
Abstract
Inflammation plays an important role in the pathogenesis of renal and cardiovascular disease in renovascular hypertension (RVH). Ccl2 is an important mediator of inflammation, and is induced within 24 hours following surgery to establish RVH in the murine 2 kidney 1 clip model, a time prior to onset of interstitial inflammation, fibrosis, or tubular atrophy. We tested the hypothesis that Ccl2 deficiency protects the stenotic kidney (STK) from development of chronic renal damage in mice with renovascular hypertension due to renal artery stenosis (RAS). RAS surgery was performed on wild type (WT) and Ccl2 knock out (KO) mice; animals were studied for four weeks. Renal blood flow was reduced to similar extent in both WT and Ccl2 KO mice with RVH. Perfusion of the stenotic kidney was significantly reduced in Ccl2 KO mice as assessed by magnetic resonance imaging (MRI). Stenotic kidney volume in WT, but not in Ccl2 KO mice, was significantly reduced following surgery. Cortical hypoxia was observed in the stenotic kidney of Ccl2 KO mice, as assessed by blood oxygen level-dependent MRI (BOLD-MRI). Ccl2 KO mice showed less cortical atrophy than WT RAS mice. Ccl2 deficiency reduced the number of infiltrating mononuclear cells and expression of Ccl5, Ccl7, Ccl8, Ccr2 and Cd206. We conclude that Ccl2 is a critical mediator of chronic renal injury in RVH.
Similar content being viewed by others


Introduction
Recent studies have linked both local and systemic inflammation in the pathogenesis of renal and cardiovascular disease in patients with renovascular hypertension (RVH)1,2. Indeed, systemic inflammation may be responsible for the significantly increased risk of mortality from cardiovascular disease in patients with chronic kidney disease (CKD)3. Chronic angiotensin II infusion promotes cardiac hypertrophy and remodeling where renal infiltration of macrophages (Mø) and T cells occurs as an early event4,5,6. In the two kidney 1 clip (2K1C) model of RVH, influx of inflammatory cells into the stenotic kidney occurs within one week and precedes the development of severe interstitial fibrosis and tubular atrophy7,8,9. Immunosuppressive therapy, at least in part, ameliorates renal damage in the angiotensin II infusion model10. These studies support the notion that therapies to target inflammation may have a role in the management of patients with RVH.
Of the many pro-inflammatory cytokines/chemokines, monocyte chemoattractant protein-1 (MCP-1) or Ccl2 has emerged as a key mediator of inflammation following tissue injury. Ccl2 is a member of the C-C chemokine family and signals at least in part through its receptor CCR2. Both Ccl2 and its receptor CCR2 are induced and are mechanistically linked to the pathogenesis of many chronic diseases, including neurodegenerative diseases11,12, inflammatory bowel disease13, rheumatoid arthritis14, inflammation-associated insulin resistance15,16, obesity, diabetes, cardiovascular diseases17, and atherosclerosis18.
Ccl2 has also been implicated as a key mediator of chronic kidney disease in animal models19,20,21,22 as well as in humans23,24. Pharmacological inhibition of Ccl2 has been shown to reduce chronic renal damage in lupus nephritis25, improve podocyte function in diabetic nephropathy26, and to improve renal function in diabetic patients with albuminuria27. Inhibition of the renin-angiotensin system reduces Ccl2 mediated inflammation in a variety of chronic injury models6,28,29.
Using the murine 2K1C model of RVH, we have previously shown that Ccl2 is induced within 24 hours of surgery to establish RAS, prior to the onset of interstitial inflammation, interstitial fibrosis, or tubular atrophy20. A CCR2 selective inhibitor protected the stenotic kidney from development of chronic renal damage in this model20. It is recognized that other ligands (such as MCP3/CCL7) signal through CCR230,31. It is also recognized that pharmacological inhibition might have “off-label” effects that may not be due to specific CCR2 inhibition. Based on these considerations, we tested the hypothesis that Ccl2 deficiency alone can protect the stenotic kidney from the development of severe interstitial fibrosis, tubular atrophy, and interstitial inflammation in mice with homozygous deletion of the Ccl2 gene (Ccl2 KO mice) subjected to unilateral renal artery stenosis.
