
Abstract
The shallow oxidized asthenosphere may contain a small fraction of potassic silicate melts that are enriched in incompatible trace elements and volatiles. Here, to determine the chemical composition of such melt, we analysed fossilized melt inclusions, preserved as multiphase solid inclusions, from an orogenic garnet peridotite in the Bohemian Massif. Garnet-poor (2 vol.%) peridotite preserves inclusions of carbonated potassic silicate melt within Zn-poor chromite (<400 ppm) in the clinopyroxene-free harzburgite assemblage that equilibrated within the hot mantle wedge (Stage 1, > 1180 °C at 3 GPa). The carbonated potassic silicate melt, which has a major element oxide chemical composition of K2O = 5.2 wt.%, CaO = 17 wt.%, MgO = 18 wt.%, CO2 = 22 wt.%, and SiO2 = 20 wt.%, contains extremely high concentrations of large ion lithophile elements, similar to kimberlite melts. Peridotites cooled down to ≅800 °C during Stage 2, resulted in the growth of garnet relatively poor in pyrope content, molar Mg/(Mg + Fe + Ca + Mn), (ca. 67 mol.%). This garnet displays a sinusoidal REE pattern that formed in equilibrium with carbonatitic fluid. Subsequently, subduction of the peridotite resulted in the formation of garnet with a slightly higher pyrope content (70 mol.%) during the Variscan subduction Stage 3 (950 °C, 2.9 GPa). These data suggest the following scenario for the generation of melt in the mantle wedge. Primarily, infiltration of sediment-derived potassic carbonatite melt into the deep mantle wedge resulted in the growth of phlogopite and carbonate/diamond. Formation of volatile-bearing minerals lowered the density and strength of the peridotite. Finally, phlogopite-bearing carbonated peridotite rose as diapirs in the mantle wedge to form carbonated potassic silicate melts at the base of the overriding lithosphere.
Similar content being viewed by others

Introduction
Subduction of carbonates stored within the oceanic and continental crust at convergent plate boundaries has played an important role in mantle refertilization and the global carbon cycle1. The total carbon input to subduction zone systems has been estimated to be as high as 24–48 megatons per year, whereby 17 megatons of crustal carbon are returned to the surface through volcanism1. Consequently, a significant quantity of carbon could be stored within the downgoing plate and returned to the deep mantle in the form of high-pressure carbonate minerals2. In warm subduction zones and collision zones, partial melting of subducting plate could remove carbon more efficiently from the downgoing crust3. Due to their low viscosity and high mobility, carbonate-rich melts (i.e. carbonatites) are expected to percolate into the overlying mantle wedge4. Carbonatite not only refertilizes peridotite in the mantle wedge, but also lowers the density of the mantle wedge by precipitating volatile-rich minerals. A reduction in density can generate Rayleigh–Taylor instabilities and initiate diapiric flow5. The presence of carbonates, graphite, diamond and carbonate-rich multiphase solid inclusions (MSIs) has been reported in mantle wedge peridotites from ultrahigh-pressure (UHP) metamorphic terranes6,7,8,9. Such C-bearing phases may provide direct geological evidence for the presence of mantle wedge melts.
Mantle wedge peridotites record polyphase metasomatism during transfer from the asthenosphere into the subducting continental crust6,7,10. The initial stages of metasomatism are induced by the infiltration of melts at temperatures (T) in excess of 1200 °C, whereas secondary metasomatism results from the addition of crustal fluids at 500–850 °C6,10. This study aims to constrain melt compositions in mantle wedge peridotites. Previous studies have characterised melt compositions using trace elements in nominally anhydrous minerals6,10 or whole-rock samples10. Both methods indicate the presence of high-temperature silicate melts that are enriched in incompatible trace elements such as light-REE (LREE), Pb, U, and Li. Here, we examine fossilized melt inclusions11 to constrain the major and trace element compositions of potassium-rich carbonatitic melts in the mantle wedge.
Mantle wedge peridotites in the Bohemiam Massif
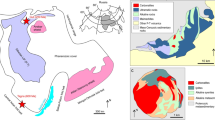
To study melts within the mantle wedge, we investigated an orogenic garnet peridotite from the Moldanubian Zone of the Bohemian Massif (BM) in the Czech Republic, which experienced corner flow in the mantle wedge and contains MSIs (i.e. fossilized melt)7,12,13. Orogenic garnet peridotite bodies within the Moldanubian Zone occur as lenticular bodies ranging in size from several centimetres to kilometres across, and are surrounded by felsic granulites and gneisses. A previous study has shown that Mg–Cr-rich garnet peridotites within granulite equilibrated in a low P/T regime (2.5–3.0 GPa, 1015–1335 °C) and were possibly derived from the asthenosphere, whereas those occurring within migmatitic orthogneiss equilibrated in a medium P/T regime (3.3–6.0 GPa, 875–1180 °C) within the lithospheric mantle wedge14. The Blanský les granulite15 in the BM has a cylindrical dome structure and contains numerous serpentinized garnet peridotite bodies that occur mainly in a wide NW–SE striking serpentinite belt in the central part of the granulite massif. At the SE margin of the serpentinite belt, fresh spinel–garnet peridotite crops out within the Plešovice quarry.
Figure 1 summarizes the thermal history of the Plešovice peridotite7,12, which may have experienced P–T conditions within the diamond stability field at the earliest stage (Stage 0, > 4 GPa). The peridotite was then uplifted to shallow depths (Stage 1)7 and subsequently cooled into the chlorite stability field (Stage 2, < 750 °C). It was then subducted into the high P–T garnet lherzolite field (Stage 3, 950 °C at 2.9 GPa). At the final stage, the peridotite was partially recrystallized to spinel lherzolite assemblage (Stage 4, 750 °C at 1.5 GPa). Previous Sm–Nd geochronological studies have yielded ages of 375 to 329 Ma for the garnet peridotites in the Moldanubian Zone of the BM14.

Panel (a) shows a pressure–temperature path of the Plešovice peridotite, which consists of four stages represented by yellow circles. The decarbonation reaction, recording the breakdown of phlogopite + magnesite (1) is from ref.32, and the chromite (Chr)-in reaction (2) for depleted lherzolite from ref.31. The graphite–diamond transition is from ref.65. The solidus for metasomatized peridotite is from ref.30 clinopyroxene (Cpx)-out curve is from ref.20 other phase boundaries are after ref.7 and references therein. Pl = Plagioclase16. In panel (b) Zn concentrations of Chr are summarized. Zn concentration of Chr1 grains included in garnet (Grt) are 300–950 ppm, and those of matrix Chr3 are 1200–5600 ppm. Zinc-in-chromite temperatures17 are shown on the upper axis. Panels (c) and (d) are results of thermobarometric calculations for Type A and B peridotites, respectively. The Grt–Cpx thermobarometers of refs21,22,23 have been used.
