary Fig. S4), suggesting that gingival epithelial cells could not undergo proliferation when cells were stimulated with cytokines. The 238 of 537 up-regulated genes were significantly enriched and reclassified into eight major GO categories, namely, “cell surface”, “stimulus response”, “cytokine production”, “developmental process”, “extracellular organization”, “signal transduction”, “cell adhesion”, and “cell motility” (Supplementary Fig. S5). In contrast to the downregulation of proliferation-related genes, genes associated with the developmental process were up-regulated in dGE/amelo. We focused on GO categories such as “cell surface”, “extracellular organization”, and “cell adhesion” for the identification of ameloblast-specific cell surface markers. By analyzing the transcriptome signatures and biological implications of DEGs, we were able to identify 20 genes known as cell surface molecules, which might be associated with amelogenesis: ACHE, AMTN, CDH3, CDH8, CELSR1, CLDN4, COL17A1, DACT2, GPR56, ITGB6, JUP, LAMA3, LAMB3, LAMC2, MMP9, MMP15, TGM2, TNF, WNT7A, and WNT10A (Fig. 5D), and their expressions were checked again through qRT-PCR (Fig. 5E). As positive markers of ameloblasts, the expressions of ameloblastin, amelogenin, and enamelin were higher in ameloblast-like cells than in gingival epithelial cells. Most of the 20 markers were increased in ameloblast-like cells, although the expression changes of COL17A1, TNF, and WNT7A were not statistically significant.

Transcriptome analysis of ameloblast-like cells derived from gingival epithelial cells. (A) Validation of molecular differences among the fibroblasts and epithelial cells. Relative mRNA expressions of ameloblastic and osteogenic markers were analyzed by qPCR in different states of gingival cells. For differentiation, cells were treated with BMP4, BMP2, and TGFβ-1 for 7 days. a, expression of amelogenin; b, expression of ameloblastin; c, expression of osteopontin. 1 & 3, gene expression in cells without cytokines; 2 & 4, gene expression in cells treated with cytokines. Statistical significance of *P < 0.1, **P < 0.01, or ***P < 0.001 was determined by using Student t-test from three attempts performed by a limited culture number of 10 individual cultures. (B) The number of up- and down-regulated genes identified from the three comparison groups (GF vs dGE, dGF vs dGE, and GE vs dGE). Overlapping areas in Venn diagram represent common genes between the comparison groups. dGE/amelo, gingival epithelial cells treated with cytokines; GE, gingival epithelial cells without cytokines; dGF, gingival fibroblasts treated with cytokines; GF, gingival fibroblasts without cytokines. See also Table S2, Figs S1 and S2. (C) Heat map of 1007-differentially expressed genes (DEGs) from the pairwise comparison. Rows and columns represent 1007 DEGs (537 up- and 470 down-regulated genes) and profiled samples, respectively. The relative expression was depicted according to the color scale. See also Figs S3, S4, and S5. (D) Gene expression profile of the 20 genes selected by association with three major GO categories ‘cell surface’, ‘extracellular structure organization’, and ‘regulation of cell adhesion’. The heat map shows log2FPKM values for 20 selected genes (rows) and four different states of cell (columns). Three GO categories were arranged by hierarchical clustering at left columns (cell surface in green, extracellular structure organization in purple, and regulation of cell adhesion in olive). (E) Confirmation of individual gene expression by qRT-PCR. Statistical significance of *P < 0.1, **P < 0.01, or ***P < 0.001 was determined by using Student t-test. GE, gingival epithelial cells without cytokines; dGE/amelo, gingival epithelial cells treated with cytokines.
Discussion
So far, it has been difficult to study amelogenic differentiation because ameloblasts disappear due to apoptosis during tooth eruption and enamel epithelium remains fully mineralized in the human tooth. For this reason, most studies on amelogenic differentiation have been performed using rat and mouse incisors. Although the epithelial cells having the capacity to differentiate into ameloblasts have been cultured from root epithelium cell lineages such as HERS/ERM26,27, these tissues are generally not easy to obtain from intact teeth extracted from human adults. To secure human ameloblasts, at first, we developed the epithelial cell culture procedure using human dental gingival tissue, which is a relatively accessible dental tissue. Gingival fibroblasts and gingival epithelial cells turned out to have completely different cellular and biochemical features (Figs 2 and 5A). Interestingly, although these gingival epithelial cells were still expressing mesenchymal markers (Fig. 2C, c & d), this phenomenon was also seen in cells originating from ERM from periodontal ligament21. For ameloblastic cytodifferentiation, gingival epithelial cells were treated with BMP2, BMP4, and/or TGFβ-1. Treatment with TGFβ-1 alone induced the epithelial-mesenchymal transition (Fig. 3A & bar 4 in B), but when co-treated with BMPs and TGFβ-1, the expression of ameloblast-specific genes was apparently increased, as was the ALP activity (Fig. 3B,C).