Materials and Methods
Animals
Male Ccl2 knockout (B6. 129S4-Ccl2tm1Rol/J) (KO) and wild type (C57BL6/J) (WT) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). WT mice (N = 27 RAS, N = 9 sham) and Ccl2 KO (N = 24 RAS, N = 9 sham) mice underwent RAS or Sham surgery at 8–10 weeks of age. RAS surgery was done by placing a polytetrafluoroethylene cuff around the right renal artery using two sutures to secure the cuff in place as described previously32; sham surgeries were performed by isolating and manipulating the right renal artery without cuff placement. All of the animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee (IACUC) prior to conducting any experiment. These animal procedures were conducted in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Imaging protocol and Analysis
MRI studies were performed on a subset of animals (N = 13 WT RAS, N = 4 WT Sham, N = 12 Ccl2 KO RAS, N = 4 Ccl2 KO Sham) employing a vertical 16.4T scanner equipped with a 38 mm inner diameter birdcage coil (Bruker Biospin, Billerica, MA). The images for the kidney volume determination were collected axially using respiratory gated 3D Fast Imaging with Steady Precession (3D-FISP) and reconstructed into 3D volumetric images. The kidney volume for cortex and medulla was then assessed from 3D-FISP using Analyze software (Biomedical Imaging Resource, Mayo Clinic, MN) and reported as sum of both cortex and medulla. Perfusion was assessed by arterial spin labelling (ASL) method by using a Flow-sensitive Alternating Inversion Recovery (FAIR) sequence with Rapid Acquisition with Relaxation Enhancement (RARE) as described earlier33. Perfusion of cortex and medulla in the stenotic kidney was calculated using the ASL tool module of Bruker-Paravision software and reported as the sum of both compartments. Blood flow for cortex and medulla was calculated by multiplying the perfusion times the volume of corresponding compartment and reported as their sum.
Renal oxygenation was assessed by BOLD-MRI imaging using a respiration-gated 3D multi-echo gradient echo sequence33. The imaging parameters used were: TR 200 ms; TE 3.5–24.5 ms; echo number 8; flip-angle 25°; slab thickness 1 mm; FOV 2.56 × 2.56 cm2; matrix size 128 × 128 × 8; number of averages 2.” 8 images were reconstructed after zero-filling the k-space data to 256 × 256 for BOLD MRI. T2* was quantified by pixel-wise mono-exponential fitting on the averaged magnitude of all 8 images over echo times. R2* (1/T2*), was used as an index of blood oxygenation level. ROIs were drawn on T2 images for BOLD analysis.
Animal Blood Pressure Measurement
Non-invasive method using the tail cuff and volume pressure recording (VPR) sensor technology was used on conscious mice to record the blood pressure measurements at baseline and following surgery (CODA 6, Kent Scientific Corporation, Torrington, Connecticut, USA).
Plasma Renin Activity
Plasma was available from 21 WT RAS, 7 WT Sham, 21 Ccl2 KO RAS, and 8 Ccl2 KO Sham mice. Blood was collected via the inferior vena cava four weeks after surgery; plasma was separated from it by centrifugation and stored at −80 °C until the time of assay. Quantitative determination of plasma renin content was done by radioimmunoassay as described earlier20.
Renal Function
Renal function was assessed by the measurement of serum creatinine and blood urea nitrogen (BUN) levels at 24 h after ischemia using a Creatinine Analyzer 2 and a BUN Analyzer 2 (Beckman Instruments, Fullerton, CA) as previously described34.
Histology and immunohistochemical analysis
Histologic and immunohistochemical analysis was performed on the subset of animals for which imaging studies were performed (see above). The kidney tissues were fixed in 10% neutral buffered formalin and processed using standard techniques. 5 µm histological sections were prepared and stained with hematoxylin-eosin (H&E). Immunohistochemical staining was done for anti-F4/80 (1:200, Abd Serotec Raleigh, NC) and anti-CD3 (1:100, Dako Agilent, Santa Clara, CA). Sections were stained with Sirius red to quantitate matrix deposition. All measurements and quantifications were performed in a blinded fashion using Olympus Bx50 microscope (Olympus Optical Co. Ltd., Buffalo Grove, IL, USA), Micropublisher 3.3 RTV camera (QImaging, Surrey, BC, Canada). The area of atrophy was calculated on H&E sections as percentage of atrophic tubules over the cortical area, as previously described9,20.