The spinel–garnet peridotite at Plešovice is a depleted lherzolite consisting mainly of olivine (Ol) and orthopyroxene (Opx) (14–16 vol.%) with minor clinopyroxene (Cpx) (1 vol.%) and calcic amphibole (Amp) (1–2 vol.%) and accessory chromian spinel (Chr), phlogopite (Phl) and apatite (Ap)7. Mineral abbreviations follows ref.16. Two lithological types of the spinel–garnet peridotite, Type A and Type B, have been distinguished by variations in the abundance of garnet (Grt) (~2 and ~5 vol.%, respectively). Garnet occurs as sporadic large grains (>5 mm) in Type A peridotites, whereas they are ubiquitous and show a range of grain sizes (0.1–30 mm) in Type B peridotites (Fig. 2a,b). We document the trace element composition of the minerals in order to correlate metasomatic events within the existing framework of P–T constraints7,12.

Panels (a) and (b) show typical Type A and B peridotites from the Plešovice quarry in the Bohemian Massif. Panel (c) shows Al2O3 weight % concentration map of an orthopyroxene porphyroclast (Opx1) stable at Stage 1 in a Type A peridotite. Crystallization stages are denoted by subscript numbers in figures. Chemical analyses along the traverse from A to B are shown in ref.7. Kelyphite (kely4) formed after garnet (Grt). Matrix consists of olivine (Ol3), Al-poor orthopyroxene (Opx3), Na2O-rich clinopyroxene (Cpx3), chromite (Chr3), phlogopite (Phl3), and calcic amphibole (Amp3) that formed during Stage 37. Panel (d) displays a centimetre-sized garnet (Grt2) with Na2O-poor (~0.35 wt.%) Cpx2 within embayments in a Type A peridotite. Plane polarized light. Panel (e) shows a Grt3 with large Opx3 inclusions within a Type B peridotite. Plane polarized light. Panel (f) shows a micrograph of kelyphite, which are separated from matrix Ol3 by an orthopyroxene corona (Opx4). Plane polarized light.
Garnet-poor (~2 vol.%) Type A peridotite contains centimetre-size crystals of orthopyroxene and garnet that formed during Stages 1 and 2. Type A peridotite contains centimetre-size orthopyroxene porphyroclasts7 (Fig. 2c), the cores of which (Opx1, where the subscript represents the crystallization stage) contain 3 wt.% Al2O3, consistent with equilibration under high-temperature Stage 1 conditions (Fig. 1a). Large garnet crystals, which crystallized at Stage 2 (i.e., Grt2), contain abundant inclusions of chromite (Chr1) (Fig. 2d). Chr1 grains potentially retain a record of temperatures during Stage 1. Here we used Zn-in-chromite thermometry17 to constrain Stage 1 temperatures. The Zn concentrations in Chr1 in garnet (250–950 ppm) are systematically lower than those in matrix Chr3 (1320–5500 ppm) (Fig. 1b). In particular, Chr1 inclusions in large Grt2 grains preserve low Zn concentrations (250–400 ppm). Calculated crystallization temperatures for such Zn-poor Chr1 crystals are >1180 °C (Fig. 1b). At this temperature, the 3 wt.% Al2O3 in orthopyroxene isopleth constrains pressure at 2.8–3.6 GPa (Fig. 1a). Opx1 shows enrichment in LILE (Rb ≈ 1 ppm, Ba ≈ 16 ppm; Fig. 3a; Supplementary Table S2), a pronounced depletion in Ti (TiN = 0.1, where TiN is the measured composition normalised against the primitive mantle value18), a fractionated REE pattern with a high LaN/YbN value (≈10) and relatively flat LREE contents (LaN/SmN ≈ 0.5) (Supplementary Fig. S1a). A negative Ti anomaly in orthopyroxene is observed only in clinopyroxene-free harzburgite19. This finding is consistent with the absence of clinopyroxene porphyroclast, which constrains Stage 1 temperatures to above the clinopyroxene-out curve in the hydrous peridotite system (>1120 °C20).

Panels (a,b) show primitive-mantle (PM) normalized18 trace element patterns of orthopyroxene (Opx) in Type A and Type B peridotites, with stages are denoted by subscript numbers in the figure. Opx1 displays a negative Ti anomaly and is rich in large ion lithophile element (LILE) and light REE (LREE). Opx3 in Type A peridotites shows a convex REE pattern with an increase in heavy REE (HREE), whereas Opx3 in Type B peridotite is depleted in HREE and shows a positive Ti anomaly. Opx4 is moderately enriched in HREE. Panels (c,d) show PM-normalized trace element patterns of clinopyroxene (Cpx2/3) and amphibole (Amp3) in Type A and B peridotites. They show positive Pb anomalies and negative anomalies in high field strength elements. The trace element pattern of Amp in the Nonsberg peridotite6 is shown for comparison. Panel (e) shows PM-normalized trace element patterns of Garnet (Grt2/3) in Type A peridotites, in which pyrope (Prp)-poor Grts (~67 mol.%) are poor in HREE and Ti. Panel (f) shows trace element patterns of Prp-rich Grts (~70 mol.%) in Type B peridotites, which are rich in HREE and Ti.
Garnets in Type A peridotites are relatively poor in pyrope (~67 mol.%, Supplementary Table S1) and Na (<5 ppm) relative to those in Type B peridotites, and exhibit a pronounced depletion in Ti (TiN < 0.1) (Supplementary Table S2) and sinusoidal REE patterns with low LREE, HREE (LaN ≈ 0.5, YbN ≈ 2) and high MREE (NdN ≈ 7) concentrations (Fig. 3e and Supplementary Fig. S1e). Cpx2 within embayments in Grt2 is depleted in Na2O (0.3 wt.%; Supplementary Table S1) and shows positive Pb anomalies, negative HFSE anomalies (TiN ≈ 0.05) (Fig. 3c), and fractionated REE pattern (LaN/YbN > 3000) (Supplementary Fig. S1c). The application of garnet–clinopyroxene geothermobarometry21,22,23 yields 810 °C at 2 GPa (Fig. 1c), corresponding to Stage 2. The peridotite matrix consists of fine-grained (~0.1 mm) Ol3, Opx3, Cpx3, Ca-Amp3, Chr3, Phl3 and Apt3 that crystallized during Stage 3. Where garnet rims are in contact with Na2O-rich (~1 wt.%) Cpx3 (Supplementary Fig. S2a), they are relatively enriched in pyrope (~70 mol.%), Na (35 ppm), Ti (TiN > 0.2), and HREE (YbN ≈ 10) (Fig. 3e, Supplementary Fig. S1e), suggesting recrystallization during Stage 3. Amp3 contains a lot of Chr3 inclusions (Supplementary Fig. S2b), and it coexists with Cpx3 in the matrix (Supplementary Fig. S2c). The trace element compositions of both Amp3 and Cpx3 are characterised by positive Pb anomalies and a marked depletion in HFSE (Nb, Ta, Zr, Hf and Ti; TiN ~ 1 in Amp3 and 0.2 in Cpx3) (Fig. 3c), and are rich in LREE (LaN ≈ 30–50 in both Amp3 and Cpx3) (Supplementary Fig. S1c). High LILE and low HFSE contents in Amp3 are similar to those in amphibole from the Nonsberg peridotite6. Matrix Opx3 in Type A peridotite exhibits a convex-downward REE pattern characterised by a relative enrichment in HREE (DyN/YbN ≈ 0.1–0.2; YbN ≈ 0.4–0.6).