Although ameloblast-like phenotypes were induced in gingival epithelial cells, we were unable to conclude that gingival epithelial cells treated BMPs/TGF could have the same molecular signature as real human ameloblasts. It is impossible for culture the real ameloblasts from adult tooth sample, but there may be another way to isolate ameloblasts from tooth germ of third molar.
A recent study demonstrated that epithelial cells undergo the epithelial-mesenchymal transition in the presence of TGFβ-1 during differentiation to hard tissues such as cementum21,27,28. Our data indicated that BMPs accelerate the epithelial-mesenchymal transition induced by TGFβ-1 and ameloblastic cytodifferentiation. TGFβ-1 and BMPs have a synergistic effect during amelogenesis (Figs 3 and 5A).
Previously, it was reported that TGFβ-1 suppresses the process of hard tissue formation stimulated by BMPs in mesenchymal progenitor cells29,30, suggesting that the BMP response might differ between mesenchymal and epithelial cells. The Activation of both Smad1/5/8 and Smad2/3 pathways is found in diverse epithelial lineages31. Smad pathways were stimulated when cells were treated with TGFβ-1/BMPs, but the phosphorylation of each Smad was not synergistically increased by co-treatment. In addition to the canonical Smad pathway, p38 phosphorylation was increased in epithelial cells treated with TGFβ-1/BMPs (Supplementary Fig. 1), suggesting that the MAPK pathway is also involved in oral epithelial and enamel formation.
Exploration of progenitor-specific cell surface markers and cell purification using these markers is important to supply a specific stem cell source for tissue regeneration. Although decoy immunization has many advantages in terms of directly obtaining the surface antibody32, this method has a downside that a large number of cells are needed. Because of this limitation, we performed RNA-Seq to analyze the gene expression profile of ameloblast-like cells. To further identify which genes are closely related to the character of the cell surface structure on ameloblasts, we focused on 20 genes associated with the cell surface, extracellular structure, and cell adhesion (Fig. 5D). ACHE, CELSR1, CLDN4, DACT2, LAMA3, LAMC2, WNT7A, and WNT10A have been reported in previous studies to have direct and indirect relevance to amelogenesis, enamel formation, epithelial cell adhesion, or stabilization of the extracellular matrix in tooth epithelium33,34,35,36,37,38,39,40,41,42,43,44,45,46. MMP9 and MMP15 are possibly involved in controlling enamel formation47,48, although information such as phenotype of MMP9 or MMP15 deficiency in animal model or mutation in humans was not reported so far. TNF is known to promote the transcription of the human amelotin (AMTN) gene encoding an enamel protein49,50,51, indicating that TNF expression is correlated with increased AMTN gene expression and leads to amelogenesis in gingival epithelial cells. Previously, an effort was made to characterize the transcriptome profile of ameloblasts, which were located in tooth buds of human fetuses and captured by laser-dissection52. This study reported that 21 genes were significantly increased in pre-secretory ameloblasts than in odontoblasts and secretory ameloblasts and 4 genes were specifically expressed in secretory ameloblasts based on RNA microarray data. Some of them were given a baseline of genes expressed by ameloblasts: dentin matrix protein-1, ameloblastin (expressed in pre-secretory ameloblasts), enamelin, and amelogenin (expressed in secretory ameloblasts), suggesting that the ameloblast-like cells that are differentiated from gingival epithelial cells in this study have the characteristics of ameloblasts (Fig. 5A,E). Although further validation is required, these 20 gene products and their specific antibodies could be useful as ameloblast-specific cell surface markers in future studies. The 20 genes selected are indeed important in amelogenesis and may be involved in amelogenesis imperfecta.