Quantitative analysis for F4/80 CD3, and Sirius red was done by calculating % positive stained area for F4/80, CD3, and Sirius red using NIS elements BR 4.13.00 64-bit image analysis system (Nikon Instruments INC., Melville, NY) at 200X magnification.
Real-time PCR
Total RNA was extracted from stenotic kidney tissues using RNeasy plus Mini kit (Qiagen, Valencia, CA) (N = 17 WT RAS, N = 6 WT Sham, N = 20 Ccl2 KO RAS, N = 8 KO Sham). RNA quantification was done using spectrophotometry (NanoDrop Technologies, Wilmington, DE). RNA quality was assessed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). First-strand cDNA was prepared from total RNA using iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). Real time PCR amplification reactions were performed on a Bio-Rad IQ5 real-time PCR detection system. The sequences for primers used in the studies were described earlier20 except Gata3 (Forward:5′-AAG CTC AGT ATC CGC TGA CG-3′ Reverse: 5′-GAC ACC TCT GCA CCG TAG CC-3′) and Rorc (Forward: 5′-CCC TAG GCT TGC CTT GTA GG-3′ Reverse:5′-CCC ATC TAC CCC ACA GCT TC-3′). Commercially available primers for Gapdh, Acta2 and Col3A were used (ThermoFisher Scientific, Waltham, MA).
Statistical analysis
Data are presented as means ± SEM. ANOVA or t-test were performed for comparison between groups and post hoc Tukey/Dunn’s correction was used for multiple comparisons. P values < 0.05 were considered as significant. Statistical analyses were performed with GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Data availability
All data generated or analyzed in this project are included within this paper.
Results
Blood pressure was elevated to a similar extent in Ccl2 KO RAS and WT RAS mice
There were no differences in the total body weight of WT RAS, WT sham, Ccl2 KO RAS and Ccl2 sham mice. The mean systolic blood pressure in both WT and KO mice following surgery to establish unilateral renal artery stenosis was significantly higher compared to sham mice. However, there was no significant difference in blood pressure measured in WT RAS and Ccl2 KO RAS mice (Fig. 1a). As expected, the weight of the cuffed kidney was significantly reduced, compared to sham (Fig. 1b). In WT mice, the contralateral kidney became hypertrophic, as evidenced by a significant increase in weight (Fig. 1c). The heart weight was also increased in the WT RAS mice (Fig. 1d). In contrast, the contralateral kidney weights of the Ccl2 KO RAS mice was similar to that of Ccl2 sham mice (Fig. 1c) and the heart weights of Ccl2 KO RAS and Ccl2 KO sham mice were similar (Fig. 1d). There were no significant differences in plasma BUN, creatinine or plasma renin activity between any of the groups.

Blood Pressure, heart, and kidney weights of Ccl2 KO and WT mice. (a) Weekly blood pressure measurements in Ccl2 KO and WT mice. (b) Weight of the stenotic (cuffed) kidney, assessed at 4 weeks following surgery (STK = stenotic kidney). (c) Weight of the contralateral kidney, assessed at 4 weeks following surgery (CLK = contralateral kidney). (d) Heart weight, assessed at 4 weeks following surgery. *p < 0.0001, **p < 0.01, ***p < 0.05.
Renal Blood flow was reduced to a similar extent in WT and Ccl2 KO RAS mice as assessed by MRI
The renal blood flow in both WT and Ccl2 KO RAS mice was reduced to similar extent following surgery to establish RVH (Fig. 2). The volume of the STK in WT RAS was significantly reduced compared to the pre-surgery baseline but no differences were observed between Ccl2 KO RAS and WT RAS mice at 3 weeks following surgery (1 week prior to the end of the experiment) (Fig. 2). Perfusion of the stenotic kidney in Ccl2 KO mice was significantly reduced, compared to its pre-surgery baseline (Fig. 3). There were no significant differences in perfusion of the stenotic kidneys of WT RAS or Ccl2 KO RAS mice. KO RAS mice showed significant elevation in cortical R2*, which is a measure of hypoxia following RAS at 3 weeks compared to its baseline. Although the stenotic kidneys of WT RAS mice showed higher R2* values compared to their baseline, this difference did not reach significance (Fig. 4).