Garnet-rich (~5 vol.%) Type B peridotites have an inequigranular texture in which large spheroidal garnet grains (5–30 mm in length) occur within a millimetre-sized matrix of Ol3 + Opx3 + Cpx3 + Chr3 + Phl3 + Amp3 that equilibrated during Stage 37 (Fig. 2e). Grt3 grains are rich in pyrope (~70 mol.%), Na (65 ppm), Ti (TiN ≈ 0.6) (Fig. 3f), and HREE (YbN ≈ 10, LaN/YbN ≈ 0.01; Supplementary Fig. S1f). Grt3 grains contain abundant Opx3 and Ol3, along with subordinate Cpx3 and Phl3 (Fig. 2e, Supplementary Fig. S2d). Grt3 contains rare chromite inclusions that have concave surfaces against their host, suggesting that Chr1 grains were consumed to grow Grt37. Opx3 grains are relatively depleted in Al2O3 (1.5 wt.%7) and LILE (Rb, Ba < 0.03 ppm; Supplementary Table S2), and show variable enrichment in LREE (LaN ≈ 0.01–0.10). A positive Ti anomaly (TiN ≈ 0.4) and low HREE (YbN ≈ 0.03–0.06) contents in Opx3 (Fig. 3b) are consistent with their crystallization in equilibrium with the garnet lherzolite assemblage. Cpx3 grains show high contents of Na2O (~1 wt.%), a positive Pb anomaly, negative anomalies in HFSE (TiN ≈ 0.4) (Fig. 3d) and fractionated REE patterns (Supplementary Fig. S1d; LaN/YbN > 70). Application of garnet–clinopyroxene geothermobarometers21,22,23 to Grt3–Cpx3(in Grt) pairs yields conditions of ~3 GPa at 950–1000 °C (Fig. 1d). Garnet crystals were transformed into kelyphite during Stage 4 (Fig. 2f), in which the kelyphite consists of Opx4, Cpx4, and spinel (Spl4) with minor Ca-Amp47. Opx4 that formed at this stage shows extremely high HREE contents (YbN > 1; Fig. 3b and Supplementary Fig. S1b), presumably inherited from the breakdown of garnet.
Reconstruction of melt compositions using multiphase solid inclusions
Multiphase solid inclusions (MSIs) have been widely observed in high-pressure minerals within UHP metamorphic rocks and orogenic garnet peridotites, and are interpreted as the crystallization products of dense, solute-rich supercritical fluids or melts11,24. Many studies have been undertaken on MSIs in order to reconstruct the original chemical composition of supercritical fluids or melts formed in subduction zones11,13,24. The Plešovice garnet peridotite preserves fossilized melts as MSIs that consist of volatile-rich minerals7,13. Based on rare accessory minerals, including priderite and burbankite, parental melts to the MSIs are speculated to have been kimberlite- or carbonatite-like13, although quantitative estimates of melt compositions are lacking.
All observed Plešovice MSIs are within Chr1 grains enclosed in Grt2/Grt3 crystals. Large grains (>3 mm in length) of Chr1 contain exsolution lamellae of diopside and enstatite12 (Fig. 4a), suggesting that chromite contained pyroxene components during Stage 012 (i.e. Chr0). However, we found no MSIs in these larger Chr1 grains. Smaller Chr0 grains recrystallized into exsolution-free chromite (Chr1) + diopside (Cpx1) + enstatite (Opx1) (Fig. 4b), and most MSIs occur in these exsolution-free Chr1 grains. Chr1 has a relatively high Cr# [molar Cr/(Cr + Al) = 0.57–0.64] but is deficient in Zn (as low as ~350 ppm; Fig. 1b) and equilibrated during Stage 1 (Fig. 1a). MSIs show negative crystal shapes and consist mainly of hydrous aluminosilicate minerals, phosphates and carbonates (Fig. 4c–f). Single-phase phlogopite inclusions also occur within Chr1. The presence of garnet in some MSIs (Supplementary Fig. S2e) suggests that the original melts were stable in the garnet lherzolite facies. For more information on variations in the minerals in MSIs, see the Supplementary Methods (Variations in multiphase solid inclusions and the cut effect on volume estimates and Supplementary Figs S3 and S4).

Panel (a) is a photomicrograph of a chromite grain (Chr1) containing pyroxene lamellae, indicating the former presence of chromite (Chr0) that grew under ultrahigh-pressure (UHP) conditions in the diamond stability field during Stage 012, plane polarized light. Subscript numbers represent crystallization stages of minerals. Panel (b) shows aggregates of chromite (Chr1) + orthopyroxene (Opx1) + clinopyroxene (Cpx1) formed from UHP Chr0 by a recrystallization under the Stage 1. They are surrounded by kelyphite (kely4) of Stage 4. The matrix consists mainly of Stage 3 olivine (Ol3) and orthopyroxene (Opx3), cross polarized light. Panels (c) and (d) show back-scattered electron (BSE) images of multiphase solid inclusions (MSIs) consisting of phlogopite (Phl), calcite (Cal), dolomite (Dol) and apatite (Ap). Panels (e) and (f) show BSE images of MSIs consisting of Cal, Dol and Ap. Mnz = monazite, Str = strontianite, Gr = graphite, Chl = chlorite. Panel (g) is a diagram of F/(F + Cl + OH) versus Cl/(F + Cl + OH) in Ap occurring as MSIs in Chr1, matrix Ap3 and Ap4-proximal kelyphite. Panel (h) is a diagram of Ti versus Na contents in Phl from MSIs within Chr1, matrix Phl3 and Phl4-proximal kelyphite.
We examined 37 polished thin sections by electron microscopy and found 243 inclusions that are free of severe secondary alteration (Supplementary Table S3). These inclusions occur mainly within Type A peridotites (187), but some occur within Type B peridotites (56). The main carbonates in the MSIs are dolomite and calcite, with a dolomite/calcite volume ratio of 2.0 ± 0.6. Rarely, carbonates are magnesite, Ba–Ca–Mg carbonate, strontianite, and burbankite13. Other minerals identified in the MSIs include Nb-rich rutile, a Mn–Fe–Ti–W–Ta phase (possibly koragoite), Cr-priderite13, monazite, a U–Th oxide and various sulphide minerals including maucherite, orcelite, millerite, galena and pentandite (Supplementary Table S3). The occurrence of such a wide variety of minerals in the MSIs can be explained by use of the phase rule (see Supplementary Discussion – application of the phase rule to multiphase solid inclusions). Some MSIs contain chlorite, calcic amphibole, talc, serpentine, brucite, and diaspore. These MSIs are connected with the matrix via annealed fractures (Supplementary Fig. S2f). We did not use such altered MSIs for the reconstruction of melts.