Materials and Methods
Cell cultures
Intact third molar teeth were collected from ten dental surgery patients aged 19~29 years old under guidelines approved by the Dankook Dental Hospital, and the informed consent for all experiments using extracted teeth was obtained from all participants. Gingival tissues were separated from gum part on extracted teeth and were treated with 3 mg/ml of collagenase (Sigma-Aldrich) and 4 mg/ml of dispase (Sigma-Aldrich). The primary gingival cells were grown outward from the sliced tissues in α-MEM containing 20% FBS (Hyclone). The gingival fibroblasts (GF) were dominantly grown out by continuous culture in the same media23. For isolation of gingival epithelial cells (GE), cells grown outward from tissues were trypsinized and washed with Keratinocyte Basal Medium (KBM, Lonza), and were transferred into Keratinocyte Growth Medium (KGM, Lonza). To collect only GE, the selective trypsinization was performed with modification in previous procedure53. GF were thrown out by treatment with low concentration of trypsin (0.062% trypsin/0.552 mM EDTA), and remaining GE were harvested by trypsinization using 0.25% trypsin/2.21 mM EDTA. Summary of cell culture procedure was shown in Fig. 1. Gingival epithelial cells collected were verified by immunophenotyping using epithelial and mesenchymal cell markers as mentioned below. The data in this report were originated from a cell line or from the average value of individual cell lines (see Figure legends). For ameloblastic cytodifferentiation, cells were treated with 100 ng/ml of BMP4 and BMP2, and/or 10 ng/ml of TGFβ-1 for 7 days in KGM.
Flow cytometric analysis
Cells were resuspended in PBA (PBS containing 0.5% BSA) and incubated with FITC- or PE-conjugated antibodies for 1 hour on ice. The antibodies used are as follows: FITC-integrin α-6, FITC-EpCAM, FITC-p75NTR, PE-CD44, PE-CD73, PE-CD90, and PE-CD146 antibodies purchased from BD Bioscience. The fluorescent signals were measured by using FACSCalibur flow cytometer (BD Bioscience), and were analyzed using WinMDI software.
Western blot analysis
Cell lysate was prepared by using NP-40 lysis buffer (20 mM Tris-HCl, pH8.0, 150 mM NaCl, 0.5% NP-40, 2 mM EGTA, 1 mM EDTA, 1 mM Na3VO4, 10 mM NaF, 20 mM p-nitrophenol phosphate, and protease inhibitors). Total proteins were separated by SDS-PAGE and were transferred to PVDF membrane. The membrane was blocked with 5% skim milk or BSA in TBST (0.1% Tween 20, 150 mM NaCl, 10 mM Tris-HCl, pH7.6). After blocking, membranes were incubated with the primary antibody and the HRP-conjugated secondary antibody. Signals were visualized using the ECLTM system (Amersham Biosciences). Anti-phospho-Smad1/5/8, -Smad1, -phospho-Smad3, and -Smad3 antibodies were purchased from Cell Signaling Technology. Anti-phospho-p38, -p38, and -vimentin antibodies were purchased from Santa Cruz Biotechnology.
Quantitative Real Time-PCR analysis
Total RNA was purified from cells using Easy-SpinTM Kit (iNtRON), and used for cDNA synthesis using ReverTra MixTM (Toyobo). The gene expression levels were analyzed by quantitative real time-PCR (qRT-PCR) using StepOnTM system (Applied Biosystems) with SYBR Green SupermixTM (Bio-Rad). Primers used were indicated at Supplementary Table S1. GAPDH was used as a control to normalize the variability in target gene expression. During qRT-PCR, a dissociation curve was constructed in the range of 65 °C to 95 °C, and the cycling parameters were followed as: 1 cycle for 1 min at 95 °C, 40 cycles for 15 sec at 95 °C, and 1 cycle for 1 min at 60 °C. The threshold cycle was obtained and the relative comparison of each target gene was analyzed.