In vivo assessment of kidney size and blood flow by MRI 3 weeks following RAS surgery. (a) Coronal cross section MR images of WT and Ccl2 KO mice showing stenotic and contralateral kidney after RAS surgery. (b) Assessment of renal volume following RAS surgery. (c) Renal blood flow measured in the stenotic kidney. (‘B’ = baseline measurement assessed prior to surgery, ‘RAS’ = renal volume measurement at 3 weeks after surgery). *p ≤ 0.001, **p < 0.05.

In vivo assessment of renal perfusion by MRI 3 weeks following RAS surgery. (a) Representative axial cross sectional MR image of the stenotic and contralateral kidney. (b) Representative perfusion map of renal cortex and medulla. (c) Assessment of renal perfusion at baseline and following RAS surgery. (‘B’ = baseline measurement prior to surgery, ‘RAS’ = renal perfusion measurement at 3 weeks after surgery). *p = 0.01.

In vivo assessment of renal oxygenation by MR BOLD 3 weeks following RAS surgery. (a) Representative R2* maps obtained by blood oxygen dependent (BOLD) MRI for WT and Ccl2 KO mice following surgery. The increase in hypoxia is indicated by change in color intensity from yellow to red. (b) R2* value of renal cortex in STK in WT and KO mice at baseline and following surgery. (‘B’ = baseline measurement prior to surgery, ‘RAS’ = renal oxygenation measurement at 3 weeks after surgery). *p < 0.05.
The cuffed kidneys from Ccl2 KO RAS mice were protected from development of chronic renal damage
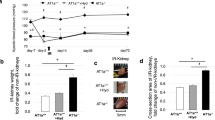
The stenotic kidney of KO mice showed protection against renal damage as assessed by “% atrophy cortical area” compared to WT mice (Fig. 5a,b) despite a similar elevation in blood pressure (Fig. 1a).

The stenotic kidney of Ccl2 KO mice is protected from development of chronic renal injury. (a) Representative histological images showing WT and KO following RAS/sham at 4 weeks following surgery (stained with H&E, 200X magnification). Scale bar represents 100 µm. (b) Semiquantitative assessment of tubular atrophy at 4 weeks following surgery. *p < 0.0001, **p < 0.05.
The extent of interstitial fibrosis in Ccl2 KO RAS mice was less than that of WT RAS mice
Quantitative assessment of interstitial fibrosis was performed on Sirius red stained sections of renal cortex. The extent of interstitial fibrosis was significantly greater in WT RAS mice than in Ccl2 KO RAS mice (Fig. 6).

Collagen deposition, as assessed by Sirius red staining, is decreased in the stenotic kidney of Ccl2 KO RAS mice compared to WT RAS mice. (a) Representative images of renal cortex stained with Sirius red highlighting matrix deposition along tubular basement membranes and the interstitium (red staining). Scale bar represents 50 µm. (b) Quantitative analysis of the percent cortical surface area staining positively for extracellular matrix. *p ≤ 0.0001, **p < 0.01
Macrophage influx is decreased in Ccl2 KO mice compared to WT mice following RAS surgery
Macrophage influx was lower in the stenotic kidney of Ccl2 KO mice than WT mice with RAS, as assessed by the percentage of cortical surface areas staining positively for the macrophage marker F4/80 (Fig. 7). We did not find any significant differences in CD3 + T cell influx between Ccl2 KO and WT mice with RAS (Fig. 8).

Macrophage influx, as assessed by F4/80 staining, is decreased in the stenotic kidney of Ccl2 KO RAS mice compared to WT RAS mice. (a) Immunohistochemical staining with anti-F4/80 antibody. The arrowheads highlight regions of positive staining for F4/80. Scale bar represents 100 µm. (b) Quantitative analysis of the percent of total cortical surface area staining positively for F4/80. *p < 0.05.

Lymphocyte influx, as assessed by immunohistochemical staining with anti CD3 antibody. (a) Representative histological images of WT and Ccl2 KO mice studied at 4 weeks following surgery. Sections are stained with anti CD3 antibody (200X magnification). The arrow highlights regions of positive staining for CD3. Scale bar represents 100 µm. (b) Quantitative analysis of the percent of total cortical surface staining positively for CD3. *p < 0.05.
