
Abstract
Pythium insidiosum is a human-pathogenic oomycete. Many patients infected with it lose organs or die. Toward the goal of developing improved treatment options, we want to understand how Py. insidiosum has evolved to become a successful human pathogen. Our approach here involved the use of comparative genomic and other analyses to identify genes with possible functions in the pathogenicity of Py. insidiosum. We generated an Oomycete Gene Table and used it to explore the genome contents and phylogenomic relationships of Py. insidiosum and 19 other oomycetes. Initial sequence analyses showed that Py. insidiosum is closely related to Pythium species that are not pathogenic to humans. Our analyses also indicated that the organism harbours secreted and adhesin-like proteins, which are absent from related species. Putative virulence proteins were identified by comparison to a set of known virulence genes. Among them is the urease Ure1, which is absent from humans and thus a potential diagnostic and therapeutic target. We used mass spectrometric data to successfully validate the expression of 30% of 14,962 predicted proteins and identify 15 body temperature (37 °C)-dependent proteins of Py. insidiosum. This work begins to unravel the determinants of pathogenicity of Py. insidiosum.
Similar content being viewed by others


Introduction
Oomycetes are a unique and evolutionarily diverse group of microorganisms that belong to the Kingdom Stramenopiles and share the microscopic hyphal morphology of fungi1. Based on phylogenetic analyses, oomycetes are closely related to diatoms and algae, and only distantly related to fungi1,2. Well-known oomycete genera include Albugo, Aphanomyces, Bremia, Hyaloperonospora, Lagenidium, Peronospora, Phytophthora, Plasmopara, Pythium and Saprolegnia1,3,4,5. Many oomycetes are pathogenic to economically important plants, while some species in the genera Aphanomyces, Lagenidium, Pythium and Saprolegnia are capable of infecting animals1,5. Only a few oomycetes (i.e. Pythium insidiosum6, Pythium aphanidermatum7,8 and Lagenidium species9,10) have been reported to cause devastating or fatal infections in humans. No commercially available drug is effective against the oomycete pathogens, making management of the infections caused by these organisms challenging.
Among the human-pathogenic oomycetes, Py. insidiosum (the causative agent of the life-threatening infectious condition called pythiosis) is the most frequently reported, with only four reported cases of infections with other species6,7,8,9,10,11. Py. insidiosum is also an invasive pathogen of nonhuman animals, such as horses, dogs, cats and cattle11. Available antifungal drugs are not useful in most cases of this infection, due to the organism’s lack of the drug-targeted enzymes for ergosterol biosynthesis12. Surgical removal of an infected organ is the main recourse to remove the infection, which often leads to permanent disabilities in surviving patients. Despite surgical intervention, many patients die from progressive and uncontrolled infection6. An effective treatment modality (i.e. anti-oomycete drug or vaccine) is thus urgently needed for the control of Py. insidiosum and related oomycetes.
A better understanding of the biology and pathogenesis of Py. insidiosum is required for the development of an efficient method for infection control. How Py. insidiosum, among the oomycetes, has evolved to become a successful human and animal pathogen is an open question that needs investigation. With the development of next-generation sequencing (NGS) technology, the genomes and transcriptomes of many oomycetes have been sequenced and are now publicly accessible3,4,13,14,15,16,17,18,19,20,21,22. Recently, we sequenced the first draft genome and transcriptome of Py. insidiosum, using the 454 and Illumina NGS platforms19,20. The genome of Py. insidiosum is 53.2 Mb in size and comprises 14,962 predicted open reading frames. This sequence information provides a useful resource for exploring the biology, evolution and pathogenicity of Py. insidiosum.
As a human and animal pathogen11, Py. insidiosum must have evolved a special set of genes that are required for this pathogenesis, which are not found in other oomycetes. In the current study, we analyse the genome content of Py. insidiosum, in comparison with that of 19 other oomycetes (including Py. aphanidermatum, which is a well-known plant pathogen, and sporadically causes human infections, including in two soldiers with severe blast injury in Afghanistan7,8) and 2 diatoms, to identify putative virulence genes, and shed light on the pathogenicity-related evolution of this pathogen. Since the ability to grow at the host’s body temperature is essential for an invasive pathogen, we also incorporated newly generated mass spectrometric data to validate and identify Py. insidiosum proteins that are differentially expressed in response to a shift in temperature from 25 °C to 37 °C. These studies have begun to reveal the factors behind the pathogenicity of Py. insidiosum.
Results
General characteristics of oomycete genomes
All genome sequences were retrieved from public databases (Table 1). The oomycete genomes differ in size (33–229 Mb) and are larger than those of diatoms (26–31 Mb). These genomes have different G + C contents (43–59%), numbers of coding sequences (CDS; 11,958–27,941 genes) and CDS density (12–75%). Albugo and Pythium harbour smaller genomes (33–53 Mb) than other oomycetes (53–229 Mb). Ph. infestans has the largest genome (229 Mb) with the lowest CDS density (12%), whereas Al. laibachii has the smallest genome (33 Mb) with the highest CDS density (75%).
Homologous gene cluster data
A total of 368,724 genes identified in 20 oomycete and 2 diatom genomes were grouped into 98,988 unique homologous gene clusters. The homologous gene cluster data used in this study can be assessed using our Oomycete Gene Table online tool, which can be found at http://www.sbi.kmutt.ac.th/cgi-bin/gt/viewer?organism=oomycetes&build=150418 (description and instructions of the Oomycete Gene Table will be reported in detail elsewhere). To narrow down the Py. insidiosum-specific gene clusters, we selectively classified these 98,988 gene clusters into subgroups based on gene content similarities, at the ‘major group’ level (including two groups, i.e. Diatoms and Oomycetes), the ‘genus’ level (including six oomycete genera, i.e. Albugo, Aphanomyces, Hyaloperonospora, Phytophthora, Pythium and Saprolegnia) and the ‘species’ level (including seven Pythium species, i.e. Py. insidiosum, Py. irregulare, Py. iwayamai, Py. ultimum, Py. vexans, Py. arrhenomanes and Py. aphanidermatum). Each ‘Core’ box (the green box in Fig. 1) represents the shared gene clusters, identified in (i) all 20 oomycetes and 2 diatoms (assigned as Core 1, containing 4,371 gene clusters), (ii) all 6 oomycete genera (assigned as Core 2, 3,893 gene clusters) and (iii) all 7 Pythium species (assigned as Core 3, 69 gene clusters). Each ‘Variable’ box (the blue box in Fig. 1) contains non-Core gene clusters, in each subgroup: Variable 1 (major groups, containing 94,617 gene clusters), Variable 2 (oomycete genera, 73,671 gene clusters) and Variable 3 (Pythium species, 5,196 gene clusters). Each ‘Unspecific’ box (the orange box in Fig. 1) represents the gene clusters, identified in at least two, but not all, major groups (assigned as Unspecific 1, containing 7,343 gene clusters), oomycete genera (assigned as Unspecific 2, 38,594 gene clusters) and Pythium species (assigned as Unspecific 3, 2,229 gene clusters). The gene clusters (those specific to a particular major group, genus or species) are grouped together (the white box in Fig. 1). Py. insidiosum contained a unique set of 997 gene clusters, not identified in other oomycete and diatom genomes.

