
Abstract
This study described an efficient and practical approach for amide synthesis. The reaction was conducted under metal- and solvent-free conditions at a mild temperature (40 °C) in air, and readily available formamides were used as an amino source. This reaction can be easily upgraded to the gram level with an excellent yield.
Similar content being viewed by others

Introduction
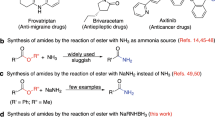
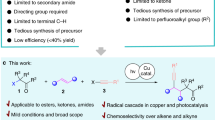
Amide is one of the primary components of biomolecules, such as proteins, and is also commonly found in natural products, pharmaceuticals, pesticides, and functional materials1,2,3. Amide synthesis has attracted continuous interest, and various methods have been developed4,5,6,7. Formamides are cheap, readily available, and versatile organic compounds that are commonly used as solvents and as a source for carbonyl, dimethylamino, and Me2NCO8,9,10,11,12. Based on the application potential and environmental benign aspects, the coupling reaction of formamide with carboxylic acid derivative shows promise as a synthesis method for amides. However, these coupling strategies are often conducted in the presence of a metal catalyst, such as Cu13,14,15,16,17,18,19,20, Ru21, Co22, and Ln23, thereby resulting to the production of metal residues and the excessive use of DMF as a solvent (Fig. 1A). Additionally, high reaction temperature of 80 °C to 150 °C is required. Several metal-free cross-coupling reactions of formamide with a carboxylic acid derivative were also developed. Recently, Wan et al. reported Bu4NI-catalyzed cross-coupling of formamide with aldehyde using TBHP as the oxidant and Cl2CHCH2Cl as the solvent; in their study, 25 equiv of DMF and a high reaction temperature of 90 °C are still required (Fig. 1B)24. Wolf also reported a metal-free oxidative amination of aldehydes to amides using TBHP as oxidant25. Mavel and Tortoioli reported that phosphorus-containing compounds promote the coupling reaction of formamide with carboxylic acid (Fig. 1C)26,27. However, the use of a large amount of phosphorus reagent, the high reaction temperature above 130 °C, and the excessive wastage of formamides are not environmentally friendly. Yoon et al. reported the coupling reaction of acid chloride with DMF, which also required a high reaction temperature and resulted to the wastage of DMF (Fig. 1D)28. In this study, we synthesized valuable amides by a metal- and solvent-free method conducted at 40 °C in air under mild conditions (Fig. 1E).

Cross-coupling reaction of formamide with a carboxylic acid derivative.
Results and Discussion
In the initial study, the reaction of tert-butyl benzoperoxoate (0.5 mmol) and DMF (1.5 mL) was investigated in the presence of KOtBu (Fig. 2, entry 1). The desired product was obtained in an 86% yield. A series of optimization reactions, including the optimization of the amount and type of base (Fig. 2, entries 1–6; Table S1, entries 1–14), the reaction time (Fig. 2, entries 7–9; Table S1, entries 15–18), the reaction temperature (Fig. 2, entries 10–12; Table S1, entries 19–22), the solvent (Fig. 2, entries 13–15; Table S1, entries 23–26), and the reaction atmosphere (Fig. 2, entry 16; Table S1, entry 27) was conducted. The reaction was also conducted under solvent-free conditions and yielded almost the same amount (85%) (Fig. 2, entry 17; Table S1, entry 28). A good yield of 88% was achieved after adjusting the amount of KOtBu to 2.5 equiv (Fig. 2, entry 19; Table S1, entry 30).

Selected optimization resultsa. aUnless otherwise noted, all reactions were conducted on a 0.5 mmol scale; Yields were determined by 1H NMR spectroscopy using nitromethane as internal standard. bUsing DMF (5 equiv, 194 μL). cUnder the atmosphere of argon. dSolvent free, using DMF (5 equiv, 194 μL).
The generalizability of this method was explored under the following optimized conditions: peroxoate, 0.5 mmol; substrate amide, 5 equiv; and KOtBu, 4 equiv at 40 °C in air for 3 h. N,N-disubstituted, N-monosubstituted, and unsubstituted amides 2 were converted into the desired product 3 with good to excellent yields (Fig. 3a–c). In addition, formyl hydrazine smoothly reacted with tert-butylbenzoperoxoate and obtained the corresponding product with a moderate yield (3d). When the hydrophobicity of the alkyl substituents on the nitrogen of amide 2 was increased, the solubility of KOtBu decreased, which is unfavorable to this conversion (3e–f). No product was detected when N-cyclohexyl formamide was used as the amino source (3 g). Other tert-butylbenzoperoxoate derivatives were also investigated and found that electron-rich or electron-deficient derivatives render this reaction smooth (3h–q). Naphthylperoxoates also showed high reactivity and obtained good isolated yields (3s–t). Heteroaromatic (including furan, thiophene, and benzothiophene) peroxoates can also be converted into corresponding products with moderate yields (3v–y). However, 3r and 3 u were either undetected or with only a poor yield, which might be caused by the aforementioned poor solubility of KOtBu on the reaction systems. The reaction of alkyl perester, such as tert-butyl ethaneperoxoate with N-benzylformamide was conducted, but no amide product could be detected.