Expression of pro-inflammatory chemokines is lower in Ccl2 KO than WT mice following RAS surgery
Ccl2 deficiency was confirmed using real time PCR in KO mice (Fig. 9a). Ccl2 expression was significantly higher in WT RAS mice, compared to Ccl2 KO RAS and WT sham mice. Similar results were observed for the expression of Ccl5, Ccl7 and Ccl8 (Fig. 9b,c,d respectively), although we did not observe any difference in Ccl8 expression between WT and Ccl2 KO RAS.

Pro-inflammatory chemokine expression in Ccl2 KO and WT RAS mice. Gene expression of (a) Ccl2, (b) Ccl5, C) Ccl7, (d) Ccl8 in WT and KO mice. *p < 0.0001, **p < 0.01, ***p < 0.05.
Ccr2 expression was significantly higher in WT RAS mice, compared to Ccl2 KO RAS and WT sham mice (Fig. 10a). There were no significant differences in expression of iNos in any group (Fig. 10b). Cd206 expression was higher in WT RAS mice, compared to WT sham and KO RAS mice (Fig. 10c).

Pro-inflammatory chemokine expression in Ccl2 KO and WT RAS mice. The gene expression of (a) Ccr2, (b) iNos, (c) Cd206 in WT and KO mice. *p < 0.0001, **p < 0.01, ***p < 0.05.
Il6 expression was significantly higher in WT RAS mice compared to WT sham (Fig. 11a). However, we did not find any differences in Il12, Gata3, Rorc expression in any of the groups (Fig. 11b,c,d respectively). Although, TNFα expression was significantly higher in WT RAS compared to sham (Fig. 12a), there were no differences in TNFα expression between WT and Ccl2 KO RAS. No difference in Tgfβ expression was observed in any group (Fig. 12b).

Pro-inflammatory chemokine expression in Ccl2 KO and WT RAS mice. The gene expression of (a) Il6, (b) IL12, (c) Gata3, (d) Rorc in WT and KO mice. *p < 0.05.

Pro-inflammatory chemokine expression in Ccl2 KO and WT RAS mice. The gene expression of (a) TNFα, (b) Tgfβ, in WT and KO mice. *p < 0.05.
Acta2 and Col3A1 expression was significantly higher in WT RAS mice compared to its sham (Fig. 13a and b), whereas, Col3A1 expression was significantly elevated in the KO RAS mice compared to the KO sham group (Fig. 13b). Acta2 expression was significantly higher in WT RAS mice than Ccl2 KO RAS mice (Fig. 13a). However, differences in Col3A1 expression between Ccl2 KO and WT RAS mice did not reach statistical significance.

Alpha-actin and collagen 3 expression in Ccl2 KO and WT RAS mice. The gene expression of (a) Acta2, (b) Col3A1 in WT and KO mice. *p < 0.01, **p < 0.05.
Discussion
Our studies provide further evidence that Ccl2 plays a key role in mediating renal inflammation in RVH and its deficiency alone can protect against the chronic renal injury. Despite a similar flow reduction in the stenotic kidneys of WT and Ccl2 KO mice, the stenotic kidney of Ccl2 KO mice showed significant protection against renal injury, as evidenced by less cortical atrophy, compared to WT RAS. The R2* value, a measure of renal hypoxia, was significantly higher in the STK of KO mice which could be due to the fact that perfusion was significantly reduced in KO RAS mice, whereas perfusion was maintained in WT RAS mice, albeit at a reduced cortical volume. Plasma BUN and creatinine were within normal limits in all groups, indicating that the contralateral kidney was able to compensate for the chronic damage which developed in the stenotic kidney. These findings are consistent with observations that BUN and creatinine are relatively normal in patients who undergo unilateral nephrectomy.
Although, blood pressure was increased to a similar extent in KO and WT RAS mice, the heart weight was significantly higher than sham in WT RAS but not Ccl2 KO RAS mice. Similarly, the contralateral (non-cuffed) kidney showed a higher weight in WT RAS but not Ccl2 KO RAS mice. Reduction of chronic renal injury in the STK of KO RAS mice was associated with less macrophage, but not T cell infiltration.
In our previous study, we found that pharmacologic inhibition of CCR2 reduced Ccl5 and TNFα expression, but not Ccl2, Il-6, Il-10, Il-12, or Tgfβ expression20. In the current study, we demonstrate that Ccl2 deficiency significantly reduces expression of other pro-inflammatory molecules, including Ccl5, Ccl7, Ccl8, Ccr2, and the M2 macrophage marker Cd206. Unlike the CCR2 inhibitor, TNFα expression was not significantly reduced in Ccl2 KO RAS mice, compared to WT RAS animals, raising the possibility that TNF expression may not directly involve Ccl2-CCR2 signaling.