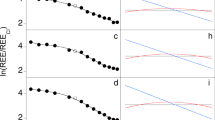
Crystals of apatites and phlogopites are ubiquitous within both the MSIs and the matrix, and their chemical compositions correlate with textures, for both major elements (Fig. 4g,h; Supplementary Table S1) and trace elements (Fig. 5a,b; Supplementary Table S2). Apatite grains in MSIs are rich in F {XF = F/(F + Cl + OH) > 0.45, XCl = Cl/(F + Cl + OH) < 0.1} and Ce relative to U (CeN/UN > 3), and are clearly distinguished from apatites in the surrounding kelyphite (XF < 0.1, XCl > 0.5; CeN/UN < 1) (Figs 4g, 5a). Apatites in the MSIs show high REE contents, with fractionated REE patterns (CeN/YbN ≈ 100) (Fig. 5a). Phlogopite grains in MSIs are strongly deficient in Na ( < 0.03 cations per 23 oxygens) and depleted in Pb and Sr, whereas phlogopite grains in both the matrix and proximal kelyphite are rich in Na (>0.1 cations per 23 oxygens), Pb and Sr (Figs 4h, 5b). The observed large compositional gaps between apatites and phlogopites in MSIs, compared with those in the matrix, suggest that the MSIs formed under different physical and chemical conditions to the matrix minerals.

Panel (a) shows a trace element patterns of apatites (Aps), in which the subscripts indicate the crystallization stage, normalized to primitive mantle values18. MSI = multiphase solid inclusion. Panel (b) shows trace element patterns of phlogopites. Panel (c) displays a trace element pattern for a carbonated potassic silicate (CPS) melt calculated using trace elements in Ap and Phl within MSIs (Supplementary Table S2) in combination with appropriate partitioning coefficients for these minerals and carbonatite melts33,34. For comparison, the trace element patterns of a sediment-derived potassic carbonatite35 are shown. Panel (d) shows a trace element pattern for a carbonatite that formed from the CPS melt by the precipitation of phlogopite. A trace element pattern for global average calcio-carbonatite36 is shown for comparison.
The bulk chemical compositions of MSIs in Type A and Type B peridotites have been reconstructed using the mineral proportions (vol.% in Supplementary Table S3) and mineral compositions (Supplementary Table S1). In spite of the variable proportions of constituent minerals in each of the MSIs due to the cut effect (Supplementary Figs. S3a and S4c), their average volumes converge to constant values (Supplementary Fig. S3b) after the accumulation of more than 80 data points. This verifies our assumption that the chemical compositions of the MSIs are scattered around a meaningful average.
The results are plotted on a ternary diagram (Fig. 6) of (SiO2 + TiO2 + Al2O3), (CaO + FeO + MgO), and (Na2O + K2O) wt.% projected from CO2 and H2O. The parental melt composition is K2O = 5.2 wt.%, CaO = 17 wt.%, MgO = 18 wt.%, CO2 = 22 wt.%, and SiO2 = 20 wt.%, giving an average K2O/Na2O ratio of ~120 (Supplementary Table S1). This chemical composition is similar to that of Group II kimberlites25 and suggests that the carbonated potassic silicate melts (hereafter referred to as CPS) formed in equilibrium with a garnet-harzburgite residue at temperatures above the carbonated peridotite solidus (i.e. T > 1200 °C25). Although the alkali contents in the CPS melts are much higher than those of melts in the peridotite–CO2 system26, the alkali contents are much lower than those of melt inclusions within diamond27 and sediment-derived carbonatites28,29. CPS melts with K2O contents of ~5 wt.%, similar to those measured in our samples, have been produced experimentally as partial melts in the peridotite–CO2 system doped with 1.5 wt.% K2O30 (Fig. 6). Therefore, a source for the CPS melt may have been mantle peridotite that was metasomatized by potassic melts. As shown in Fig. 1, the CPS melt could have been stable within the Plešovice peridotite under the conditions of Stage 1 (~1300 °C and 3 GPa), under which chromite is stable in garnet harzburgite31 (Fig. 1). Upon cooling, the rock would have crossed reaction curve (1) at ~1220 °C32, at which point the melt would have changed to a calcio-carbonatite. We hypothesize that CPS melt crystallizes to form roughly equal proportions of carbonatite melt and phlogopite, resulting in production of a melt with the following composition: MgO = 12 wt.%, CaO = 35 wt.%, CO2 = 47 wt.%, and SiO2 < 1 wt.% (Supplementary Table S1).

This panel shows a summary of carbonated silicate melts preserved as multiphase solid inclusions (MSIs) in the Plešovice peridotite in comparison with those produced by experiments26,27,28,29,30,66,67,68,69. Ternary (wt.%) diagram projected from CO2 and H2O. The average compositions of group II kimberlites25 (GII) and of phlogopite (Phl) are shown. The thick curve represents the solvus for a silicate–carbonatite liquid at 1100 °C and 3.7–5.0 GPa28. The mean value of carbonated melt reconstructed from MSIs in the Plešovice peridotite is represented as a yellow star which is close to the composition of GII kimberlites.
The trace element compositions of the CPS melts were calculated using the compositions of apatite and phlogopite (Supplementary Table S2) using the following relation: Ci (melt) = Di (melt/crystal) × Ci (crystal), where Di represents the partition coefficient of element i33,34 (see Methods). The calculated trace element pattern for the CPS melt is similar to those of sediment-derived carbonatites35 (Fig. 5c), showing significant enrichment in LILE such as Rb, Ba, Th, and U, and light REE. However, the quantity of HFSE in the CPS melt is much lower than in sediment-derived carbonatites35 (Fig. 5c). Fractional crystallization of phlogopite transformed the CPS melt to a carbonatite, whose trace element pattern is represented by the blue line in Fig. 5d. This composition closely resembles that of average calcio-carbonatite lavas worldwide36 (Fig. 5d). Subsequently, the calcio-carbonatite was likely transformed into a carbonatitic fluid through the reaction of carbonatite and enstatite to diopside, forsterite and CO237.
Equilibrium modelling of REE between minerals and melts
Here we aim to decipher the metasomatic signatures recorded in silicate minerals that were stable under the conditions of Stages 1 and 2, and we examined whether the CPS melt and carbonatite were in equilibrium with the residual silicate minerals. Firstly, orthopyroxene and olivine in Type A peridotite have high Mg# [Mg/(Mg + Fe2+)] (Mg#Opx ≈ 92 in Table S1; Mg#Ol ≈ 91.57). Due to their relative insensitivity to changes in T and P, the Mg# of orthopyroxene and olivine serve as indicators of melt depletion19. At ~3 GPa, an olivine Mg# of 91.5 equates broadly to the point of complete clinopyroxene consumption at ~25% partial melting19,38. Modelling that considers batch melting has shown that orthopyroxene in a residual peridotite acquires a REE pattern with a high HREE/LREE ratio19. Therefore, the observed high LREE content in Opx1 cannot be formed as a result of partial melting events, but it requires the metasomatic addition of LREE after partial melting. Here we examine the hypothesis that a CPS melt coexisted with Opx1 during Stage 1. To test this hypothesis, we calculate REE patterns of a hypothetical melt that coexisted with Opx1, utilizing partitioning coefficients determined by an experiment at 1300 °C and 6 GPa (Run#: M35539). As shown in Fig. 7a, the calculated REE pattern of melt coexisting with Opx1 matches fairly well with that of the CPS melt. This suggests that the CPS melt was a metasomatic agent during Stage 1.

