
Abstract
Polymorphisms within NF-κB pathway genes may be linked to hepatitis C virus (HCV) infection susceptibility and outcomes. We investigated the associations between single nucleotide polymorphisms (SNPs) in NF-κB and the susceptibility as well as resolution of HCV infection. A Chinese population, including 1125 uninfected control cases, 558 cases with spontaneous viral clearance and 898 cases with persistent HCV infection, was genotyped for four SNPs (rs11820062, rs230530, rs1056890 and rs3774963) using a TaqMan assay. Our logistic analyses indicate that the subjects carrying RelA rs11820062 A allele had a significantly increased risk of HCV susceptibility (P Bonferroni < 0.003125 in a dominant or additive model). In stratified analysis, the increased risk associated with rs11820062 A allele on HCV susceptibility remained in some case subgroups. This study demonstrates that a genetic variant involved in the NF-κB pathway gene (rs11820062 A allele) is associated with an increased HCV susceptibility within a high-risk Chinese population.
Similar content being viewed by others

Introduction
More than 185 million people are estimated to be chronically infected with hepatitis C virus (HCV). In China, the prevalence of HCV is approximately 1.6% of the population1,2. Over 70% of HCV infected patients who fail to eliminate the virus go on to develop chronic hepatitis C (CHC), cirrhosis and hepatocellular carcinoma3. Variable HCV infection outcomes are related to both host genetic factors and virological factors. Recently, novel direct-acting antiviral agents (DAAs) have dramatically risen HCV cure rates to 95% and have served as alternatives to PEGylated interferon (IFN) plus ribavirin therapy for chronic hepatitis C4,5. However, the efficacy and safety of DAAs in real-world settings are not well understood. Genetic factors related to the occurrence and clearance of HCV infection need to be explored.
Nuclear factor κ-light-chain-enhancer of activated B cells (NF-κB) exists in almost all mammalian cell types, and controls the transcription of many target genes by binding κB response elements. Thus activated, it can influence cell differentiation, apoptosis, inflammation processes and immune response6. NF-κB is not a single transcription factor but rather a human protein family containing five members, including NF-κB1(p50), NF-κB2 (p52), RelA (p65), RelB, c-Rel6,7. These members can form several kinds of homodimers or heterodimers. In unstimulated cells, inactive NF-κB proteins exist in the cytoplasm in association with inhibitors of κB (IκB), including IκBα, IκBβ, IκBγ (p105), IκBδ (p100), IκBε and Bcl-3. Different dimers of NF-κB proteins can be activated in different ways depending on their association with IκBα or IκBβ, in which IκB is prone to phosphorylation and degradation6,7,8. The predominant activated form of NF-κB is a p50/p65 dimer that is associated with IκBα6. In the canonical pathway, after stimulation with a large number of NF-κB inducers, IκBα is phosphorylated by IκB kinase (IKK) at serines 32 and 36 and then becomes ubiquitylated by a specific E3 ligase, followed by the degradation by the 26S proteasome6. The released NF-κB dimer can subsequently translocate into the nucleus where its target gene is activated by binding to gene promoters with high affinity to NF-κB elements6.
NF-κB activity is necessary for both innate and adaptive immunity. It can be induced by a variety of stimulants such as bacterial and viral antigens, cytokines, oxidative stress, and triggers the transcription of many inflammatory mediators, including chemokines, pro-inflammatory cytokines, and adhesion molecules6,9,10. Consequently, aberrant activation or dysregulation of NF-κB signaling pathway genes contributes to the development of some autoimmune diseases, inflammation, and malignant disorders11; it is also involved with viral evasion and subversion with some types of infections via the biphasic regulation of NF-κB activity12.
Single nucleotide polymorphisms (SNPs) in the NF-κB signaling pathway genes have been associated with many diseases. SNP rs11820062 in RelA is not only significantly associated with kidney function and chronic kidney disease susceptibility13, but also with susceptibility to schizophrenia and may affect androgen receptor transcription factor (TF) binding14. Previous studies have also identified SNP rs230530 in NF-κB1 as associated with liver cancer15 and alcohol addiction16. Furthermore, NFκB2 rs1056890 has been related to the development of multiple myeloma and response to bortezomib therapy17, and related to the inflammatory response among patients with secondary lymphedema following breast cancer surgery18 as well as other immune-related diseases19,20.