The lack of cardiac hypertrophy in KO RAS mice contrasts with our studies of other signaling pathways culminating in renal atrophy in RAS. The STK of RAS mice treated with the p38 inhibitor SB203580 was protected from development of renal inflammation, tubular atrophy, and interstitial fibrosis. However, SB203580 had no effect on blood pressure, plasma renin activity, or heart weights compared to vehicle treated RAS mice35. Similarly, the STK of mice with deficiency of Smad3, a critical intermediate of TGF-β signaling, was protected from development of renal atrophy, interstitial fibrosis, and tubular atrophy. Although, blood pressure was increased to a similar extent in Smad3 KO and WT RAS mice at 3 days after surgery, blood pressure after that time point was significantly lower in Smad3 KO RAS mice than WT mice32,36. Despite this, the heart weight to body weight ratio was significantly higher in the Smad3 KO RAS mice than WT RAS mice. Furthermore, the Smad3 KO RAS mice had a severe cardiac phenotype characterized by myocardial inflammation with extensive myocyte necrosis, which was associated with sudden death in Smad3 KO RAS mice36. Based on these considerations, we propose that Ccl2 may provide a better therapeutic target to reduce chronic renal damage and to protect the heart than p38 or Smad3 inhibition.
Although, our studies support a central role for Ccl2 in reducing inflammatory responses to renal injury, there are several limitations. We demonstrated that mice with global Ccl2 deficiency are protected from development of renal atrophy in RVH; we have not established whether Ccl2 signaling in parenchymal cells or in bone marrow derived infiltrating inflammatory cells is responsible for this protective effect. Studies to address this issue will be important to determine whether more specific therapy should target immune/inflammatory cells or parenchymal cells to prevent development of chronic renal and cardiovascular disease in RVH. Not all studies support a protective role for Ccl2 deficiency in renal disease. In a renal ischemia-reperfusion model, Ccl2 deficiency exacerbated renal damage37. With that caveat in mind, human studies in patients with chronic renal disease show a strong correlation between urinary Ccl2 excretion, albuminuria, and renal infiltration of macrophages23. We conclude that therapy directed towards Ccl2 signaling may provide a novel therapeutic target to prevent progression renal and possibly cardiovascular disease in RVH.
References
Eirin, A. et al. Inflammatory and injury signals released from the post-stenotic human kidney. Eur Heart J 34, 540–548a, https://doi.org/10.1093/eurheartj/ehs197 (2013).
Harrison, D. G. et al. Inflammation, immunity, and hypertension. Hypertension 57, 132–140, https://doi.org/10.1161/HYPERTENSIONAHA.110.163576 (2011).
Kalantar-Zadeh, K., Ikizler, T. A., Block, G., Avram, M. M. & Kopple, J. D. Malnutrition-inflammation complex syndrome in dialysis patients: causes and consequences. Am J Kidney Dis 42, 864–881, https://doi.org/10.1016/j.ajkd.2003.07.016 (2003).
Mai, M. et al. Early interstitial changes in hypertension-induced renal injury. Hypertension 22, 754–765, https://doi.org/10.1161/01.hyp.22.5.754 (1993).
Ozawa, Y., Kobori, H., Suzaki, Y. & Navar, L. G. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. Am J Physiol Renal Physiol 292, F330–339, https://doi.org/10.1152/ajprenal.00059.2006 (2007).
Hilgers, K. F., Hartner, A., Porst, M., Veelken, R. & Mann, J. F. Angiotensin II type 1 receptor blockade prevents lethal malignant hypertension: relation to kidney inflammation. Circulation 104, 1436–1440, https://doi.org/10.1161/hc3601.095576 (2001).
De Miguel, C., Das, S., Lund, H. & Mattson, D. L. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol 298, R1136–1142, https://doi.org/10.1152/ajpregu.00298.2009 (2010).
De Miguel, C., Guo, C., Lund, H., Feng, D. & Mattson, D. L. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol 300, F734–742, https://doi.org/10.1152/ajprenal.00454.2010 (2011).