Scope of amide synthesisa. aUnless otherwise noted, all reactions were conducted on a 0.5 mmol scale, amide compounds (5 equiv), KOtBu (2.5 equiv, 140 mg) in a sealed tube under an atmosphere of air for 3 h. Isolated yield was showed out brackets, 1H NMR yield were showed in brackets. b10 equiv of amide 2 was used, temperature is 70 °C. c10 equiv of amide 2 was used, temperature 80 °C. d1.5 mL DMF was used, 4 equiv KOtBu was used, temperature is 80 °C. ePeroxide (0.1 mmol), KOtBu (5 equiv), HCONH2 (25 equiv), temperature is 80 °C.
Several control experiments were conducted to investigate the reaction mechanism. First, we used benzoyl peroxide instead of tert-butylbenzoperoxoate and obtained a poor yield of 32% (Fig. 4, eq. 1). When one of the benzene rings is converted into the product, the remaining ring would probably lose its reactivity. The reaction of other peroxide acids such as 3-chloroperoxybenzoic acid and peroxyacetic acid with formamide derivatives were also tried, but no desired product could be detected (Fig. 4, eqs 2 and 3).

Control experiments.
From a general perspective, this transformation was considered as radical process because peroxide was used as a substrate. Hence, the radical block reaction was conducted using TEMPO as a radical inhibitor. The addition of TEMPO showed no evident effect, and the product yield was 79% (Fig. 4, eq. 4). Good and moderate yields were still obtained when BHT and benzoquinone were used as inhibitors, respectively (Fig. 4, eqs 5 and 6). These results excluded the radical process of this transformation. Afterwards, the hydrogen of aldehyde was replaced by methyl, and almost no desired product was detected (Fig. 4, eq. 7). This result indicated the need for decarbonylation, which was blocked by methyl. Then, an isotope labeling experiment was conducted to confirm the source of carbonyl on the target molecules (T.M.) (Fig. 4, eq. 8). Almost all carbon molecules in the carbonyl group of the product were identified as isotope 13C. Afterward, the decomposition reaction of formamide derivative was tested, and it was found morpholine-4-carbaldehyde could be decomposed to morpholine under the standard condition (Fig. 4, eq. 9). Followed, the reaction of tert-butyl benzoperoxoate and amine were conducted, and the desired product was found with a yield of 38% (Fig. 4, eq. 10). These results indicated that the decomposition of formamide derivative and the corresponding decomposition product amine might be played an important role in this amide synthesis procedure. Based on these control experiments and previous studies13,29, a possible mechanism was proposed (Fig. 5).

Proposed mechanism.
Initially, dimethylamine anion (I) was formed via the decarbonylation of formamide with the release of CO in the presence of KOtBu30. The nucleophilic addition of (I) to tert-butylbenzoperoxoate subsequently occurred while gathering the T.M.
A gram-scale reaction of 1c was conducted to verify its potential in industrial production, and an excellent isolated yield of 90% was achieved (Fig. 6, eq. 1). Amides are known to have great potential application. For example, amides can be easily converted into amines (including primary, secondary, and tertiary amines) through reduction reactions (Fig. 6, eq. 2)31,32,33. Another example is that product 3f can be converted into bioactive molecules (Fig. 6, eq. 3)34,35,36.