Mineralization and alkaline phosphatase (ALP) assay
Cells were treated with the BMPs and TGF for 7 days, followed by treatment with osteogenic reagents (50 μg/ml ascorbic acid, 10 mM β-glycerophosphate, and 5 μM dexamethasone) for another 7 days. After 7 days of osteogenic induction, ALP activity was analyzed using ALP assay kit (BioVision). Briefly, cells were collected and disrupted by sonication, and after centrifugation, the supernatant incubated with pNPP substrate at 25 °C in dark. Absorbance was measured at 405 nm. For mineralization analysis, alizarin red staining was performed after the osteogenic induction. Cells were fixed with 4% paraformaldehyde for 15 min, washed with PBS, and treated with 2% alizarin red S (pH 4.5, Sigma). For quantification, plate was incubated with 10% acetic acid for 30 min at RT and heated for 10 min at 85 °C. Then, supernatants were neutralized by 10% ammonium hydroxide.
Library construction and analyses of differentially expressed genes (DEGs) in RNA-Seq analysis
After purification of total RNA, RNA purity was estimated by using Agilent 2100 Bioanalyzer (Agilent Technologies), and mRNA was enriched by oligo-dT magnetic bead. After cDNA synthesis, the samples were sequentially subjected to end-repair and addition with poly-A and adaptors using TruSeqTM RNA prep Kit (Illumina). cDNA fragments of 400–500 bps were separated on BluePippinTM system (Sage Science) and constructed into final library. Sequencing was performed in the paired-end sequencing mode using an Illumina Hiseq2500 sequencer (Illumina). Prior to read mapping, the raw reads with low-quality were filtered through in-house scripts as previous reported54. The qualified reads were implicated in alignment to the human genome (Ensembl release 72) using TopHat version 2.1.055,56, and uniquely mapped read pairs were used for the further analysis. The levels of gene expression with fragments per kilobase of exon per million fragments (FPKM) were calculated, and DEGs were determined using CUFFLINKS v.2.2.157. For each of the three pairwise comparisons, genes displaying log2FC(fold-change) ≥1 and p-value -v0.05 were handled. Gene ontology (GO) analysis of DEGs was performed using Metascape (http://metascape.org/gp/index.html). Further, clustering analysis of DEGs was performed based on the log2FPKM values and the heat map was generated using Pheatmap v1.0.8 with the popular clustering and hierarchical clustering method functions.
References
Fincham, A. G., Moradian-Oldak, J. & Simmer, J. P. The structural biology of the developing dental enamel matrix. Journal of structural biology 126, 270–299, https://doi.org/10.1006/jsbi.1999.4130 (1999).
Fukumoto, S. & Yamada, Y. Review: extracellular matrix regulates tooth morphogenesis. Connective tissue research 46, 220–226, https://doi.org/10.1080/03008200500344017 (2005).
Gallon, V., Chen, L., Yang, X. & Moradian-Oldak, J. Localization and quantitative co-localization of enamelin with amelogenin. Journal of structural biology 183, 239–249, https://doi.org/10.1016/j.jsb.2013.03.014 (2013).
Simmer, J. P. & Hu, J. C. Expression, structure, and function of enamel proteinases. Connective tissue research 43, 441–449 (2002).
Honda, M. J., Shinmura, Y. & Shinohara, Y. Enamel tissue engineering using subcultured enamel organ epithelial cells in combination with dental pulp cells. Cells, tissues, organs 189, 261–267, https://doi.org/10.1159/000151743 (2009).
Shinmura, Y., Tsuchiya, S., Hata, K. & Honda, M. J. Quiescent epithelial cell rests of Malassez can differentiate into ameloblast-like cells. Journal of cellular physiology 217, 728–738, https://doi.org/10.1002/jcp.21546 (2008).
Jayasudha, B., Navin, H. K. & Prasanna, K. B. Enamel regeneration - current progress and challenges. Journal of clinical and diagnostic research: JCDR 8, ZE06–09, https://doi.org/10.7860/JCDR/2014/10231.4883 (2014).
Thesleff, I. Epithelial-mesenchymal signalling regulating tooth morphogenesis. Journal of cell science 116, 1647–1648 (2003).
Thesleff, I. & Mikkola, M. The role of growth factors in tooth development. International review of cytology 217, 93–135 (2002).
Fawzy El-Sayed, K. M. et al. Effect of Emdogain enamel matrix derivative and BMP-2 on the gene expression and mineralized nodule formation of alveolar bone proper-derived stem/progenitor cells. Journal of cranio-maxillo-facial surgery: official publication of the European Association for Cranio-Maxillo-Facial Surgery 42, 568–576, https://doi.org/10.1016/j.jcms.2013.07.028 (2014).