Flow chart of the 98,988 homologous gene clusters of the oomycetes and diatoms, selectively subgrouped into 2 major groups, 6 oomycete genera and 7 Pythium species. The ‘Core’ boxes (green) represent the shared gene clusters, identified in all major groups (assigned as Core 1), all oomycete genera (Core 2) and all Pythium species (Core 3). The ‘Variable’ boxes (blue) contain non-Core gene clusters, that is, Variable 1 (major groups), Variable 2 (oomycete genera) and Variable 3 (Pythium species). The ‘Unspecific’ boxes (orange) represent the gene clusters identified in at least two, if not all, major groups (assigned as Unspecific 1), oomycete genera (Unspecific 2) and Pythium species (Unspecific 3). The gene clusters (those specific to a particular major group, genus or species) are grouped together in the white boxes.
BLAST searches against the Clusters of Orthologous Groups of Proteins (COGs) database23,24 demonstrated that 62% of the Core 1 gene clusters (belonging to oomycetes and diatoms) can be assigned to 22 out of 23 COG groups, with no gene matches for ‘Extracellular structures’ (Supplementary Fig. S1). Of the Core 2 gene clusters (belonging to the oomycetes), 23% can be allocated to 21 COG groups, with none in ‘Nuclear structure’ or ‘Extracellular structures’. Approximately 3% of the Core 3 gene clusters (belonging to the genus Pythium) had matches in two COG groups: ‘Cytoskeleton’ and ‘Post-translational modification, protein turnover, chaperones’.
Gene content comparison
In Fig. 2, the gene content similarity is the proportion of the gene clusters identified in each organism, listed on the left of the figure, that matched the gene clusters identified in each organism, listed at the top. For example, the gene content similarity of Py. insidiosum (listed on the left of Fig. 2) vs. Ph. infestans (listed at the top) is 77%, indicating that 77% of the Py. insidiosum gene clusters matched those of Ph. infestans, whereas the gene content similarity of Ph. infestans (listed on the left of Fig. 2) vs. Py. insidiosum (listed at the top) is 70%, indicating that 70% of the Ph. infestans gene clusters matched those of Py. insidiosum. Less than 25% of gene clusters present in the oomycetes matched those of the diatoms. Within oomycetes of the same genus, there were at least 70% shared gene clusters, for example: Albugo (≥72%), Phytophthora (≥74%), Pythium (≥75%), Aphanomycete (≥81%) and Saprolegnia (≥94%). Among 6 animal-pathogenic oomycetes, there is a broad range of gene content similarities (50–94%; the red box in Fig. 2), as well as among 14 plant-pathogenic oomycetes (34–95%). When the pair-wise comparison was performed at the genus level, the genus Pythium, unlike other genera, demonstrated lower gene content similarities against itself on average (Pythium vs. Pythium; average, 82%; range, 75–90%; the yellow box in Fig. 2), compared with that against the genus Phytophthora (Pythium vs. Phytophthora; average, 84%; range, 77–90%; the green box in Fig. 2).

Pair-wise comparison of gene contents identified in 20 oomycete and 2 diatom genomes. The gene cluster number of each organism is shown in parentheses. The numbers and colour shadings indicate the percentage of the genes in each organism, listed on the left, that matched the genes in each organism, listed at the top. Pair-wise comparisons of the genera Pythium vs. Phytophthora (similarities: average, 84%; range, 77–90%) and the genera Pythium vs. Pythium (similarities: average, 82%; range, 75–90%) are depicted in the green and yellow boxes, respectively. Gene content similarities among six animal-pathogenic oomycetes (50–94%) are shown in the red box.
Phylogenomic relationship
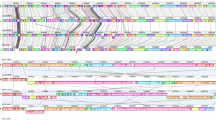
To investigate the evolutionary relatedness and differences between oomycetes, a phylogenomic analysis was performed using a set of 2,073 core genes found across 20 oomycete genomes (Supplementary Table S1). These core genes were found to produce phylogenetic trees with the best likelihood when the Generalized Time Reversal (GTR) evolutionary model was used (see the Methods). Concatenation of 2,073 individual core gene sequences produced a final multiple sequence alignment that is 2,455,151 positions in length. The concatenated multiple sequence alignment was then used to create a maximum likelihood phylogenomic tree using GTR as an evolutionary model. The resulting phylogenomic tree is shown in Fig. 3. Our phylogenomic tree separated almost all oomycetes according to their genera, with an exception to Py. vexans, which was separated from the other Pythium species, and phylogenetically proximal to the Phytophthora species and Hy. arabidopsis. For the genus Pythium, our phylogenomic analysis categorized all Pythium species, except Py. vexans, into two different clades: Group-A species (Py. insidiosum, Py. aphanidermatum, and Py. arrhenomanes), and Group-B species (Py. irregulare, Py. iwayamai, and Py. ultimum).

A maximum-likelihood phylogenomic tree generated by a set of 2,073 core genes found across all 20 oomycete genomes (total concatenated multiple sequence alignment length of 2,455,151). Descriptions of all 2,073 core genes are shown in Supplementary Table S1. The tree categorizes all Pythium species, except Pythium vexans (currently reclassified as Phytopythium vexans; indicated by asterisk), into clades A and B. The reliability of the trees tested by bootstrap analysis to support each branch is indicated.
We also analyzed oomycete relatedness based on the hierarchical clustering of their gene presence profiles. In contrast to the 2,073 core gene-based phylogenomic tree, the resulting hierarchical clustering of gene presence profile-based tree showed that all oomycetes (including Py. vexans) were grouped according to their genera (Fig. 4).