Panel (a) shows a calculated REE pattern of a carbonated melt in equilibrium with the core of an orthopyroxene porphyroclast, normalized to primitive mantle (PM) values18. The calculated REE pattern of the melt agrees well with that of the reconstructed REE pattern for the MSIs. Panel (b) shows a result of REE modelling of a reaction between pre-metasomatic garnet40 and a calculated carbonatite melt. The red-coloured curves are calculated patterns for varying volumes of carbonatite melt that reacted with garnet. Garnet–melt partition coefficients are after ref.39. Only 0.5–1.0% carbonatite melt relative to garnet is needed to generate the observed sinuous REE pattern in Type A peridotite.
After Stage 1, the CPS melt crystallized phlogopite, was transformed to carbonatite, and then may have been converted into a carbonaceous fluid during cooling to Stage 2. It has been shown that carbonaceous fluids derived from carbonatite have similar trace element compositions to the source carbonatite40. Both are strongly enriched in incompatible trace elements and have the potential to significantly alter the trace element signatures of Stage 2 minerals. Grt2 in Type A peridotite shows sinuous REE patterns (Fig. 7b) that may have been the product of metasomatism by LREE-rich metasomatic agents41. Here we attempt to reproduce the sinuous REE patterns in Grt2 by modelling interaction between a pre-metasomatic garnet40 and the calculated composition of the carbonatite melt. Firstly, we assume various proportions of melt (0.01, 0.1, 0.5, 2, and 5 wt.%) relative to the mass of pre-metasomatic garnet40. To simulate the reaction between carbonatite melt and garnet, we solved following two equations simultaneously, (1) CREE (garnet) + p × CREE (carbonatite) = constant (mass-balance equation), and (2) CREE (garnet)/CREE (carbonatite) = DREE (garnet/carbonatite)39, where p represents proportion of melt. This modelling shows that only 0.5–1.0 wt.% melt relative to garnet can produce the observed sinuous REE pattern observed in Grt2 grains of Type A peridotites (Fig. 7b). As Type A peridotite contains 2 vol.% garnet, the required melt fraction is only about 0.02–0.04 vol.% (the densities of pyrope and carbonatite are assumed to be 3.7 and 2.2 gcm–3, respectively).
Discussion
We have reconstructed the major and trace element composition of carbonated potassic silicate (CPS) melt inferred to have been present in the mantle wedge during Stage 1 (~1300 °C at 3 GPa). This melt could have been sourced from sedimentary rocks subducted to depths of > 250 km28. Infiltration of the sediment-derived carbonatite (i.e. K2O > 27 wt.%, CaO = 17 wt.%, CO2 = 38 wt.%, SiO2 = 0.3 wt.% in run am-5B28) into the mantle produces a phlogopite-bearing peridotite. Due to the highly reduced conditions in the deep mantle wedge, the carbonatite may have recrystallized to diamond through the reduction of carbon dioxide during Stage 012,42 (Fig. 1). Subsequent convective upwelling of the volatile-rich peridotite induced melting beneath the overriding lithosphere by oxidation42, producing a CPS melt during Stage 1 (Fig. 1). The low abundances of HFSE in the CPS melts can be explained by the persistence of ilmenite in a residual peridotite to temperatures 100–120 °C above the solidus30.
It has been proposed that the P–T paths of mantle wedge peridotites are controlled by corner flow43, which can be envisaged as two types (Supplementary Fig. S5). Models of slab-induced flow in the mantle wedge show that the flow lines are sub-horizontal and directed towards the slab in the uppermost part of the mantle wedge44, consistent with the initial P–T paths observed for the Nonsberg43 and Higashi-Akaishi peridotites45,46. However, this model cannot account for initial upward flow, such as that inferred for the Plešovice and Zhimafang peridotites8. The volatile contents in the mantle wedge potentially control the style of corner flow5, as well as directly influencing the chemical compositions of mantle wedge melts. Mantle wedge peridotites originating in the spinel lherzolite facies record magmatic activity producing high-temperature silicate melts6,45,46, whereas those originating in the garnet lherzolite facies preserve volatile-rich melts as MSIs. In the Zhimafang peridotite, an alkali-rich hydrous silicate melt stable at ~5 GPa has been identified based on the study of MSIs (consisting of calcic amphibole, chlorite, phlogopite, and talc) enclosed within primary garnet11. Thus, we propose that volatile-rich partial melts can create upward flow in the mantle wedge.
When the CPS melt cooled below 1220 °C it started to crystallise phlogopite. Thus, phlogopite crystallization should have taken place near the base of the lithosphere or a short distance above the thermal boundary layer (~1300–1400 °C)47. Recent high-resolution seismic studies have demonstrated a substantial reduction in shear wave velocities (VS) at depths of 60–150 km in the cratonic lithosphere48,49, in the oceanic lithosphere–asthenosphere boundary (LAB)50,51 and in the mantle wedge52. Several hypotheses have been proposed to explain this reduction in VS: 1) the presence of partial melt50,51 2) elastically accommodated grain boundary sliding53 and 3) the presence of hydrous minerals produced through metasomatism48,49. Phlogopite is seismically slow and can cause a reduction in VS. For example, the formation of 5–7 vol.% phlogopite in the lithosphere can reduce VS by up to 6%49. This velocity reduction matches values observed at the oceanic LAB (a 7–9% reduction50,54) and can explain modest velocity reductions observed in the cratonic lithosphere (a 2%–24% reduction48,49).
Recently petrologists have proposed that physical mixing of subducted sediment and mantle wedge peridotites may result in the formation of volatile-rich hybrid rocks in low-temperature subduction zones55. Such hybrid rocks could create low-VS regions in the subduction mélange. In warmer subduction/collision zones, subducted sediment would produce potassic carbonatites28. Interaction between potassic carbonatites and mantle peridotites results in the creation of phlogopite-bearing carbonated peridotite5 or a phlogopite-rich metasomatic layer (i.e. glimmerite56). In both cases, blebs of hybrid rocks or phlogopite-bearing carbonated peridotite/glimmerite will rise buoyantly from the surface of the subducting slab into the sub-arc mantle, producing low seismic anomalies along their ascent pathways55. Finally, these diapirs would melt to produce potassic melts. Melting of phlogopite in the mantle wedge generates ultrapotassic magmas with high K2O and MgO (>3 wt.%)57, such as those observed in the Bohemian massif. Orogenic garnet peridotite in the Bohemian Massif (Austria) contains glimmerite veins58 as well as CPS melts. Both are key features that give rise to orogenic ultrapotassic magmatism.
In this study, we constrained the composition of mantle wedge melts. High contents of LILE, LREE and carbon in the CPS melts were likely derived from subducted carbonate-rich sediments28,56. Although the presence of this type of potassium- and volatile-enriched melt in the asthenosphere has been speculated to exist59,60, and is sometimes referred to as “failed kimberlite”25, this study is the first to report a natural example from mantle wedge samples. Such CPS melts may be ubiquitous in the deep mantle wedge, but they are rarely present in sufficient quantity to reach the Earth’s surface. Due to small melt fractions, the CPS melts are expected to have differentiated to carbonaceous fluid within the lithosphere, with carbon dioxide returned to the surface through arc volcanism1.