Xu, X. et al. Developmental expression of Smad1-7 suggests critical function of TGF-beta/BMP signaling in regulating epithelial-mesenchymal interaction during tooth morphogenesis. The International journal of developmental biology 47, 31–39 (2003).
Wang, X. P. et al. Follistatin regulates enamel patterning in mouse incisors by asymmetrically inhibiting BMP signaling and ameloblast differentiation. Developmental cell 7, 719–730, https://doi.org/10.1016/j.devcel.2004.09.012 (2004).
Yang, Z. et al. Cessation of epithelial Bmp signaling switches the differentiation of crown epithelia to the root lineage in a beta-catenin-dependent manner. Molecular and cellular biology 33, 4732–4744, https://doi.org/10.1128/MCB.00456-13 (2013).
Klopcic, B. et al. TGF-beta superfamily signaling is essential for tooth and hair morphogenesis and differentiation. European journal of cell biology 86, 781–799, https://doi.org/10.1016/j.ejcb.2007.03.005 (2007).
Kobayashi-Kinoshita, S., Yamakoshi, Y., Onuma, K., Yamamoto, R. & Asada, Y. TGF-beta1 autocrine signalling and enamel matrix components. Scientific reports 6, 33644, https://doi.org/10.1038/srep33644 (2016).
Miyazono, K., Kamiya, Y. & Morikawa, M. Bone morphogenetic protein receptors and signal transduction. Journal of biochemistry 147, 35–51, https://doi.org/10.1093/jb/mvp148 (2010).
Mou, H. et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell stem cell 19, 217–231, https://doi.org/10.1016/j.stem.2016.05.012 (2016).
Shi, Y. & Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700 (2003).
Wu, M., Chen, G. & Li, Y. P. TGF-beta and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone research 4, 16009, https://doi.org/10.1038/boneres.2016.9 (2016).
Xing, Y. et al. Comparison of P75 NTR-positive and -negative etcomesenchymal stem cell odontogenic differentiation through epithelial-mesenchymal interaction. Cell proliferation 49, 185–194, https://doi.org/10.1111/cpr.12248 (2016).
Xiong, J., Mrozik, K., Gronthos, S. & Bartold, P. M. Epithelial cell rests of Malassez contain unique stem cell populations capable of undergoing epithelial-mesenchymal transition. Stem cells and development 21, 2012–2025, https://doi.org/10.1089/scd.2011.0471 (2012).
Hwang, H. I., Lee, T. H. & Jang, Y. J. Cell proliferation-inducing protein 52/mitofilin is a surface antigen on undifferentiated human dental pulp stem cells. Stem cells and development 24, 1309–1319, https://doi.org/10.1089/scd.2014.0387 (2015).
Min, J. H., Ko, S. Y., Cho, Y. B., Ryu, C. J. & Jang, Y. J. Dentinogenic potential of human adult dental pulp cells during the extended primary culture. Human cell 24, 43–50, https://doi.org/10.1007/s13577-011-0010-7 (2011).
Dominici, M. et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8, 315–317, https://doi.org/10.1080/14653240600855905 (2006).
Pivoriuunas, A. et al. Proteomic analysis of stromal cells derived from the dental pulp of human exfoliated deciduous teeth. Stem cells and development 19, 1081–1093, https://doi.org/10.1089/scd.2009.0315 (2010).
Tummers, M. & Thesleff, I. Observations on continuously growing roots of the sloth and the K14-Eda transgenic mice indicate that epithelial stem cells can give rise to both the ameloblast and root epithelium cell lineage creating distinct tooth patterns. Evolution & development 10, 187–195, https://doi.org/10.1111/j.1525-142X.2008.00226.x (2008).
Akimoto, T. et al. Establishment of Hertwig’s epithelial root sheath cell line from cells involved in epithelial-mesenchymal transition. Biochemical and biophysical research communications 404, 308–312, https://doi.org/10.1016/j.bbrc.2010.11.112 (2011).
Lee, J. H. et al. Upregulation of GM-CSF by TGF-beta1 in epithelial mesenchymal transition of human HERS/ERM cells. In vitro cellular & developmental biology. Animal 50, 399–405, https://doi.org/10.1007/s11626-013-9712-3 (2014).