Gram-scale reaction and application of amides.
Conclusions
In summary, an efficient and practical approach for the synthesis of amide has been developed. The reaction is conducted in air at a mild temperature (40 °C) under metal- and solvent-free conditions, and the readily available substitute formamides were used as an amino source. This transformation can easily be upgraded to the gram level, thereby providing an avenue for the synthesis of valuable amides.
Materials and Methods
General information
Preparative thin-layer chromatography was performed for product purification using Sorbent Silica Gel 60 F254 TLC plates and visualized with ultraviolet light. IR spectra were recorded on a new Fourier transform infrared spectroscopy. 1H, 13C and 19F NMR spectra were recorded on 400, 100, 377 MHz NMR spectrometer using CDCl3 as solvent unless otherwise stated. HRMS were made by means of ESI. Melting points were measured on micro melting point apparatus and uncorrected. Unless otherwise noted, all reagents were weighed and handled in air, and all reactions were carried out in a sealed tube under an atmosphere of air. Unless otherwise noted, all reagents were purchased from reagent company, and used without further purifications.
Experimental Section
A typical experimental procedure for transamidation was conducted as follows: A solution of peroxoate (0.5 mmol), KOtBu (2.5 equiv, 140 mg) and amide (5 equiv or 10 equiv) were stirred in a sealed tube under an atmosphere of air at 40 °C for 3 h. The reaction mixture was then extracted with ethyl acetate. Afterward, the solution was evaporated under vacuum. The residue was purified by preparative thin-layer chromatography (TLC) on silica gel with petroleum ether and ethyl acetate (5% triethylamine) to achieve the pure product.
References
Humphrey, J. M. & Chamberlin, A. R. Chemical synthesis of natural product peptides: coupling methods for the incorporation of noncoded amino acids into peptides. Chem. Rev. 97, 2243–2266 (1997).
Carey, J. S., Laffan, D., Thomson, C. & Williams, M. T. Analysis of the reactions used for the preparation of drug candidate molecules. Org. Biomol. Chem. 4, 2337–2347 (2006).
Kuranaga, T., Sesoko, Y. & Inoue, M. Cu-mediated enamide formation in the total synthesis of complex peptide natural products. Nat. Prod. Rep. 31, 514–532 (2014).
Figueiredo, R. M. D., Suppo, J. S. & Campagne, J. M. Nonclassical routes for amide bond formation. Chem. Rev. 116, 12029–12122 (2016).
Valeur, E. & Bradley, M. Amide bond formation: beyond the myth of coupling reagents. Chem. Soc. Rev. 38, 606–631 (2009).
Miyamura, H. & Kobayashi, S. Tandem oxidative processes catalyzed by polymer-incarcerated multimetallic nanoclusters with molecular oxygen. Acc. Chem. Res. 47, 1054–1066 (2014).
Allen, C. L. & Williams, J. M. J. Metal-catalysed approaches to amide bond formation. Chem. Soc. Rev. 40, 3405–3415 (2011).
Muzart, J. N,N-dimethylformamide: much more than a solvent. Tetrahedron 65, 8313–8323 (2009).
Ding, S. T. & Jiao, N. N,N-dimethylformamide: a multipurpose building block. Angew. Chem. Int. Ed. 51, 9226–9237 (2012).
He, T., Li, H., Li, P. & Wang, L. Direct amidation of azoles with formamides via metal-free C-H activation in the presence of tert-butyl perbenzoate. Chem. Commun. 47, 8946–8948 (2011).
Niu, Z. H. et al. Otherwise inert reaction of sulfonamides/carboxamides with formamides via proton transfer-enhanced reactivity. Org. Biomol. Chem. 11, 2460–2465 (2013).
Gao, L. H., Tang, H. M. & Wang, Z. Y. Oxidative coupling of methylamine with an aminyl radical: direct amidation catalyzed by I2/TBHP with HCl. Chem. Commun. 50, 4085–4088 (2014).
Liu, H. Q., Liu, J., Zhang, Y. H., Shao, C. D. & Yu, J. X. Copper-catalyzed amide bond formation from formamides and carboxylic acids. Chin. Chem. Lett. 26, 11–14 (2015).
Albert-Soriano, M. & Pastor, I. M. Metal-organic framework based on copper and carboxylate-imidazole as robust and effective catalyst in the oxidative amidation of carboxylic acids and formamides. Eur. J. Org. Chem. 2016, 5180–5188 (2016).
Kumar, P. S., Kumar, G. S., Kumar, R. A., Reddy, N. V. & Reddy, K. R. Copper-catalyzed oxidative coupling of carboxylic acids with N,N-dialkylformamides: an approach to the synthesis of amides. Eur. J. Org. Chem. 2013, 1218–1222 (2013).
Priyadarshini, S., Joseph, P. J. A. & Kantam, M. L. Copper catalyzed cross-coupling reactions of carboxylic acids: an expedient route to amides, 5-substituted gamma-lactams and alpha-acyloxy esters. RSC Adv. 3, 18283–18287 (2013).