A phylogenomic tree based on hierarchical clustering of the gene presence profile of all 20 oomycetes. The heat map indicates the gene presence profile of 178 representative genes (green and black represent gene presence and absence, respectively). The tree categorises all Pythium species, including Pythium vexans (currently reclassified as Phytopythium vexans; indicated by an asterisk), into clades A and B. The reliability of the trees tested by bootstrap analysis to support each branch is indicated.
Elicitin gene family in oomycetes
Because of their association with pathogenicity, we were interested in investigating elicitin-encoding genes in the oomycete genomes. A total of 121 elicitin gene clusters, defined by the Oomycete Gene Table, were differentially present in all oomycetes, but absent in diatoms (Fig. 5). The organisms in the genera Pythium and Phytophthora harboured more elicitin gene clusters (average, 48 clusters/species; range, 34–55) than other genera (average, 13 clusters/species; range, 4–29). The elicitin gene clusters 88, 89 and 90 were found across all oomycetes (the ‘All’ box in Fig. 5). Some elicitin gene clusters appeared to be genus-specific, that is, clusters 1–29 of Pythium (the ‘Py’ box in Fig. 5), 38–42 of Phytophthora (the ‘Ph’ box) and 107–121 of Saprolegnia (the ‘Sa’ box).

The Oomycete Gene Table shows a total of 121 elicitin gene clusters that are differently present in the oomycetes, but absent from the diatoms. The elicitin gene clusters 88, 89 and 90 are found across all oomycetes (as indicated by the ‘All’ box). Some elicitin gene clusters appear to be genus-specific: clusters 1–29 for the genus Pythium (the ‘Py’ box), 38–42 for the genus Phytophthora (the ‘Ph’ box) and 107–121 for the genus Saprolegnia (the ‘Sa’ box). The rightmost column shows the number of species that contain each of the elicitin gene clusters. The colour shadings correlate with percent completeness of each predicted gene, in relation to the longest gene of each gene cluster. The light grey box indicates the presence of a similar sequence (that does not fulfil our gene-defining criteria; see the Methods) and the black box indicates that neither a similar sequence nor gene is found.
Genome annotation of Py. insidiosum
BLAST searches of the 14,962 predicted proteins of Py. insidiosum against the NCBI database showed that 2,811 proteins (19%) matched proteins with known functions, 10,606 (71%) matched hypothetical proteins and 1,545 (10%) had no significant sequence similarity. BLAST2GO assigned 1,405 Gene Ontology (GO) terms to 7,822 proteins (52% of all proteins) in three categories: (i) 550 GO terms in the Biological Processes (4,404 proteins), (ii) 162 GO terms in the Cellular Components (2,261 proteins) and (iii) 693 GO terms in the Molecular Functions (6,946 proteins). The top 40 GO terms assigned in each category are shown in Supplementary Figure S2. BLAST2GO also allocated 1,177 proteins (8% of all proteins) with 1,296 different enzyme commission (EC) numbers into six classes: Oxidoreductases (193 proteins), Transferases (442 proteins), Hydrolases (425 proteins), Lyases (68 proteins), Isomerases (65 proteins) and Ligases (103 proteins) (Supplementary Table S2). Based on InterProScan, 5,113 different domains were detected in 10,242 proteins (68% of all proteins) and the top 40 domains are shown in Supplementary Table S3. SignalP identified signal peptides (average length, 23 amino acids; range, 11–56) in 1,208 proteins (8% of all proteins). Transmembrane helices were predicted in 3,142 proteins (21% of all proteins).
Validation of predicted proteins of Py. insidiosum
Py. insidiosum protein extracts (SABHs) were prepared in four replicates, two each from the organism grown at either 25 °C or 37 °C (designated as 25 °C and 37 °C SABHs, respectively), and subjected to LC-MS/MS analysis. The set of 14,962 predicted proteins of Py. insidiosum, theoretically digested with trypsin, was used as an in-house Mascot library for LC-MS/MS search. Any peptide identified by LC-MS/MS in at least two replicates was recruited for protein validation, resulting in a pool of 120,265 peptides. A total of 4,445 genome-derived proteins (30% of all proteins), mapped with at least two different recruited peptides, were validated for their actual expression in Py. insidiosum. The average number of mapped peptides per protein was 27 (range, 2–200).
Effect of temperature on protein expression in Py. insidiosum
Changes in Py. insidiosum protein abundance, upon exposure to increased temperature, were assessed using LC-MS/MS data of the 25 °C and 37 °C SABHs (two replicates each). Unquantifiable proteins, assigned as ‘no emPAI’ (see the Methods), were excluded. There were 344 validated proteins recruited for comparison of protein abundances. Py. insidiosum exhibited 212 relatively high- and 124 relatively low-abundance proteins in the 37 °C SABHs, compared with the 25 °C SABHs. Of these differentially expressed proteins, 53 and 40 were at least twofold up- and downregulated, respectively (Supplementary Table S4). The top upregulated proteins included cyclophilin-A (45-fold increase), peroxiredoxin-2 (15-fold), inositol-3-phosphate synthase (13-fold), alanine aminopeptidase (10-fold) and proteasome subunit beta type-6 (10-fold), whereas the markedly downregulated ones included 6-phosphogluconate dehydrogenase (40-fold decrease), a hypothetical protein (22-fold) and glucokinase (10-fold).
Py. insidiosum-specific genes
Based on the Oomycete Gene Table, which included 98,988 unique homologous gene clusters, 997 gene clusters were found only in Py. insidiosum (Fig. 1). From these Py. insidiosum-specific gene clusters, we identified 1,194 predicted genes. Of these, 85% (1,013 predicted proteins) had no positive BLAST hits in the NCBI database and the other 15% (181 proteins) matched proteins with unknown function. The LC-MS/MS analyses detected peptides that map to 116 of these Py. insidiosum-specific proteins. Signal peptide and transmembrane helices were predicted in 194 and 199 proteins, respectively. Thirty-three adhesin-like proteins were predicted by FungalRV.
Putative virulence genes of Py. insidiosum
All predicted proteins were used as queries for a BLAST search against the MvirDB database, a collection of pathogen virulence factors25. A total of 1,820 proteins (12% of all proteins) found matches, which can be classified into five groups: (i) virulence factors (887 proteins), (ii) pathogenicity islands (584 proteins), (iii) toxins (240 proteins), (iv) antibiotic resistance-related proteins (99 proteins) and (v) a miscellaneous group (10 proteins). The top 40 protein hits for the defined virulence factors are listed in Table 2. The matched virulence factors with highest probability (E-value, 0.0) were heat shock protein 70, carbamoyl phosphate synthetase II, chaperone protein ClpB, oligopeptidase B, tripeptidyl peptidase 2 and urease.