Ehnert, S. et al. Transforming growth factor beta1 inhibits bone morphogenic protein (BMP)-2 and BMP-7 signaling via upregulation of Ski-related novel protein N (SnoN): possible mechanism for the failure of BMP therapy? BMC medicine 10, 101, https://doi.org/10.1186/1741-7015-10-101 (2012).
Kawahara, T. et al. TGF-Beta Negatively Regulates the BMP2-Dependent Early Commitment of Periodontal Ligament Cells into Hard Tissue Forming Cells. PloS one 10, e0125590, https://doi.org/10.1371/journal.pone.0125590 (2015).
Guyot, B. & Maguer-Satta, V. Blocking TGF-beta and BMP SMAD-dependent cell differentiation is a master key to expand all kinds of epithelial stem cells. Stem cell investigation 3, 88, https://doi.org/10.21037/sci.2016.11.15 (2016).
Hwang, H. I., Lee, T. H., Kang, K. J., Ryu, C. J. & Jang, Y. J. Immunomic Screening of Cell Surface Molecules on Undifferentiated Human Dental Pulp Stem Cells. Stem cells and development 24, 1934–1945, https://doi.org/10.1089/scd.2014.0493 (2015).
Liu, C. et al. Cell cycle control, DNA damage repair, and apoptosis-related pathways control pre-ameloblasts differentiation during tooth development. BMC genomics 16, 592, https://doi.org/10.1186/s12864-015-1783-y (2015).
Sorkin, B. C., Wang, M. Y., Dobeck, J. M., Albergo, K. L. & Skobe, Z. The cadherin-catenin complex is expressed alternately with the adenomatous polyposis coli protein during rat incisor amelogenesis. The journal of histochemistry and cytochemistry: official journal of the Histochemistry Society 48, 397–406, https://doi.org/10.1177/002215540004800309 (2000).
Inai, T., Sengoku, A., Hirose, E., Iida, H. & Shibata, Y. Differential expression of the tight junction proteins, claudin-1, claudin-4, occludin, ZO-1, and PAR3, in the ameloblasts of rat upper incisors. Anat Rec (Hoboken) 291, 577–585, https://doi.org/10.1002/ar.20683 (2008).
Kettunen, P. et al. Dact1-3 mRNAs exhibit distinct expression domains during tooth development. Gene expression patterns: GEP 10, 140–143, https://doi.org/10.1016/j.gep.2010.02.002 (2010).
Ko, S. O., Kim, T. H., Lee, H. K., Lee, J. C. & Cho, E. S. Temporospatial localization of acetylcholinesterase activity in the dental epithelium during mouse tooth development. Life sciences 81, 1235–1240, https://doi.org/10.1016/j.lfs.2007.08.035 (2007).
Obara, N., Suzuki, Y., Irie, K. & Shibata, S. Expression of planar cell polarity genes during mouse tooth development. Archives of oral biology 83, 85–91, https://doi.org/10.1016/j.archoralbio.2017.07.008 (2017).
Poulter, J. A. et al. A missense mutation in ITGB6 causes pitted hypomineralized amelogenesis imperfecta. Human molecular genetics 23, 2189–2197, https://doi.org/10.1093/hmg/ddt616 (2014).
Ryan, M. C., Lee, K., Miyashita, Y. & Carter, W. G. Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. The Journal of cell biology 145, 1309–1323 (1999).
Suomalainen, M. & Thesleff, I. Patterns of Wnt pathway activity in the mouse incisor indicate absence of Wnt/beta-catenin signaling in the epithelial stem cells. Developmental dynamics: an official publication of the American Association of Anatomists 239, 364–372, https://doi.org/10.1002/dvdy.22106 (2010).
Otsu, K. & Harada, H. Rho GTPases in ameloblast differentiation. The Japanese dental science review 52, 32–40, https://doi.org/10.1016/j.jdsr.2015.09.001 (2016).
Asaka, T. et al. Type XVII collagen is a key player in tooth enamel formation. The American journal of pathology 174, 91–100, https://doi.org/10.2353/ajpath.2009.080573 (2009).
Kim, J. W. et al. LAMB3 mutations causing autosomal-dominant amelogenesis imperfecta. Journal of dental research 92, 899–904, https://doi.org/10.1177/0022034513502054 (2013).