Methods
Major element analyses were performed using an EPMA (JEOL JXA-8900L at the University of Tokyo) and a FE-EPMA (JEOL JSM-8350F, at the University of Tokyo). Most silicate minerals, carbonates, phlogopite, and phosphates were analysed by EPMA. The analytical conditions were 15 kV, a surface beam current of 12 nA [1–3 µm beam diameter (b.d.)] for silicates and phosphates, and 3 nA (scanning mode with magnification of 20,000) for carbonate with a ZAF correction. The sulphides, priderite, burbankite, Ba-rich carbonates, and monazite were analysed by FE-EPMA. Analytical conditions were 15 kV and a surface beam current of 6 nA (3 μm b.d.) for priderite, 15 kV and 12 nA (1 um b.d.) for sulphides, and 25 kV and 20 nA (10 μm beam diameter) for Ba-rich carbonates, burbankite, and monazite. Oxides, silicate minerals, and natural and synthetic carbonates with known compositions were used as analytical standards.
Trace element analyses were performed on an iCAP Q inductively coupled plasma mass spectrometer (Thermo Fisher Scientific) attached to an LSX-213 G2 + Nd:YAG laser ablation system (Cetac Technologies) at the University of Tokyo. Each analysis consisted of a 25-sec baseline measurement followed by a ~35-sec measurement with laser ablation sampling. The data were obtained from 15–25 µm ablation pits with a laser repetition rate of 4–8 Hz. An external calibration was made against NIST 611 SRM, and an internal standard calibration was applied using the Ca (apatite and garnet), Mn (chromite and orthopyroxene) and K (phlogopite) concentrations determined by EPMA.
Trace element concentrations in melts were calculated using those in apatite and phlogopite in multiphase solid inclusions (MSIs). According to the phase diagram and crystal shapes, the first mineral to crystallize from parental melt is phlogopite followed by apatite. The phlogopite crystallization reaction is as follows: Melt → 0.474 residual melt-1 + 0.526 phlogopite (wt.% basis). The concentrations of Na, Rb, Cs, Ba, Ta, Nb, Hf, Zr and Ni in a residual melt-1 were calculated using those in phlogopite grains in conjunction with trace element partitioning coefficients between phlogopite and carbonatite34. The apatite crystallization reaction is as follows: Melt → 0.442 residual melt-2 + 0.526 phlogopite + 0.032 apatite. Some trace elements (Pb, Th, U, and REE) in the residual melt (melt-2) were estimated using those in the apatite grains in combination with trace element partitioning coefficients between apatite and carbonatite33.
Partitioning coefficients of Rb, Ba, Nb, and Zr between phlogopite and carbonatite are from ref.34, and those of Cs and Ta are from ref.61. The partitioning coefficients of Hf was assumed to be 0.1962, which agrees with the average value in the literature63. In order to reduce the error of partitioning coefficients between apatite and carbonatite33, we calculated the standard error by dividing σ by the square root of the number of data (n = 60) (i.e. σ/√n)33.
The calibration of Zn in chromite geothermometer of ref.17 {T = h (Zn ppm)} adopted the temperature based on the Ni-in-garnet geothermometer of ref.17 {T = f (Ni ppm)}, which has been shown to be inconsistent with the calibration proposed by ref.64 {T = g (Ni ppm)}. Therefore, we corrected Zn-in-chromite thermometer as follows: T = g {f −1 {h (Zn ppm)}} to ensure that it is consistent with the calibration proposed by ref.64.
Change history
12 February 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Hayes, J. M. & Waldbauer, J. R. The carbon cycle and associated redox processes through time. Phil. Trans. R. Soc. Lond. B. 361, 931–950 (2006).
Gorman, P. J., Kerrick, D. M. & Connolly, J. A. D. Modeling open system metamorphic decarbonation of subducting slabs. Geochem. Geophys. Geosyst. 7, https://doi.org/10.1029/2005GC001125 (2006).
Poli, S. Carbon mobilized at shallow depths in subduction zones by carbonatitic liquids. Nature Geoscience 8, 633–636 (2015).
Tsuno, K., Dasgupta, R., Danielson, L. & Righter, K. Flux of carbonate melt from deeply subducted pelitic sediments: Geophysical and geochemical implications for the source of Central American volcanic arc. Geophys. Res. Lett. L16307, https://doi.org/10.1029/2012GL052606 (2012).
Tumiati, S., Fumagalli, P., Tiraboschi, C. & Poli, S. An experimental study on COH-bearing peridotite up to 3·2GPa and implications for crust-mantle recycling. J. Petrology 54, 453–479 (2012).
Scambelluri, M., Hermann, J., Morten, L. & Rampone, E. Melt- versus fluid-induced metasomatism in spinel to garnet wedge peridotites (Ulten Zone, Eastern Italian Alps): clues from trace element and Li abundances. Contrib. Mineral. Petrol. 151, 372–394 (2006).
Naemura, K., Hirajima, T. & Svojtka, M. The Pressure-Temperature Path and the Origin of Phlogopite in Spinel-Garnet Peridotites from the Blanský Les Massif of the Moldanubian Zone, Czech Republic. J. Petrology 50, 1795–1827 (2009).
Ye, K. et al. Multistage metamorphism of orogenic garnet–lherzolite from Zhimafang, Sulu UHP terrane, E. China: Implications for mantle wedge convection during progressive oceanic and continental subduction. Lithos 109, 155–175 (2009).
Scambelluri, M., Pettke, T. & van Roermund, H. L. M. Majoritic garnets monitoring deep subduction fluid flow and mantle dynamics. Geology 36, 59–62 (2008).
Marocchi, M., Hermann, J. & Morten, L. Evidence for multi-stage metasomatism of chlorite-amphibole peridotites (Ulten Zone, Italy): Constraints from trace element compositions of hydrous phases. Lithos 99, 85–104 (2007).
Malaspina, N., Hermann, J. & Scambelluri, M. Fluid/Mineral interaction in UHP garnet peridotite. Lithos 107, 38–52 (2009).
Naemura, K. et al. Diamond and other possible ultra-deep evidence discovered in the orogenic spinel-garnet peridotite from the Moldanubian zone of the Bohemian Massif, Czech Republic. In Dobrzhinetskaya, R., Faryad, S.W., Wallis, S. & Cuthbert, S. (Eds.), Ultrahigh-Pressure metamorphism: 25 Years After The Discovery of Coesite and Diamond 77–111 (Elsevier, 2011).
Naemura, K., Shimizu, I., Svojtka, M. & Hirajima, T. Accessory priderite and burbankite in multiphase solid inclusions in the orogenic garnet peridotite from the Bohemian Massif, Czech Republic. J. Mineral. Petrol. Sci. 110, 20–28 (2015).
Medaris, G. Jr., Wang, H., Jelínek, E., Mihaljevič, M. & Jakeš, P. Characteristics and origins of diverse Variscan peridotites in the Gföhl Nappe, Bohemian Massif, Czech Republic. Lithos 82, 1–23 (2005).