Given these findings about the critical role of NF-κB in the host immune response against infection, any genetic variation in NF-κB signaling pathway genes may affect the progression and outcomes of HCV infection. However, no research has addressed the association between the NF-κB genetic polymorphisms and HCV infection. Thus, this study aims to examine the relationships between NF-κB signaling pathway genes SNP rs1056890, rs11820062, rs230530 and rs3774963, and HCV infection outcomes within a high-risk Chinese population.
Materials and Methods
Ethics statement
The study was performed in accordance with the World Medical Association Declaration of Helsinki on ethical principles in medical research involving human subjects, and was approved by the medical ethics committee of Nanjing Medical University. All study subjects provided signed written informed consent.
Study subjects
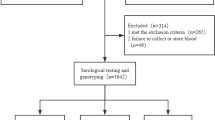
A total of 2581 subjects were recruited between October 2008 and May 2015 including 816 hemodialysis (HD) subjects from nine hospital hemodialysis centers in southern China, 510 intravenous drug users from Nanjing Compulsory Detoxification Center and 1255 paid blood donors from six villages within Zhenjiang City. These paid blood donors had become infected with HCV when they engaged in paid blood donation multiple times, some as often as 100 times or more. Subjects were excluded if they were co-infected with hepatitis B virus or human immunodeficiency virus (HIV), suffered from other liver diseases (including autoimmune, alcoholic or metabolic liver diseases), or were treated with any antiviral medications before or during the trial. All subjects were divided into three groups for further analysis: the HCV-uninfected controls (Group A) were subjects seronegative for anti-HCV and HCV-RNA; those in the spontaneous clearance group (Group B) were subjects with seropositive anti-HCV and seronegative HCV-RNA; and those in the persistent HCV infection group (Group C) were subjects with seropositive anti-HCV and HCV RNA. Group B and Group C were combined as the HCV infected group. All serological results were confirmed by three separate experiments within the 6 month follow-up period. The control subjects in group A were matched by age, gender and the village of recruitment (5-year intervals) with infected subjects belonging to group B or group C.
An interview was carried out with each participant, and a structured questionnaire was administered by well-trained interviewers to collect demographic information, an environmental exposure history and a medical history of HCV infection. Quality control procedures were established to guarantee the reliability of all data obtained.
Viral testing
A 10 mL venous blood sample was collected from each participant after the interview. White blood cells were isolated by centrifugation and subsequently stored at −80 °C until use. Anti-HCV antibodies were tested using a third-generation enzyme-linked immunosorbent assay (ELISA: Diagnostic Kit for Antibody to HCV 3.0 ELISA, Intec Products Inc, Xiamen, China) according to the manufacturer’s instructions. HCV RNA was extracted from patient serum using Trizol LS Reagent (Takara Biotech, Tokyo, Japan), and reverse transcription PCR (Takara Biotech) was performed. The Murex HCV Serotyping 1–6 Assay ELISA (Abbott, Wiesbaden, Germany) was used to determine the type-specific antibodies of various HCV genotypes21.
SNPs selection
Tag SNPs (rs230530 and rs3774963) were selected using Haploview software (version 4.2; Broad Institute, Cambridge, MA, USA) based on the linkage disequilibrium (LD) data of HapMap Phase II CHB (Chinese in Beijing) obtained from the HapMap database (http://www.hapmap.org/) or the 1000 Genomes Project database (http://www.1000genomes.org/). We used minor allele frequency (MAF) > 5% as the criteria among the Chinese Han population. Considering the potential regulatory effects of adjacent sequences, 2000 bp upstream and downstream of NF-κB gene transcription initiation sites were included in the analysis. SNPs located on 5′-UTR, 5′ flanking region (rs11820062), 3′-UTR (rs1056890), and exons with missense substitutions were considered, and combined with SNPs with some connection to the liver15,16 or or immune-related disorders13,14,17,18,19,20 reported in the literature. Utilizing the above strategies, four candidate SNPs, rs1056890, rs11820062, rs230530 and rs3774963, were selected for further study.