Among the top-ranking putative virulence proteins of Py. insidiosum (Table 2), the urease (Ure1), which is 844 amino acids long (Fig. 6a), was selected for further characterisation. Forty LC-MS/MS peptides map to eight different regions in the Ure1 sequence (Fig. 6b). The Py. insidiosum urease, Ure1, had a high degree of sequence homology (E-value, 0.0; identity, 57%; similarity, 71%; Fig. 6c) and shared domain architecture (Fig. 6a) with the Cryptococcus neoformans urease URE1 (accession number, AAC62257). Three different clinical isolates (Pi19, Pi20 and Pi-S) of Py. insidiosum gave a positive urease reaction (Fig. 6d).

Characterisation of the Pythium insidiosum urease protein Ure1: (a) Comparison of urease domain architecture of Py. insidiosum Ure1 and Cr. neoformans URE1 (the bars indicate the length of each protein). (b) LC-MS/MS-derived peptides map eight different regions in the Ure1 sequence. (c) Two-sequence alignment of the Ure1 and the Cryptococcus neoformans urease URE1 (accession number, AAC62257). (d) Results of the urease test of three different Py. insidiosum isolates (i.e. Pi19, Pi-S and Pi20; the test is read as positive (+) if the colour turns pink and negative (−) if it remains yellow; the controls C1 and C2 were tested with an agar plug without the growing organism).
Discussion
The genomes of 20 oomycetes and 2 diatoms altogether contained 368,724 genes, which can be grouped into 98,988 homologous gene clusters (Fig. 1). Of all of the unique gene clusters, 4,371 were assigned to ‘Core 1’ as they were found in every genome. However, since the genome data of Py. arrhenomanes and Py. iwayamai appeared to have lower quality (i.e. a larger number of contigs and shorter N50 length; Table 1), this number of core gene clusters was probably lower than it actually was. The core gene clusters (i.e., Cores 1, 2 and 3) generally contained housekeeping genes, whereas most of the other gene clusters contained genes with no defined function (Fig. 1 and Supplementary Fig. S1). Pair-wise comparison of the gene contents, at the genus level, showed an unexpected finding, where the similarities of Pythium vs. Pythium (average, 82%) were lower than those of Pythium vs. Phytophthora (average, 84%) (Fig. 2). This finding suggests that the vast majority of gene contents present in the genus Pythium are present in the genus Phytophthora (which generally harbours larger genomes than Pythium; average genome size: 101 Mb vs. 43 Mb; Table 1). How evolution contributed to this paradoxical observation in Phytophthora and Pythium is an interesting issue, and requires further investigation.
We explored the evolutionary relationships among oomycetes, at the genome scale, by generating hierarchical clustering of the gene presence profile-based tree (which summarises the relatedness of genomes according to the gene contents) and a 2,073 core gene-based tree (which focuses on the relatedness of genomes according to the core gene set). The former tree, based on the gene content, showed that all oomycete organisms were nicely grouped according to their genera (Fig. 4). However, the latter tree, based on the core genes, placed the organisms into expected clades, according to their genera, with an exception for Py. vexans (which appeared in a separate clade, apart from Pythium, but closer to the Phytophthora species and Hy. arabidopsis) (Fig. 3). Recently, Py. vexans has been reclassified into the genus ‘Phytopythium’, and its current binomial name is Phytopythium vexans, based on phylogenomic analyses26. Taken together, our results suggest that species of Pythium and Phytopythium share a common ancestor, but may have secondarily gained or lost different sets of genes as they went through their evolutionary journey. For the genus Pythium, our phylogenetic trees separated various Pythium species into two different clades (Figs 3 and 4), which were correlated with their pathogenic characteristics. Specifically, two of the Group A species (i.e. Py. insidiosum and Py. aphanidermatum) are known to have the ability to infect humans, whereas all Group B species are strictly plant pathogens6,7,8,11.
Elicitins form a unique protein family found in oomycetes (mainly Phytophthora and Pythium) and are not present in other organisms27,28,29. The proteins function as sterol carriers and act as ‘pathogen-associated molecular patterns’ in plants30,31,32,33,34. We used the Oomycete Gene Table to search for elicitins in 22 genomes (Table 1). Elicitins were absent from diatoms, but present in all oomycetes (Fig. 5). Phytophthora and Pythium harboured a more extensive set of elicitins than the other genera. The elicitin family, thus, could be an example of genes that existed in a common ancestor and subsequently underwent differential gene expansion, gene loss and gene modification during evolution.
There are 14,962 predicted genes in the genome of Py. insidiosum19, most of which (~80%) had no match to any given genes with defined function. Predicting protein sequences based on predicted eukaryotic genes, especially those without homologous sequences in the database, can be complicated by the presence of introns and six possible reading frames. Peptide mapping can circumvent this obstacle, and it enabled us to successfully validate ~30% of the theoretically translated proteins of Py. insidiosum. Peptide mapping is thus a useful method that can increase the reliability of gene annotation. We also utilised the mass spectrometric data to initially explore proteins that might be associated with the growth of Py. insidiosum at body temperature. Fifteen proteins exhibited ≥5-fold greater abundance upon a shift to a higher temperature (25 °C to 37 °C; Supplementary Table S4). The SignalP program35 identified no signal peptide among these 15 upregulated proteins, suggesting that they are nonsecreted proteins and could function inside the cell. In addition, based on the COG analysis23,24, the majority of these proteins were predicted to be involved in: (i) metabolism and transportation of amino acids, nucleotides, carbohydrates or lipids (eight proteins); and (ii) post-translational modification, protein turnover and chaperones [five proteins, including cyclophilin-A, which was the most highly (45-fold) upregulated]. Such upregulated proteins might prove to be necessary for the temperature tolerance and infectivity of Py. insidiosum.