Seymen, F. et al. Novel ITGB6 mutation in autosomal recessive amelogenesis imperfecta. Oral diseases 21, 456–461, https://doi.org/10.1111/odi.12303 (2015).
Caccamo, D. et al. Expression pattern of transglutaminases in the early differentiation stage of erupting rat incisor. Amino acids 36, 49–56, https://doi.org/10.1007/s00726-008-0025-x (2009).
Feng, J. et al. Binding of amelogenin to MMP-9 and their co-expression in developing mouse teeth. Journal of molecular histology 43, 473–485, https://doi.org/10.1007/s10735-012-9423-1 (2012).
Simmer, J. P. et al. Ameloblast transcriptome changes from secretory to maturation stages. Connective tissue research 55(Suppl 1), 29–32, https://doi.org/10.3109/03008207.2014.923862 (2014).
Yamazaki, M. et al. Tumor necrosis factor-alpha stimulates human amelotin gene transcription in gingival epithelial cells. Inflammation research: official journal of the European Histamine Research Society… [et al.] 67, 351–361, https://doi.org/10.1007/s00011-017-1126-3 (2018).
Abbarin, N., San Miguel, S., Holcroft, J., Iwasaki, K. & Ganss, B. The enamel protein amelotin is a promoter of hydroxyapatite mineralization. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research 30, 775–785, https://doi.org/10.1002/jbmr.2411 (2015).
Nunez, S. M. et al. Maturation stage enamel malformations in Amtn and Klk4 null mice. Matrix biology: journal of the International Society for Matrix Biology 52-54, 219–233, https://doi.org/10.1016/j.matbio.2015.11.007 (2016).
Hu, S., Parker, J. & Wright, J. T. Towards unraveling the human tooth transcriptome: the dentome. PloS one 10, e0124801, https://doi.org/10.1371/journal.pone.0124801 (2015).
Yamanaka, T. et al. Isolation and serum-free culture of epithelial cells derived from epithelial rests of Malassez in human periodontal ligament. In vitro cellular & developmental biology. Animal 36, 548–553, https://doi.org/10.1290/1071-2690(2000)036<0548:IASFCO>2.0.CO;2 (2000).
Martin, J. A. & Wang, Z. Next-generation transcriptome assembly. Nature reviews. Genetics 12, 671–682, https://doi.org/10.1038/nrg3068 (2011).
Flicek, P. et al. Ensembl 2012. Nucleic acids research 40, D84–90, https://doi.org/10.1093/nar/gkr991 (2012).
Ghosh, S. & Chan, C. K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods Mol Biol 1374, 339–361, https://doi.org/10.1007/978-1-4939-3167-5_18 (2016).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics 12, 323, https://doi.org/10.1186/1471-2105-12-323 (2011).
Acknowledgements
This research was supported by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (NRF-2015M3A9C6029130).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: S.-Y.H. and Y.-J.J. Performed selective culture of epithelial cells and cytodifferentiation: S.-Y.H. and K.-J.K. Performed RNA preparation and analyses of DEGs in RNA-Seq analysis: S.-Y.H. and S.M. Contributed reagents/materials/analysis tools: K.-J.K., J.-C.L. and S.-Y.K. Analyzed the data: S.-Y.H., S.M., K.H. and Y.-J.J. Wrote the paper: S.-Y.H., S.M. and Y.-J.J.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hyun, SY., Mun, S., Kang, KJ. et al. Amelogenic transcriptome profiling in ameloblast-like cells derived from adult gingival epithelial cells. Sci Rep 9, 3736 (2019). https://doi.org/10.1038/s41598-019-40091-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40091-x
This article is cited by
-
Single-cell transcriptomics reveals cell atlas and identifies cycling tumor cells responsible for recurrence in ameloblastoma
International Journal of Oral Science (2024)
-
Regulatory role of N-myc downregulated genes in amelogenesis in rats
Journal of Molecular Histology (2024)
-
Regulatory role of insulin-like growth factor-binding proteins in odontogenic mineralization in rats
Journal of Molecular Histology (2021)
-
Expression dynamics of integrin α2, α3, and αV upon osteogenic differentiation of human mesenchymal stem cells
Stem Cell Research & Therapy (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