Genotyping Assays
Protease K digestion was used to extract genomic DNA from subject peripheral blood leukocytes, followed by phenol–chloroform extraction and ethanol precipitation. Genotyping of the four SNPs was performed with a TaqMan allelic discrimination assay on an ABI 7900HT Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The primers and probe sequences for selected SNPs are shown in Table 1. The technicians who performed the genotyping were blinded to subject clinical data. For quality confirmation, two negative controls were set in each 384-well plate, and a 100% concordance was achieved in 10% random samples. The success rates of genotyping for the selected four SNPs were all above 95%. Samples that failed genotyping were excluded in the statistical analyses.
In silico analysis
The function of SNPs in genes was predicted using the bioinformatics tool SNP function prediction online server (SNPinfo; http://snpinfo.niehs.nih.gov/). To detect and characterize the influence of the polymorphism sites on the secondary structure of genes, RNA secondary structures were predicted using the Vienna RNA Web Servers based on the latest Vienna RNA Package (Version 2.3.1) (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Secondary structures with the lowest free energies of minimum free energy (MFE) and centroid secondary structures (a structure with minimal base pair distance) for wild-type and mutant-type mRNA sequences were calculated and compared. Moreover, Human Splicing Finder (http://www.umd.be/HSF/) was used to identify possible splicing signal in sequences containing the SNP sites.
Statistical analysis
χ2-test, Kruskal–Wallis test or one-way analysis of variance were used to appropriately analyze the distribution general demographic, clinical, virological features, and genotype frequencies among the three groups. Hardy–Weinberg equilibrium (HWE) for each SNP was estimated by χ2 goodness of fit test among controls. LD parameters (r2 and D′) were calculated using Haploview software. The associations between SNPs and the HCV infection outcomes were estimated by calculating the odds ratios (ORs) and 95% confidence intervals (CI), adjusted for age, gender and route of infection using binary logistic regression models. A P-value < 0.05 in a two-sided test was considered statistically significant. Bonferroni correction was used to correct for multiple comparisons. All statistical analyses were carried out with the Statistical Package for the Social Sciences (SPSS: version 20.0; SPSS Institute, Chicago, IL, USA) and Statistical Analysis System software (SAS: version 9.1.3; SAS Institute, Cary, NC, USA).
Results
Demographic and selected variables of participants
The demographic and other selected characteristics for a total of 2581 subjects are summarized in Table 2. There were no significant differences in the distribution of age or gender among the three groups, while ALT, AST, routes of infection and HCV genotype (all P < 0.001) showed significant differences.
All four SNPs’ allele distributions in the control group were in accordance with the expectations of the Hardy–Weinberg equilibrium. Group A was considered as the control group when compared with group B + C (group A: P = 0.834 for rs1056890, P = 0.179 for rs11820062, P = 0.063 for rs230530, P = 0.303 for rs3774963) and group B was considered as the control group when compared to group C (group B: P = 0.512 for rs1056890, P = 0.479 for rs11820062, P = 0.366 for rs230530, P = 0.579 for rs3774963).
Association between NF-κB genes polymorphisms and the susceptibility to HCV infection
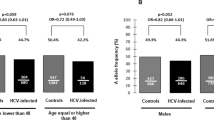
The distributions of genotypes rs1056890, rs11820062, rs230530 and rs3774963 among the control group, spontaneous clearance group, and persistent HCV infection group are shown in Table 3. Three genetic models, including additive, dominant and recessive models, were used to analyze the association between each SNP and susceptibility to HCV infection. After adjusting for gender, age, and the route of infection, the results of logistic regression analysis demonstrated that individuals carrying the rs11820062 AA genotype had a significantly higher risk of HCV infection (P = 0.002) after Bonferroni correction at the level 0.05/16. In the additive and dominant statistical models, the relationship between the rs11820062 A allele and susceptibility to HCV infection remained statistically significant after Bonferroni correction (P = 0.002 and P = 0.003, respectively).
No associations between rs230530, rs1056890, rs3774963 and HCV susceptibility remained statistically significant after Bonferroni correction (all P > 0.003).
Further stratified analysis was conducted based on age, gender and routes of infection. Subjects were divided into two subgroups by average age (age < 50 and age ≥ 50) for analysis. As shown in Table 4, the correlation between rs11820062 A allele and the susceptibility to HCV infection was still statistically significant in the young (age < 50), female and blood donors subject subgroups (all P < 0.05).