Vrána, S. & Šrámek, J. Geological interpretation of detailed gravity survey of the granulite complex in southern Bohemia and its structure. Vĕstník Českého geologického ústavu = Bulletin of the Geological Society, Prague 74, 261–278 (1999).
Whitney, D. L. & Evans, B. W. Abbreviation of rock-forming minerals. American Mineralogist 95, 185–187 (2010).
Ryan, C. G., Griffin, W. L. & Pearson, N. J. Garnet geotherms: Pressure-temperature data from Cr-pyrope garnet xenocrysts in volcanic rocks. Journal of Geophysical Research: Solid Earth 101, 5611–5625 (1996).
McDonough, W. F. & Sun, S. S. The composition of the Earth. Chemical Geology 120, 223–253 (1995).
Scott, J. M., Liu, J., Pearson, D. G. & Waight, T. E. Mantle depletion and metasomatism recorded in orthopyroxene in highly depleted peridotites. Chemical Geology 441, 280–291 (2016).
Kessel, R., Fumagalli, P. & Pettke, T. The behavior of incompatible elements during hydrous melting of metasomatized peridotite at 4–6 GPa and 1000 °C–1200 °C. Lithos 236/237, 141–155 (2015).
Nakamura, D. A new formulation of garnet–clinopyroxene geothermometer based on accumulation and statistical analysis of a large experimental data set. Journal of Metamorphic Geology 27, 495–508 (2009).
Nimis, P. & Taylor, W. R. Single clinopyroxene thermobarometry for garnet peridotites. Part I. Calibration and testing of a Cr-in-Cpx barometer and and En-in-Cpx thermometer. Contrib. Mineral. Petrol. 139, 541–554 (2000).
Ashchepkov, I. V., André, L., Downes, H. & Belyatsky, B. A. Pyroxenites and megacrysts from Vitim picrite-basalts (Russia): Polybaric fractionation of rising melts in the mantle? J. Asian Earth Sci. 42, 14–37 (2011).
Frezzotti, M. & Ferrando, S. The chemical behavior of fluids released during deep subduction based on fluid inclusions. American Mineralogist 100, 352–377 (2015).
Ulmer, P. & Sweeney, R. J. Generation and differentiation of group II kimberlites: Constraints from a high-pressure experimental study to 10 GPa. Geochim. Cosmochim. Acta 66, 2139–2153 (2002).
Ghosh, S., Litasov, K. & Ohtani, E. Phase relations and melting of carbonated peridotite between 10 and 20 GPa: a proxy for alkali- and CO2-rich silicate melts in the deep mantle. Contrib. Mineral. Petrol. 167, https://doi.org/10.1007/s00410-014-0964-z (2014).
Klein-BenDavid, O., Izraeli, E. S., Hauri, E. & Navon, O. Fluid inclusions in diamonds from the Diavik mine, Canada and the evolution of diamond-forming fluids. Geochim. Cosmochim. Acta 71, 723–744 (2007).
Grassi, D. & Schmidt, M. W. Melting of carbonated pelites at 8–13 GPa: Generating K-rich carbonatites for mantle metasomatism. Contrib. Mineral. Petrol. 162, 169–191 (2011).
Brey G. et al. Reduced sediment melting at 7.5–12 GPa: phase relations, geochemical signals and diamond nucleation. Contrib. Mineral. Petrol. 170, https://doi.org/10.1007/s00410-015-1166-z (2015).
Foley, S. F. et al. The composition of near solidus melts of peridotite in the presence of CO2 and H2O between 40 and 60 kbar. Lithos 112, 274–283 (2009).
Ziberna, L., Klemme, S. & Nimis, P. Garnet and spinel in fertile and depleted mantle: insights from thermodynamic modelling. Contrib. Mineral. Petrol. 166, 411–421 (2013).
Enggist, A., Chu, L. & Luth, R. W. Phase relations of phlogopite with magnesite from 4 to 8 GPa. Contrib. Mineral. Petrol. 163, 467–481 (2012).
Guzmics, T. et al. LA-ICP-MS study of apatite- and K feldspar-hosted primary carbonatite melt inclusions in clinopyroxenite xenoliths from lamprophyres, Hungary: Implications for significance of carbonatite melts in the Earth’s mantle. Geochim. Cosmochim. Acta 72, 1864–1886 (2008).
Sweeney, R. J., Prozesky, V. & Przybylowicz, W. Selected trace and minor element partitioning between peridotite minerals and carbonatite melts at 18-46 kb pressure. Geochim. Cosmochim. Acta 59, 3671–3683 (1995).
Grassi, D., Schmidt, Max, W. & Günther, D. Element partitioning during carbonated pelite melting at 8, 13 and 22 GPa and the sediment signature in the EM mantle components. Earth Planet. Sci. Lett. 327/328, 84–96 (2012).
Wooley, A.R. and Kemp, D.R.C. Carbonatites: nomenclature, average chemical compositions, and element distribution. In Bell, K. (Ed.) Carbonatites, Genesis and Evolution 1-14, Unwin Hyman, London (1989).
Dasgupta, R. et al. Carbon-dioxide-rich silicate melt in the Earth’s upper mantle. Nature 493, 211–215 (2013).
Walter, M. J. Melting of Garnet Peridotite and the Origin of Komatiite and Depleted Lithosphere. Journal of Petrology 39, 29–60 (1998).
Girnis, A. V., Bulatov, V. K., Brey, G. P., Gerdes, A. & Höfer, H. E. Trace element partitioning between mantle minerals and silico-carbonate melts at 6–12 GPa and applications to mantle metasomatism and kimberlite genesis. Lithos 160/161, 183–200 (2013).
Stachel, T. et al. The trace element composition of silicate inclusions in diamonds: a review. Lithos 77, 1–19 (2004).
Shu, Q. & Brey, P. G. Ancient mantle metasomatism recorded in subcalcic garnet xenocrysts: Temporal links between mantle metasomatism, diamond growth and crustal tectonomagmatism. Earth Planet. Sci. Lett. 418, 27–39 (2015).
Thomson, A. R., Walter, M. J., Kohn, S. C. & Brooker, R. A. Slab melting as a barrier to deep carbon subduction. Nature 529, 76–79 (2016).
Nimis, P. & Morten, L. P-T. evolution of ‘crustal’ garnet peridotites and included pyroxenites from Nonsberg area (upper Austroalpine), NE Italy: from the wedge to the slab. Journal of Geodynamics 30, 93-115 (2000).
Peacock, S. M. Thermal and petrological structure of subduction zones. In Bebout, D.W., Scholl, S.H., Kirby, J.P. & Platt, X.X. (Ed.) Subduction: Top to Bottom Vol. 96, G.E. 119–133 (AGU Geophys. Monogr., 1996).
Mizukami, T., Wallis, S. R. & Yamamoto, J. Natural examples of olivine lattice preferred orientation patterns with a flow-normal a-axis maximum. Nature 427, 432–436 (2004).
Hattori, K., Wallis, S., Enami, M. & Mizukami, T. Subduction of mantle wedge peridotites: Evidence from the Higashi-akaishi ultramafic body in the Sanbagawa metamorphic belt. Island Arc 19, 192–207 (2010).