Yan, H. et al. Copper-catalyzed synthesis of α,β-unsaturated acylamides via direct amidation from cinnamic acids and N-substituted formamides. Tetrahedron 69, 7258–7263 (2013).
Li, H. M., Pan, C. D., Cheng, Y. X. & Zhu, C. J. Copper-catalyzed oxidative coupling of carboxylic acids with formamides for the synthesis of α,β-unsaturated amides. Tetrahedron Lett. 54, 6679–6681 (2013).
Mohammad, A., Chandra, P., Ghosh, T., Carraro, M. & Mobin, S. M. Facile access to amides from oxygenated or unsaturated organic compounds by metal oxide nanocatalysts derived from single source molecular precursors. Inorg. Chem. 56, 10596–10608 (2017).
Saberi, D., Mahdudi, S., Cheraghi, S. & Heydari, A. Cu(II)−acetylacetone complex covalently anchored onto magnetic nanoparticles: Synthesis, characterization and catalytic evaluation in amide bond formation via oxidative coupling of carboxylic acids with N,N-dialkylformamides. J. Organomet. Chem. 772–773, 222–228 (2014).
Bi, X. J. et al. Ru-catalyzed direct amidation of carboxylic acids with N-substituted formamides. Tetrahedron 72, 8210–8214 (2016).
Bai, C. H., Yao, X. F. & Li, Y. W. Easy access to amides through aldehydic C-H bond functionalization catalyzed by heterogeneous Co-based catalysts. ACS Catal. 5, 884–891 (2015).
Chen, W. F. et al. Utility of dysprosium as a reductant in coupling reactions of acyl chlorides: the synthesis of amides and diaryl-substituted acetylenes. Organometallics 30, 2026–2030 (2011).
Liu, Z. J. et al. Cross coupling of acyl and aminyl radicals: direct synthesis of amides catalyzed by Bu4NI with TBHP as an oxidant. Angew. Chem. Int. Ed. 51, 3231–3235 (2012).
Ekoue-Kovi, K. & Wolf, C. Metal-free one-pot oxidative amination of aldehydes to amides. Org. Lett. 9, 3429–3432 (2007).
Bannwart, L., Abele, S. & Tortoioli, S. Metal-free amidation of acids with formamides and T3P®. Synthesis 48, 2069–2078 (2016).
Mavel, S., Meheux, N., Guilloteau, D. & Emond, P. Synthesis and in vitro evaluation of fluorinated diphenyloxide derivatives and sulfur analogs as serotonin transporter ligands. Bioorg. Med. Chem. 18, 236–241 (2010).
Lee, W. S., Park, K. H. & Yoon, Y. J. N,N-dimethylamination of acid chlorides with DMF. Synth. Commun. 30, 4241–4245 (2000).
Zhang, M. Z., Guo, Q. H., Sheng, W. B. & Guo, C. C. Potassium tert-butoxide-mediated amine acyl exchange reaction of N,N-disubstituted formamides with aromatic carbonyl derivatives via sequential C-N bond cleavage/formation: an approach to aromatic amides. Adv. Synth. Catal. 357, 2855–2861 (2015).
Perrin, D. D., Armarego, W. L. F. Purification of Laboratory Chemicals, 3 rd ed., Pergamon: Oxford (1988).
Giannis, V. A. & Sandhoff, K. LiBH4 (NaBH4)/Me3SiCl, ein ungewöhnlich starkes und vielseitig einsetzbares reduktionsmittel. Angew. Chem. Int. Ed. 101, 220–222 (1989).
Das, S., Join, B., Junge, K. & Beller, M. A general and selective copper-catalyzed reduction of secondary amides. Chem. Commun. 48, 2683–2685 (2012).
Blom, B. et al. Bis-N-ceterocyclic carbine (NHC) stabilized η6-arene iron(0) complexes: synthesis, structure, reactivity, and catalytic activity. J. Am. Chem. Soc. 135, 18108–18120 (2013).
Ramachandran, P. V. et al. Synthetic α-(aminomethyl)-γ- butyrolactones and their anti-pancreatic cancer activities. Bioorg. Med. Chem. Lett. 23, 6911–6914 (2013).
Gao, Y. Z. et al. Direct transformation of amides into α-amino phosphonates via a reductive phosphination process. Org. Lett. 15, 4214–4217 (2013).
Beers, S. A. et al. Phosphatase inhibitors-III. benzylaminophosphonic acids as potent inhibitors of human prostatic acid phosphatase. Bioorg. Med. Chem. 4, 1693–1701 (1996).
Acknowledgements
We are grateful to the National Natural Science Foundation of Hunan Province (No. 2018JJ2389), National Natural Science Foundation of China (Nos 21402168) and Hunan 2011 Collaborative Innovation Center of Chemical Engineering & Technology with Environmental Benignity and Effective Resource Utilization for their support of our research.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: L.L. and J.Z. performed the experiments. H.G. and F.Z. supervised all research. H.G. also wrote the manuscript. All authors contributed to reagents/materials/technical support to this study.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, F., Li, L., Zhang, J. et al. Metal- and solvent-free synthesis of amides using substitute formamides as an amino source under mild conditions. Sci Rep 9, 2787 (2019). https://doi.org/10.1038/s41598-019-39240-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-39240-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