Association between NF-κB polymorphisms and spontaneous clearance of HCV infection
No association was observed between the four SNPs (rs1056890, rs11820062, rs230530 and rs3774963) and spontaneous clearance of HCV in our logistic regression analysis (all P > 0.003, Table 2), or using additive, dominant, recessive models (all P > 0.003, Table 2).
In silico Analysis of SNPs function
Rs11820062 is located on intron 1 or near the 5′ end of the RelA gene (from two different mapping pipelines for rs11820062 on NCBI dbSNP: https://www.ncbi.nlm.nih.gov/snp/), which contains 11 exons and is mapped to chromosome 11q13.1. Using the SNPinfo web server, rs11820062 was predicted to have a TF binding site. Additionally, provided that this SNP is located near the 5′ end of RelA, the mutation in this region could also alter the binding of TF and transcriptional regulation. Therefore, the effect of this polymorphism site on the mRNA secondary structure was further analyzed using the RNAfold web server. The local structure changes are shown in Fig. 1. The minimum free energy of the centroid mRNA secondary structure (a structure with minimal base pair distance) for mutant T allele of rs11820062 (corresponded to A allele in this research, −27.70 kcal/mol) was lower than that of wild C allele (corresponded to G allele in this research, −22.50 kcal/mol).

The influence of rs11820062 on mRNA centroid secondary structures of RelA near the 5′ end region. Changes in the local structure were illustrated by the RNAfold Web Server. The arrow indicates the position of the mutation (50 bases upstream and 50 bases downstream from the mutation). The minimum free energy of the mRNA centroid secondary structure (a structure with minimal base pair distance) for wild type and mutant rs11820062 were estimated to be −22.50 kcal/mol (Fig. 1: left figure) and −27.70 kcal/mol (Fig. 1: right figure), respectively. The wild-type and mutant-type sequences are listed below. Underline bold type indicates the overlapping nucleotide letter that are unreadable in figures. The framed type indicates the nucleotide difference between the wild and mutant allele. Wild-type sequence: CAGAGGGAAGCUGAAUCAGGGCCUGUUGUACUUUCUUAAGGAAAA UGAGGGAGGGCACGCCCCACCUCCCUCCAGAGAGGAAACUGAAUC Mutant-type sequence: CAGAGGGAAGCUGAAUCAGGGCCUGUUGUACUUUCUUAAGGAAAA
UGAGGGAGGGCACGCCCCACCUCCCUCCAGAGAGGAAACUGAAUC Mutant-type sequence: CAGAGGGAAGCUGAAUCAGGGCCUGUUGUACUUUCUUAAGGAAAA UGAGGGAGGGCACGCCCCACCUCCCUCCAGAGAGGAAACUGAAUC.
UGAGGGAGGGCACGCCCCACCUCCCUCCAGAGAGGAAACUGAAUC.
Rs230530 and rs3774963 are located on the NF-κB1 gene, which contains 24 exons and is mapped to chromosome 4q23-q24. Both SNPs are located in the intron region of NF-κB1 (rs230530: deep region of intron 3, rs3774963: intron 15). The Human Splicing Finder web server and the SNPinfo web server were used to analyze the effect of the intron variants on splicing or other functions. However, no changes were predicted for either SNPs.
Rs1056890 is located in the 3′-UTR region of the NF-κB2 gene, which contains 25 exons and is mapped to chromosome 10q24.32. According to the SNPinfo web server, rs1056890 is located within the miRNA binding site and affected gene expression regulation at the post-translational level. However, this SNP was not found to be relevant to HCV infection. The mutation’s influence at this site on the secondary structures of NF-κB2 3′-UTR messenger ribonucleic acid (mRNA) was predicted using the RNAfold web server. No differences in the secondary structures between wild-type and mutant type were found (Supplementary Figure 1). The lowest free energy secondary structure for the C allele of rs1056890 is nearly identical to that of the T allele as estimated by MFE (−48.58 kcal/mol versus −46.77 kcal/mol, respectively; Supplementary Figure 1). Additionally, the minimum free energy of the mRNA centroid secondary structures for both mutant type and wild-type of rs1056890 were estimated at −22.20 kcal/mol (Supplementary Figure 1). Therefore, a mutation at this site can not influence the stability and function of the mRNA, NF-κB2 gene expression or HCV infection.