McKenzie, D., Jackson, J. & Priestly, K. Thermal structure of oceanic and continental lithosphere. Earth and Planetary Science Letters 233, 337–349 (2005).
Chen, C. W., Rondenay, S., Evans, R. L. & Snyder, D. B. Geophysical Detection of Relict Metasomatism from an Archean (~3.5 Ga) Subduction Zone. Science 326, 1089–1091 (2009).
Saha, S, Dasgupta, R. & Tsuno, K. High Pressure Phase Relations of a Depleted Peridotite Fluxed by CO2-H2O bearing Siliceous Melts and the Origin of Mid-Lithospheric Discontinuity. Geochemistry, Geophysics, Geosystems, https://doi.org/10.1002/2017GC007233 (2017).
Kawakatsu, S. et al. Seismic Evidence for Sharp Lithosphere-Asthenosphere Boundaries of Oceanic Plates. Science 324, 499–502 (2009).
Rychert, C., Harmon, N. & Ebinger, C. Receiver function imaging of lithospheric structure and the onset of melting beneath the Galápagos Archipelago. Earth and Planetary Science Letters 388, 156–165 (2014).
Nakajima, J., Takei, Y. & Hasegawa, A. Quantitative analysis of the inclined low-velocity zone in the mantle wedge of northeastern Japan: A systematic change of melt-filled pore shapes with depth and its implications for melt migration. Earth and Planetary Science Letters 234, 59–70 (2005).
Karato, S., Olugboji, T. & Park, J. Mechanisms and geologic significance of the mid-lithosphere discontinuity in the continents. Nature Geoscience 8, 509–514 (2015).
Karato, S. On the origin of the asthenosphere. Earth and Planetary Science Letters 321-322, 95–103 (2012).
Marschall, H. R. & Schumacher, J. C. Arc magmas sourced from mélange diapirs in subduction zones. Nature Geoscience 5, 862–867 (2012).
Avanzinelli, R., Lustrino, M., Mattei, M., Melluso, L. & Conticelli, S. Potassic and ultrapotassic magmatism in the circum-Tyrrhenian region: Significance of carbonated pelitic vs. pelitic sediment recycling at destructive plate margins. Lithos 113, 213–227 (2009).
Foley, S. F., Venturelli, G., Green, D. H. & Toscani, L. The ultrapotassic rocks: Characteristics, classification, and constraints for petrogenetic models. Earth Science Reviews 24, 81–134 (1987).
Becker, H., Wenzel, T. & Volker, F. Geochemistry of glimmerite veins in peridotites from lower Austria—Implications for the origin of K-rich magmas in collision zones. J. Petrology 40, 315–338 (1999).
Gaillard, F., Malki, M., Iacono-Marziano, G. & Pichavant, M. & Scaille, B. Carbonatite Melts and Electrical Conductivity in the Asthenosphere. Nature 322, 1363–1365 (2008).
McKenzie, D. Some remarks on the movement of small melt fractions in the mantle. Earth Planet. Sci. Lett. 95, 53–72 (1989).
Foley, S. F., Jackson, S. E., Fryer, B. J., Greenough, J. D. & Jenner, G. A. Trace element partition coefficients for clinopyroxene and phlogopite in an alkaline lamprophyre from Newfoundland by LAM-ICP-MS. Geochem. Cosmochim. Acta 60, 629–638 (1996).
La Tourette, T., Hervig, R. L. & Holloway, J. R. Trace element partitioning between amphibole, phlogopite, and basanite melt. Earth Planet. Sci. Lett. 135, 13–30 (1995).
Reguir, E. P., Chakhmouradian, A. R., Halden, N. M., Malkovets, V. G. & Yang, P. Major- and trace element compositional variation of phlogopite from kimberlites and carbonatites as a petrogenetic indicator. Lithos 112S, 372–384 (2009).
Canil, D. The Ni-in-garnet geothermometer: calibration at natural abundances. Contrib. Mineral. Petrol. 136, 240–246 (1999).
Day, H. W. A revised diamond-graphite transition curve. American Mineralogist 97, 52–62 (2012).
Dasgupta, R. & Hirschmann, M. M. Effect of variable carbonate concentration on the solidus of mantle peridotite. American Mineralogist 92, 370–379 (2007).
Dasgupta, R. & Hirschmann, M. M. A modified iterative sandwich method for determination of near-solidus partial melt compositions. II. Application to determination of near-solidus melt compositions of carbonated peridotite. Contrib. Mineral. Petrol. 154, 647–661 (2007).
Dasgupta, R. et al. Trace element partitioning between garnet lherzolite and carbonatite at 6.6 and 8.6 GPa with applications to the geochemistry of the mantle and of mantle-derived melts. Chemical Geology 262, 57–77 (2009).
Ghosh, S., Ohtani, E., Litasov, K. & Terasaki, H. Solidus of carbonated peridotite from 10 to 20 GPa and origin of magnesiocarbonatite melt in the Earth’s deep mantle. Chemical Geology 262, 17–28 (2009).
Acknowledgements
We thank Hideto Yoshida and Koji Ichimura for their assistance with electron probe microanalysis at the University of Tokyo. KN sincerely thanks Prof. Kazuhito Ozawa for his position as JSPS Postdoctoral Fellow at the University of Tokyo. We are grateful to Hisao Tsutsumi and Masaki Takaya for preparing thin sections. This manuscript benefited greatly from the constructive and significant comments from two anonymous reviewers, and we thank the Editor Prof. Hiroaki Ohfuji. This work was supported by a Grant-in-Aid (248040) to KN, Grants-in-Aid (14403013 & 25257208) to TH, by the Institute of Geology of the Czech Academy of Sciences institutional support RVO67985831 to MS, a Grant-in-Aid (21109005) to IS, and a Grant-in-Aid (15H05833) to TI.
Author information
Authors and Affiliations
Contributions
K.N. designed the study, carried out the analyses, and wrote the manuscript. T.H. and M.S. collected the samples, which are referred to as Type A peridotite in the manuscript. I.S. helped identify minerals via SEM-EDS. T.I. carried out LA–ICP–MS measurements. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Naemura, K., Hirajima, T., Svojtka, M. et al. Fossilized Melts in Mantle Wedge Peridotites. Sci Rep 8, 10116 (2018). https://doi.org/10.1038/s41598-018-28264-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-28264-6
This article is cited by
-
Crystallisation of trapped carbonate–silicate melts terminating at the carbonated solidus ledge: a record of carbon immobilisation mechanism in the lithospheric mantle
Contributions to Mineralogy and Petrology (2024)
-
Experimental production of K-rich metasomes through sediment recycling at the slab-mantle interface in the fore-arc
Scientific Reports (2023)
-
Decoupling of Mg from Sr–Nd isotopic compositions in Variscan subduction-related plutonic rocks from the Bohemian Massif: implications for mantle enrichment processes and genesis of orogenic ultrapotassic magmatic rocks
International Journal of Earth Sciences (2022)
-
Ultrapotassic magmatism in the heyday of the Variscan Orogeny: the story of the Třebíč Pluton, the largest durbachitic body in the Bohemian Massif
International Journal of Earth Sciences (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