Most pathogenic oomycetes infect plants. Py. aphanidermiatum, a well-known plant pathogen, has been recently reported to infect humans7,8. Unlike Py. insidiosum, the human infection caused by Py. aphanidermatum is extremely rare (only two cases) and has only been reported in patients with an unusual status (soldiers with severe blast injury), living in a specific geographic location (i.e. Afghanistan). Differences in host range, underlying status of the host and clinical features suggest different mechanisms behind the pathogenicity of Py. insidiosum and Py. aphanidermatum. In the current study, we focus on how Py. insidiosum developed the ability to infect humans. It would be expected that Py. insidiosum has gained a special set of genes necessary for human infection during its evolution. We began to address this hypothesis by looking for Py. insidiosum-specific genes (based on comparative genomic analysis) and putative virulence factors (based on an MvirDB database search)25. We found 1,194 Py. insidiosum-specific proteins, without homology to any reported proteins. Of these proteins, ~10% can be readily validated (by peptide mapping) in terms of their expression in Py. insidiosum. Secretory and surface proteins are important for microbial pathogenesis, as they act at the interface of host–pathogen interactions. A number of secretory and adhesin-like proteins (~200 each) were predicted, and could serve as a subset of virulence factor candidates, to be the subject of further analysis. The life cycle of Py. insidiosum has already been reported by Mendoza and colleagues36. Zoospores are considered to be infective units that could initiate infection, upon direct exposure to humans or other animals. In conjunction with the genome study, future transcriptome analysis of the zoospores could lead to the identification of genes responsible for the initial stage of Py. insidisoum infection.
Different pathogens may employ similar pathogenic strategies, using shared virulence factors. Py. insidiosum contained 1,820 proteins that matched known virulence factors25. Among the top-ranking putative virulence proteins of Py. insidiosum (E-value, 0.0; Table 2), only the urease (Ure1) was unmatched in the NCBI reference proteins of humans (taxid: 9606), suggesting Ure1 as a potential diagnostic and therapeutic target. The LC-MS/MS-derived peptides mapped throughout the Ure1 sequence (Table 2; Fig. 6b), indicating that Ure1 was actually expressed. Because Py. insidiosum had one copy of the urease-encoding gene (Ure1) in its genome, the positive result of the urease test (Fig. 6d) was probably due to urease activity of the Ure1 protein. In pathogenic bacteria and fungi (i.e. Helicobacter pylori and Cr. neoformans), urease hydrolyses host urea to ammonia, in order to counteract the acidic environment, inhibit immune responses or destroy host tissue37,38,39. The Py. insidiosum urease Ure1 protein shares significant sequence similarity and domain architecture with the Cr. neoformans URE1 (Fig. 6a,c). Functional characterisation of Ure1 (i.e. gene cloning and expression, loss-of-function study by gene silencing and pathogenicity study in an animal model) is underway to investigate whether this enzyme is involved in the pathogenesis of Py. insidiosum.
In summary, we generated an Oomycete Gene Table to explore the genome contents and phylogenomic relationships of Py. insidiosum and 19 other oomycetes. Changes in gene contents, during the course of evolution, could have contributed to phenotypes that might be associated with speciation, host specificity and virulence of the oomycetes. Py. insidiosum is a prominent human pathogenic oomycete that appeared to be closely related to Pythium species that are not pathogenic to humans. The human specificity of Py. insidiosum should depend on a set of acquired pathogenicity genes. Indeed, a handful of Py. insidiosum-specific, body temperature-dependent and putative virulence proteins have been identified, and could serve as initial targets for investigating the mechanisms behind the pathogenesis of this species. The Py. insidiosum urease Ure1 is a putative virulence factor, as it shared sequence similarity and domain architecture with the virulence factor urease, URE1, of Cr. neoformans. Since humans do not contain urease39, Ure1 is a potential diagnostic and therapeutic target of Py. insidiosum.
Methods
Ethics statement
This study was approved by the Committee on Human Rights Related to Research Involving Human Subjects, at the Faculty of Medicine, Ramathibodi Hospital, Mahidol University (approval number MURA2011/412). All methods were performed in accordance with the relevant guidelines and regulations.
Genome sequences of Py. insidiosum and related oomycetes
The genome sequences of Py. insidiosum (strain Pi-S) and 19 other oomycetes, including 6 Pythium species, 6 Phytophthora species, 2 Albugo species, 2 Aphanomyces species, 2 Saprolegnia species and a Hyaloperonospora species, were retrieved from public databases (Table 1). Genome sequences of two diatoms were also recruited to serve as outgroups (Table 1).
Grouping of genes into homologous gene clusters for gene content comparison
All genes found in the 22 genomes (Table 1) were processed in a sequence similarity-based gene grouping to compare gene content across multiple genomes using a Gene Table protocol, previously published by Kittichotirat and colleagues40. For each sequence comparison, we used the following thresholds to group genes into the same cluster: BLAST E-value of 10−6, pairwise sequence identity of at least 30% and pairwise sequence alignment coverage for both query and subject of at least 50%. The final homologous gene cluster results are presented in the Oomycete Gene Table, where each row represents a gene and each column represents a genome used in this study. Each cell in the table contains information regarding homologous genes or genomic regions that were found in the corresponding genome.
Identification and alignment of core genes across oomycete genomes
Core gene were identified by scanning through the homologous gene cluster data for genes that appeared to be present in all 20 oomycete genomes. For each core homologous gene cluster, the longest gene member was used as a query and orthologous sequences from other oomycete genomes were identified by using NCBI TBLASTN41 with e-value cutoff of 1e-6 and coverage of more than 50%. We obtained orthologous core gene sequences in this manner to avoid cases where a gene was not originally identified in a genome even though the sequence or a part of the sequence is present in the genome sequence data. The orthologous sequences of each gene were then aligned using the PhylomeDB pipeline (https://github.com/Gabaldonlab/phylomizer)42. Briefly, three different software, which are MUSCLE v3.743, MAFFT v7.31344 and Kalign v2.0445 were used to create multiple sequence alignments in both forward and reverse directions (also known as Head or Tail approach46). M-Coffee47 was then used to combine all six multiple sequence alignment results into a consensus alignment. Finally, trimAl v1.4rev2248 was used to trim poorly aligned regions present in the consensus alignment result with the consistency and gap score cut-off of 0.1667 and 0.1 respectively.