Discussion
Our results indicated that rs11820062 A allele was independently associated with the risk effects of HCV infection. Host genetic variation plays a critical role in the pathogenesis and development of HCV infection. Many genes have been reported to affect HCV infection immune response resulting in different disease outcomes. Our previous studies have found that genetic variants of Toll-like receptor 7 22,23, interleukin-18 24, human leukocyte antigen class II 25,26, vitamin D receptor 27 and estrogen receptor α 28 genes can affects the susceptibility and resolution of HCV infection. It is believed that NF-κB acts as a switch or a sensor in a wide variety of cellular processes and signaling pathways responsible for inflammatory response to viral infections9,12. Polymorphisms in NF-κB pathway genes have also been linked to a range of diseases. A growing number of studies suggest that NF-κB polymorphisms are related to many immunity-associated diseases, such as hepatocellular carcinoma29, colorectal cancer30, chronic hepatitis C and liver disease progression31, rheumatoid arthritis32, asthma33 as well as other diseases13,14,15,16,17,18,19,20.
In this study, the A allele (mutant type) of rs11820062 (located on intron 1 or near the 5′ end of RelA) was found to be linked to susceptibility to HCV infection. RelA protein, also known as p65, is involved in p50/65 complex formation, nuclear translocation and NF-κB activation. Previous research has shown that the SNP rs11820062 confers increased susceptibility to chronic kidney disease13 and schizophrenia14. Firstly, provided that this SNP is located on intron 1, a multitude of research has confirmed that many regulatory elements, including enhancers, silencers, or other elements that modulate the function of the main upstream promoter, are often within the first intron closest to the 5′ region34. This 5′-most intron can stimulate transcription initiation35 and is related to mRNA expression levels34. Previous in silico genotype-gene expression analysis found that this SNP can alter mRNA expression of RELA in immortalized B lymphocytes and the binding to androgen receptor14. The risk allele (mutant type: A allele in this research) was especially associated with the lower transcriptional activity of RELA 14. Secondly, according to predictions regarding this SNP location near the 5′ end of RELA gene, the minimum free energy of the centroid mRNA secondary structure for mutant type is lower than that of the wild-type. Therefore, the genetic variation of rs11820062 may affect the RELA gene transcription process and regulate gene expression, which ultimately affects NF-κB activation and the susceptibility to HCV infection.
In stratification analyses based on age, gender and route of infection, we found that rs11820062 A allele also tracked the risk of HCV susceptibility among young, female, and blood donor subgroups. In general, being young and female are regarded as the common protective factors for hepatitis C because of the youth- and estradiol-related more effective immune response36,37. The mechanism of the risk effects among young, female subgroups is not clear in this study. According to in silico analysis, a possible cause of the risk associated with rs11820062 T allele showed in young and female subgroups might be related to the disruption of the consensus transcription factor binding sequences (wild allele to risk mutant allele) in the androgen receptor14. Furthermore, from 1980 to 1990, blood donors were monetarily compensated, and subsequently numerous donors were found to be infected with HCV. The plasma of some paid donors was separated and collected by plasmapheresis, and the other blood components that contained cross-contamination were returned to the donor. A mandatory HCV screening policy for bloodborne diseases in donated blood was implemented in China in 199338. In this study, the blood donation subjects, especially regarding the unsafe paid blood donors, may result in a high HCV inoculum than that of hemodialysis patients or intravenous drug abuser. Therefore, the subjects in the blood donors subgroup carried the rs11820062 A allele had a significantly increased risk of HCV infection.
Our study was limited because the exact age and time of HCV infection could not be deduced for many patients, which may have affected the evaluation of host immune response and outcomes of HCV infection. Additionally, to reduce the inevitable selection bias in this study, confounding factors were accounted for by matching individuals according to these factors and then adding them as covariates in the logistic regression model.
In conclusion, this study revealed that genetic variants of the NF-κB pathway genes (rs11820062 T allele) are associated with an increased risk of HCV susceptibility within a high-risk Chinese population. This finding will inform novel preventive, predictive and therapeutic ideas for HCV infection. However, further functional research on SNP rs11820062 is required.
References
Kao, J. H. et al. Urgency to treat patients with chronic hepatitis C in Asia. Journal of gastroenterology and hepatology 32, 966–974 (2017).