Cheng, Y. et al. Immunoregulatory effects of sinomenine on the T-bet/GATA-3 ratio and Th1/Th2 cytokine balance in the treatment of mesangial proliferative nephritis. Int Immunopharmacol 9, 894–899, https://doi.org/10.1016/j.intimp.2009.03.014 (2009).
Muller, D. N. et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol 161, 1679–1693, https://doi.org/10.1016/S0002-9440(10)64445-8 (2002).
Adler, M. W., Geller, E. B., Chen, X. & Rogers, T. J. Viewing chemokines as a third major system of communication in the brain. AAPS J 7, E865–870, https://doi.org/10.1208/aapsj070484 (2006).
Bose, S. & Cho, J. Role of chemokine CCL2 and its receptor CCR2 in neurodegenerative diseases. Arch Pharm Res 36, 1039–1050, https://doi.org/10.1007/s12272-013-0161-z (2013).
Spoettl, T. et al. Monocyte chemoattractant protein-1 (MCP-1) inhibits the intestinal-like differentiation of monocytes. Clin Exp Immunol 145, 190–199, https://doi.org/10.1111/j.1365-2249.2006.03113.x (2006).
Ljung, L. et al. Treatment with tumor necrosis factor inhibitors and the risk of acute coronary syndromes in early rheumatoid arthritis. Arthritis Rheum 64, 42–52, https://doi.org/10.1002/art.30654 (2012).
Kamei, N. et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 281, 26602–26614, https://doi.org/10.1074/jbc.M601284200 (2006).
Rull, A., Camps, J., Alonso-Villaverde, C. & Joven, J. Insulin resistance, inflammation, and obesity: role of monocyte chemoattractant protein-1 (or CCL2) in the regulation of metabolism. Mediators Inflamm 2010, 11, https://doi.org/10.1155/2010/326580 (2010).
Panee, J. Monocyte Chemoattractant Protein 1 (MCP-1) in obesity and diabetes. Cytokine 60, 1–12, https://doi.org/10.1016/j.cyto.2012.06.018 (2012).
Coll, B., Alonso-Villaverde, C. & Joven, J. Monocyte chemoattractant protein-1 and atherosclerosis: is there room for an additional biomarker? Clin Chim Acta 383, 21–29, https://doi.org/10.1016/j.cca.2007.04.019 (2007).
Nath, K. A., Kanakiriya, S. K., Grande, J. P., Croatt, A. J. & Katusic, Z. S. Increased venous proinflammatory gene expression and intimal hyperplasia in an aorto-caval fistula model in the rat. Am J Pathol 162, 2079–2090, https://doi.org/10.1016/S0002-9440(10)64339-8 (2003).
Kashyap, S. et al. Blockade of CCR2 reduces macrophage influx and development of chronic renal damage in murine renovascular hypertension. Am J Physiol Renal Physiol 310, F372–384, https://doi.org/10.1152/ajprenal.00131.2015 (2016).
Matsunaga, N. et al. Inhibition of G0/G1 Switch 2 Ameliorates Renal Inflammation in Chronic Kidney Disease. EBioMedicine 13, 262–273, https://doi.org/10.1016/j.ebiom.2016.10.008 (2016).
Kitagawa, K. et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol 165, 237–246, https://doi.org/10.1016/S0002-9440(10)63292-0 (2004).
Eardley, K. S. et al. The relationship between albuminuria, MCP-1/CCL2, and interstitial macrophages in chronic kidney disease. Kidney Int 69, 1189–1197, https://doi.org/10.1038/sj.ki.5000212 (2006).
Tesch, G. H. MCP-1/CCL2: a new diagnostic marker and therapeutic target for progressive renal injury in diabetic nephropathy. Am J Physiol Renal Physiol 294, F697–701, https://doi.org/10.1152/ajprenal.00016.2008 (2008).
Kulkarni, O. et al. Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J Am Soc Nephrol 18, 2350–2358, https://doi.org/10.1681/ASN.2006121348 (2007).
Boels, M. G. S. et al. Systemic Monocyte Chemotactic Protein-1 Inhibition Modifies Renal Macrophages and Restores Glomerular Endothelial Glycocalyx and Barrier Function in Diabetic Nephropathy. Am J Pathol 187, 2430–2440, https://doi.org/10.1016/j.ajpath.2017.07.020 (2017).