Reconstruction of oomycete phylogenomic tree
The phylogenomic tree reconstruction was done by using the approach described by Capella-Gutierrez and co-workers49. Briefly, the process started by reconstructing a phylogenetic tree from each alignment using the Neighbor Joining (NJ) method50. Seven different evolutionary models (HKY85, JC69, K80, F81, F84, TN93, and GTR) were then used to calculate the likelihood of the NJ tree topology, with branch-length optimization permitted, using PhyML v3.051. Next, the AIC criterion52 was used to compare the likelihood of the used models to identify the model that fitted best with each alignment data. This process was applied to all core gene alignments and the alignments were grouped based on the best fitted evolutionary model. The individual alignments in the largest group were then concatenated and PhyML v3.0 was used to construct the Maximum Likelihood tree from the concatenated alignment using the corresponding best fitted model with the following options: (i) Tree topology search operation set to Subtree Pruning and Regrafting (SPR), (ii) Nucleotide frequencies estimated by counting the occurrence of the different bases in the alignment, (iii) Proportion of invariable sites estimated by the maximum likelihood approach, (iv) Value of the gamma shape parameter estimated by the maximum likelihood approach, (v) Number of relative substitution rate categories set to 8, (vi) Branch supports set to Chi2-based parametric method, (vii) Parameter optimizations carried out on tree topology, branch length and substitution rate parameters. Finally, the FigTree software v1.4.0 (http://tree.bio.ed.ac.uk/software/figtree/) was used to draw the phylogenomic tree result where tree rooting was done using midpoint approach.
Genome relatedness based on hierarchical clustering of gene presence profile
The homologous gene cluster result was used to create the gene presence profile data. A value of 0 or 1 was assigned to denote the absence or presence of a gene in each genome, respectively. A value of 1 was also assigned to cases in which a gene was not predicted but a homologous genomic region was found in that particular genome. Hierarchical clustering of gene presence profiles based on relative Manhattan distance and bootstrapping analysis was performed using the R programming language.
Annotation of Py. insidiosum genome
The 53.2-Mb draft genome sequence of Py. insidiosum (generated from the Illumina HiSeq. 200 and 454 FLX Titanium NGS platforms)19 underwent gene prediction, using the transcriptome of Py. insidiosum20, the predicted proteome of Py. ultimum18 and an array of bioinformatics tools53, including RepeatMasker (http://www.repeatmasker.org), CEGMA 2.554, GeneMark-ES55, Augustus 2.5.556 and MAKER257. To annotate the genome of Py. insidiosum, all deduced protein sequences were subjected to (i) a BLAST search against the NCBI nonredundant protein database (E-value cut-off, <−6), (ii) GO assignment using BLAST2GO58 and (iii) domain identification using InterProScan 559, Conserved Domain Database60, TMHMM 2.061 and SignalP35. Putative virulence genes of Py. insidiosum were predicted based on a sequence homology search against the MvirDB database25. Adhesin-like proteins were predicted using FungalRV62. Selected proteins were aligned for sequence similarity and checked for protein architecture using MUSCLE63, NCBI BLAST and CD-search programs (https://blast.ncbi.nlm.nih.gov/Blast.cgi).
Extraction of Py. insidiosum proteins
Ten small pieces (5 × 5 mm) of Sabouraud dextrose agar with actively growing Py. insidiosum (strain Pi-S), cut from a one-week-old colony, were transferred to two 500-ml Erlenmeyer flasks containing 100 ml of Sabouraud dextrose broth and incubated with shaking (150 rpm) for 10 days at 25 °C and 37 °C. Hyphae were harvested by filtration through Whatman filter paper No. 1 (pore size, 0.75 µm). The organism was ground in a precooled mortar, in the presence of liquid nitrogen. Ruptured hyphae were transferred to a 50-ml conical tube with 30 ml of cold sterile distilled water and mixed by inversion. The resulting cell lysate was centrifuged at 4,000 × g for 1 h (Beckman Coulter) to collect supernatant, which contained water-soluble proteins called soluble antigens from broken hyphae (SABHs)64,65. SABHs were concentrated 80-fold using an Amicon centrifugation tube (10,000 nominal-molecular-weight limit; Millipore), before storage at −20 °C. Overall, SABHs were prepared in four replicates (two each from Py. insidiosum grown at 25 °C and 37 °C).
Preparation of tryptic-digested proteins
Ten micrograms of SABH was mixed with 2× protein loading buffer, containing 2× Laemmli sample buffer (Bio-Rad Laboratories, USA) and 2-mercaptoethanol, and denatured by heating at 65 °C for 10 min. The sample was subjected to one-dimensional SDS-PAGE, using 5% stacking and 12% resolving gels, and a mini-vertical electrophoresis system (Bio-Rad Laboratories, USA). The gels were stained with Coomassie brilliant blue G250. An individual lane of the SDS-PAGE gel was segmentally cut along its length into small pieces. To perform in-gel digestion, gel pieces were destained until colourless, using acetonitrile (50%) in 50 mM NH4HCO3. After the destaining solution had been removed, 10 mM dithiothreitol was added to the gel pieces, followed by incubation at 60 °C for 15 min. Proteins were alkylated using 55 mM iodoacetamide in 50 mM NH4HCO3 at room temperature for 30 min in the dark. Following the removal of all solution, the gel pieces were dehydrated by 100% ACN (Sigma-Aldrich, USA) and dried at room temperature. Protein digestion was performed overnight at 37 °C, using 0.1 mg/mL trypsin (Sigma-Aldrich, USA) in 50 mM ammonium bicarbonate. Peptides were extracted in 50% ACN. The resulting supernatant was transferred to microcentrifuge tubes and dried in a centrifugal concentrator (TOMY, Japan) at 45 °C.
Liquid chromatography–mass spectrometric analysis
Each tryptic-digested fraction was resuspended in 0.1% formic acid and introduced to an Ultimate 3000 nano-LC system (Dionex, Surrey, UK). The separation was performed at a flow rate of 300 nL/min under a 45-min gradient. The Acclaim PepMap RSLC 75 μm × 15 cm nanoviper C18, 2-μm particle size, 100-Å pore size (Thermo Scientific, Waltham, MA), was used as a reverse-phase chromatography column. Mobile phase A was 2% (v/v) acetonitrile and 0.1% (v/v) formic acid in HPLC-grade water, and mobile phase B was 0.1% (v/v) formic acid in HPLC-grade acetonitrile. The eluate was sprayed to a MicroToF Q II mass spectrometer (Bruker, Bremen, Germany). Data were acquired using Hystar software (Bruker Daltonics, Germany). The survey scan mode covered the mass range of m/z 400–2500 and the MS/MS spectra covered the mass range of m/z 50–1500.