Menne, J. et al. C-C motif-ligand 2 inhibition with emapticap pegol (NOX-E36) in type 2 diabetic patients with albuminuria. Nephrol Dial Transplant 32, 307–315, https://doi.org/10.1093/ndt/gfv459 (2017).
Fukuda, D., Enomoto, S., Nagai, R. & Sata, M. Inhibition of renin-angiotensin system attenuates periadventitial inflammation and reduces atherosclerotic lesion formation. Biomed Pharmacother 63, 754–761, https://doi.org/10.1016/j.biopha.2009.02.006 (2009).
Combadiere, C. et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 117, 1649–1657, https://doi.org/10.1161/circulationaha.107.745091 (2008).
Tsou, C. L. et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest 117, 902–909, https://doi.org/10.1172/JCI29919 (2007).
Sozzani, S. et al. Receptors and transduction pathways for monocyte chemotactic protein-2 and monocyte chemotactic protein-3. Similarities and differences with MCP-1. J Immunol 152, 3615–3622 (1994).
Warner, G. M. et al. Genetic deficiency of Smad3 protects the kidneys from atrophy and interstitial fibrosis in 2K1C hypertension. Am J Physiol Renal Physiol 302, F1455–1464, https://doi.org/10.1152/ajprenal.00645.2011 (2012).
Ebrahimi, B. et al. Evolution of cardiac and renal impairment detected by high-field cardiovascular magnetic resonance in mice with renal artery stenosis. J Cardiovasc Magn Reson 15, 98, https://doi.org/10.1186/1532-429X-15-98 (2013).
Nath, K. A., Croatt, A. J., Warner, G. M. & Grande, J. P. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am J Physiol Renal Physiol 301, F436–442, https://doi.org/10.1152/ajprenal.00162.2011 (2011).
Wang, D. et al. Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in a murine renal artery stenosis model. Am J Physiol Renal Physiol 304, F938–947, https://doi.org/10.1152/ajprenal.00706.2012 (2013).
Kashyap, S. et al. Cardiovascular phenotype in Smad3 deficient mice with renovascular hypertension. Plos One 12, e0187062, https://doi.org/10.1371/journal.pone.0187062 (2017).
Stroo, I. et al. Deficiency for the chemokine monocyte chemoattractant protein-1 aggravates tubular damage after renal ischemia/reperfusion injury. Plos One 10, e0123203, https://doi.org/10.1371/journal.pone.0123203 (2015).
Acknowledgements
This study was supported by NIH/NIAID R01 AI100911-01 and in part by Department of Laboratory Medicine and Pathology. Hui Tang’s assistance with plasma renin content assay and Medical Genome Facility Gene Expression Core Mayo clinic’s assistance with RNA quality assessment are acknowledged. NMR Core facility Mayo Clinic is acknowledged. Secretarial assistance of Jennifer Scott is acknowledged.
Author information
Authors and Affiliations
Contributions
S.K. and J.P.G. wrote the main manuscript and performed data analysis, S.K., M.C.N. and M.O. performed the experiments, K.R.L., M.O. and B.R. performed histology and analysis, K.A.N., V.D.G., L.O.L., J.P.G. and S.K. did the study design. C.M.F. prepared Figures 2–4.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kashyap, S., Osman, M., Ferguson, C.M. et al. Ccl2 deficiency protects against chronic renal injury in murine renovascular hypertension. Sci Rep 8, 8598 (2018). https://doi.org/10.1038/s41598-018-26870-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-26870-y
This article is cited by
-
CCL7 as a novel inflammatory mediator in cardiovascular disease, diabetes mellitus, and kidney disease
Cardiovascular Diabetology (2022)
-
Cellular distribution of C-C motif chemokine ligand 2 like immunoreactivities in frontal cortex and corpus callosum of normal and lipopolysaccharide treated animal
BMC Neuroscience (2022)
-
Renal tubular PAR2 promotes interstitial fibrosis by increasing inflammatory responses and EMT process
Archives of Pharmacal Research (2022)
-
Effects of tumor necrosis factor-α inhibition on kidney fibrosis and inflammation in a mouse model of aristolochic acid nephropathy
Scientific Reports (2021)
-
RNA sequencing reveals induction of specific renal inflammatory pathways in a rat model of malignant hypertension
Journal of Molecular Medicine (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