The ‘.d’ files from the microTOFQ II mass spectrometer were converted to ‘.mgf’ files by Compass DataAnalysis software (Bruker Daltonics, Germany) and used as queries for database sequence searches against the Py. insidiosum proteome19, using an in-house Mascot server (version 2.3.0; Matrix Science, USA). Trypsin was set as the enzyme. Only one missed cleavage site was allowed in the search. Variable modifications were set as carbamidomethyl (C) and oxidation (M). MS peptide and MS/MS tolerances were set at 0.6 Da and 0.8 Da, respectively. To reduce false-positive identification, only peptides with 95% confidence were reported in this study. Abundances of the proteins were calculated, based on the exponentially modified protein abundance index (emPAI)66,67. Each protein match was reported as (i) actual amount in emPAI units, (ii) ‘no emPAI’ (if the protein was detectable, but unquantifiable) or (iii) ‘n/a’ (if the protein was undetectable).
Urease assay
A 5-mm-diameter Sabouraud dextrose agar plug with actively growing Py. insidiosum mycelia from three different isolates (Pi19, Pi20 and Pi-S) was transferred to a urease test tube (CLINAG, Thailand) and incubated at 37 °C for 7 days. Positivity for urease was identified if the urea-containing agar turned pink, while negativity was assigned if it remained yellow.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files) and are available from the corresponding author on reasonable request. The DNA sequence of the putative urease gene (Ure1) present in the genome of Py. insidiosum (strain Pi-S) has been submitted to the DNA Data Bank of Japan database, under accession number LC317047.
References
Kamoun, S. Molecular genetics of pathogenic oomycetes. Eukaryot. Cell 2, 191–199 (2003).
Kwon-Chung, K. J. Phylogenetic spectrum of fungi that are pathogenic to humans. Clin. Infect. Dis. 19(Suppl 1), S1–7 (1994).
Ascunce, M. S. et al. Phylogenomic analysis supports multiple instances of polyphyly in the oomycete peronosporalean lineage. Mol. Phylogenet. Evol. 114, 199–211 (2017).
McCarthy, C. G. P. & Fitzpatrick, D. A. Phylogenomic Reconstruction of the Oomycete Phylogeny Derived from 37 Genomes. mSphere 2 (2017).
Jiang, R. H. Y. & Tyler, B. M. Mechanisms and Evolution of Virulence in Oomycetes. Annu. Rev. Phytopathol. 50, 295–318 (2012).
Krajaejun, T. et al. Clinical and epidemiological analyses of human pythiosis in Thailand. Clin. Infect. Dis. 43, 569–576 (2006).
Calvano, T. P. et al. Pythium aphanidermatum infection following combat trauma. J. Clin. Microbiol. 49, 3710–3713 (2011).
Farmer, A. R. et al. Combat-Related Pythium aphanidermatum Invasive Wound Infection: Case Report and Discussion of Utility of Molecular Diagnostics. J. Clin. Microbiol. 53, 1968–1975 (2015).
Reinprayoon, U. et al. Lagenidium sp. ocular infection mimicking ocular pythiosis. J. Clin. Microbiol. 51, 2778–2780 (2013).
Grooters, A., Proia, L., Sutton, D. & Hodgin, E. Characterization of a previously undescribed Lagenidium pathogen associated with soft tissue infection: initial description of a new human oomycosis. 14th Focus Fungal Infect. New Orleans (2004).
Gaastra, W. et al. Pythium insidiosum: an overview. Vet. Microbiol. 146, 1–16 (2010).
Lerksuthirat, T. et al. Evolution of the Sterol Biosynthetic Pathway of Pythium insidiosum and Related Oomycetes Contributes to Antifungal Drug Resistance. Antimicrob. Agents Chemother. 61 (2017).
Baxter, L. et al. Signatures of adaptation to obligate biotrophy in the Hyaloperonospora arabidopsidis genome. Science 330, 1549–1551 (2010).
Haas, B. J. et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461, 393–398 (2009).
Lamour, K. H. et al. Genome Sequencing and Mapping Reveal Loss of Heterozygosity as a Mechanism for Rapid Adaptation in the Vegetable Pathogen Phytophthora capsici. Mol. Plant-Microbe Interact. MPMI 25, 1350–1360 (2012).
Waugh, M. et al. The phytophthora genome initiative database: informatics and analysis for distributed pathogenomic research. Nucleic Acids Res. 28, 87–90 (2000).
Tyler, B. M. et al. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science 313, 1261–1266 (2006).
Lévesque, C. A. et al. Genome sequence of the necrotrophic plant pathogen Pythium ultimum reveals original pathogenicity mechanisms and effector repertoire. Genome Biol. 11, R73 (2010).
Rujirawat, T. et al. Draft Genome Sequence of the Pathogenic Oomycete Pythium insidiosum Strain Pi-S, Isolated from a Patient with Pythiosis. Genome Announc. 3, (2015).
Krajaejun, T. et al. Transcriptome analysis reveals pathogenicity and evolutionary history of the pathogenic oomycete Pythium insidiosum. Fungal Biol. 118, 640–653 (2014).
Adhikari, B. N. et al. Comparative Genomics Reveals Insight into Virulence Strategies of Plant Pathogenic Oomycetes. PLoS ONE 8, e75072 (2013).
Jiang, R. H. Y. et al. Distinctive Expansion of Potential Virulence Genes in the Genome of the Oomycete Fish Pathogen Saprolegnia parasitica. PLoS Genet. 9, e1003272 (2013).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003).
Galperin, M. Y., Makarova, K. S., Wolf, Y. I. & Koonin, E. V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–269 (2015).
Zhou, C. et al. MvirDB—a microbial database of protein toxins, virulence factors and antibiotic resistance genes for bio-defence applications. Nucleic Acids Res. 35, D391–D394 (2007).
de Cock, A. W. A. M. et al. Phytopythium: molecular phylogeny and systematics. Persoonia 34, 25–39 (2015).
Jiang, R. H. Y., Tyler, B. M., Whisson, S. C., Hardham, A. R. & Govers, F. Ancient origin of elicitin gene clusters in Phytophthora genomes. Mol. Biol. Evol. 23, 338–351 (2006).
PanabiÈRes, F., Ponchet, M., Allasia, V., Cardin, L. & Ricci, P. Characterization of border species among Pythiaceae: several Pythium isolates produce elicitins, typical proteins from Phytophthora spp. Mycol. Res. 101, 1459–1468 (1997).
Lerksuthirat, T. et al. The elicitin-like glycoprotein, ELI025, is secreted by the pathogenic oomycete Pythium insidiosum and evades host antibody responses. PloS One 10, e0118547 (2015).
Mikes, V., Milat, M.-L., Ponchet, M., Ricci, P. & Blein, J.-P. The fungal elicitor cryptogein is a sterol carrier protein. FEBS Lett. 416, 190–192 (1997).
Mikes, V. et al. Elicitins, proteinaceous elicitors of plant defense, are a new class of sterol carrier proteins. Biochem. Biophys. Res. Commun. 245, 133–139 (1998).
Osman, H. et al. Mediation of elicitin activity on tobacco is assumed by elicitin-sterol complexes. Mol. Biol. Cell 12, 2825–2834 (2001).
Boissy, G. et al. The 2.1 A structure of an elicitin-ergosterol complex: a recent addition to the Sterol Carrier Protein family. Protein Sci. Publ. Protein Soc. 8, 1191–1199 (1999).
Nürnberger, T., Brunner, F., Kemmerling, B. & Piater, L. Innate immunity in plants and animals: striking similarities and obvious differences. Immunol. Rev. 198, 249–266 (2004).
Petersen, T. N., Brunak, S., von Heijne, G. & Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011).
Mendoza, L., Hernandez, F. & Ajello, L. Life cycle of the human and animal oomycete pathogen Pythium insidiosum. J. Clin. Microbiol. 31, 2967–2973 (1993).
Cox, G. M., Mukherjee, J., Cole, G. T., Casadevall, A. & Perfect, J. R. Urease as a virulence factor in experimental cryptococcosis. Infect. Immun. 68, 443–448 (2000).
Mora, D. & Arioli, S. Microbial urease in health and disease. PLoS Pathog. 10, e1004472 (2014).
Rutherford, J. C. The emerging role of urease as a general microbial virulence factor. PLoS Pathog. 10, e1004062 (2014).
Kittichotirat, W., Bumgarner, R. E., Asikainen, S. & Chen, C. Identification of the pangenome and its components in 14 distinct Aggregatibacter actinomycetemcomitans strains by comparative genomic analysis. PloS One 6, e22420 (2011).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Huerta-Cepas, J. et al. PhylomeDBv3.0: an expanding repository of genome-wide collections of trees, alignments and phylogeny-based orthology and paralogy predictions. Nucleic Acids Res. 39, D556–560 (2011).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Lassmann, T., Frings, O. & Sonnhammer, E. L. L. Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 37, 858–865 (2009).
Landan, G. & Graur, D. Heads or tails: a simple reliability check for multiple sequence alignments. Mol. Biol. Evol. 24, 1380–1383 (2007).
Wallace, I. M., O’Sullivan, O., Higgins, D. G. & Notredame, C. M-Coffee: combining multiple sequence alignment methods with T-Coffee. Nucleic Acids Res. 34, 1692–1699 (2006).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinforma. Oxf. Engl. 25, 1972–1973 (2009).
Capella-Gutierrez, S., Kauff, F. & Gabaldón, T. A phylogenomics approach for selecting robust sets of phylogenetic markers. Nucleic Acids Res. 42, e54 (2014).
Gascuel, O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Akaike, H. Information Theory and an Extension of the Maximum Likelihood Principle. in Selected Papers of Hirotugu Akaike (eds Parzen, E., Tanabe, K. & Kitagawa, G.) 199–213, https://doi.org/10.1007/978-1-4612-1694-0_15 (Springer New York, 1998).
Yandell, M. & Ence, D. A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 13, 329–342 (2012).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Lomsadze, A., Ter-Hovhannisyan, V., Chernoff, Y. O. & Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 33, 6494–6506 (2005).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 33, W465–467 (2005).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinforma. Oxf. Engl. 21, 3674–3676 (2005).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinforma. Oxf. Engl. 30, 1236–1240 (2014).
Marchler-Bauer, A. et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–229 (2011).
Krogh, A., Larsson, B., von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Chaudhuri, R., Ansari, F. A., Raghunandanan, M. V. & Ramachandran, S. FungalRV: adhesin prediction and immunoinformatics portal for human fungal pathogens. BMC Genomics 12, 192 (2011).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Mendoza, L., Kaufman, L., Mandy, W. & Glass, R. Serodiagnosis of human and animal pythiosis using an enzyme-linked immunosorbent assay. Clin. Diagn. Lab. Immunol. 4, 715–718 (1997).
Krajaejun, T., Kunakorn, M., Niemhom, S., Chongtrakool, P. & Pracharktam, R. Development and evaluation of an in-house enzyme-linked immunosorbent assay for early diagnosis and monitoring of human pythiosis. Clin. Diagn. Lab. Immunol. 9, 378–382 (2002).
Ishihama, Y. et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics MCP 4, 1265–1272 (2005).
Dowle, A. A., Wilson, J. & Thomas, J. R. Comparing the Diagnostic Classification Accuracy of iTRAQ, Peak-Area, Spectral-Counting, and emPAI Methods for Relative Quantification in Expression Proteomics. J. Proteome Res. 15, 3550–3562 (2016).
Acknowledgements
This research project was supported by the Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Thailand, Research Fund (grant number, BRG5980009) and Mahidol University (grant number, ngor-por 09/2560). We are grateful to Angkana Chaiprasert and Piriyaporn Chongtrakool for material support. We thank Thomas D. Sullivan for reviewing the manuscript. We also thank Edanz Group for editing a draft of this manuscript.
Author information
Authors and Affiliations
Contributions
W.K. and T.K. conceived the project. T.R., P.P., T.L., W.Y., Y.K., P.P., S.T. and O.R. performed the experiments. T.R., P.P., P.S., O.R., G.G., W.K. and T.K. analysed the data. T.R., P.P., O.R., W.K. and T.K. wrote the manuscript. All authors reviewed the manuscript. T.K. acquired the research funds.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rujirawat, T., Patumcharoenpol, P., Lohnoo, T. et al. Probing the Phylogenomics and Putative Pathogenicity Genes of Pythium insidiosum by Oomycete Genome Analyses. Sci Rep 8, 4135 (2018). https://doi.org/10.1038/s41598-018-22540-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-22540-1
This article is cited by
-
MGI short-read genome assemblies of Pythium insidiosum (reclassified as Pythium periculosum) strains Pi057C3 and Pi050C3
BMC Research Notes (2023)
-
PacBio long read-assembled draft genome of Pythium insidiosum strain Pi-S isolated from a Thai patient with pythiosis
BMC Research Notes (2023)
-
Dilution of specialist pathogens drives productivity benefits from diversity in plant mixtures
Nature Communications (2023)
-
Rapid differentiation of soil and root microbiomes in response to plant composition and biodiversity in the field
ISME Communications (2023)
-
Genome data of four Pythium insidiosum strains from the phylogenetically-distinct clades I, II, and III
BMC Research Notes (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