Cox, A. L. Global control of hepatitis C virus. Science 349, 790–791 (2015).
Hoshida, Y., Fuchs, B. C., Bardeesy, N., Baumert, T. F. & Chung, R. T. Pathogenesis and prevention of hepatitis C virus-induced hepatocellular carcinoma. Journal of hepatology 61, S79–S90 (2014).
European Association for the Study of the Liver. EASL Recommendations on Treatment of Hepatitis C 2016. Journal of hepatology 66, 153 (2017).
Omata, M. et al. APASL consensus statements and recommendations for hepatitis C prevention, epidemiology, and laboratory testing. Hepatology international 10, 681–701 (2016).
Bowie, A. & O’Neill, L. A. Oxidative stress and nuclear factor-κB activation: a reassessment of the evidence in the light of recent discoveries. Biochemical pharmacology 59, 13–23 (2000).
Wietek, C. & O’Neill, L. A. Diversity and regulation in the NF-κB system. Trends in biochemical sciences 32, 311–319 (2007).
Li, Q. & Verma, I. M. NF-[kappa] B regulation in the immune system. Nature reviews. Immunology 2, 725–734 (2002).
O’Neill, L. A. & Bowie, A. G. Sensing and signaling in antiviral innate immunity. Current Biology 20, R328–R333 (2010).
Miggin, S. M. et al. NF-κB activation by the Toll-IL-1 receptor domain protein MyD88 adapter-like is regulated by caspase-1. Proceedings of the National Academy of Sciences 104, 3372–3377 (2007).
Park, M. H. & Hong, J. T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 5, 15 (2016).
Bowie, A. G. & Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nature reviews. Immunology 8, 911–922 (2008).
O’Brown, Z. K., Van Nostrand, E. L., Higgins, J. P. & Kim, S. K. The inflammatory transcription factors NFκB, STAT1 and STAT3 drive age-associated transcriptional changes in the human kidney. PLoS genetics 11, e1005734 (2015).
Hashimoto, R. et al. Variants of the RELA gene are associated with schizophrenia and their startle responses. Neuropsychopharmacology 36, 1921–1931 (2011).
Gao, J. et al. Genetic polymorphism of NFKB1 and NFKBIA genes and liver cancer risk: a nested case–control study in Shanghai, China. BMJ open 4, e004427 (2014).
Edenberg, H. J. et al. Association of NFKB1, which encodes a subunit of the transcription factor NF-κB, with alcohol dependence. Human molecular genetics 17, 963–970 (2008).
Du, J. et al. Polymorphisms of NF-κB family genes are associated with development of multiple myeloma and treatment outcome in patients undergoing bortezomib-based regimens. Haematologica 96, 729–737 (2011).
Leung, G. et al. Cytokine candidate genes predict the development of secondary lymphedema following breast cancer surgery. Lymphatic research and biology 12, 10–22 (2014).
Byun, E. et al. Cytokine polymorphisms are associated with daytime napping in adults living with HIV. Sleep Medicine 32, 162–170 (2017).
Aouizerat, B. E. et al. Phenotypic and molecular evidence suggests that decrements in morning and evening energy are distinct but related symptoms. Journal of pain and symptom management 50, 599–614.e3 (2015).
Bhattacherjee, V. et al. Use of NS-4 peptides to identify type-specific antibody to hepatitis C virus genotypes 1, 2, 3, 4, 5 and 6. Journal of General Virology 76, 1737–1748 (1995).
Yue, M. et al. Toll-like receptor 7 variations are associated with the susceptibility to HCV infection among Chinese females. Infection, Genetics and Evolution 27, 264–270 (2014).
Yue, M. et al. Sex-specific association between X-linked Toll-like receptor 7 with the outcomes of hepatitis C virus infection. Gene 548, 244–250 (2014).
Yue, M. et al. Association of interleukin-18 gene polymorphisms with the outcomes of hepatitis C virus infection in high-risk Chinese Han population. Immunology letters 154, 54–60 (2013).
Yue, M. et al. Human leukocyte antigen class II alleles are associated with hepatitis C virus natural susceptibility in the Chinese population. International journal of molecular sciences 16, 16792–16805 (2015).
Xu, X. et al. Genetic variants in human leukocyte antigen-DP influence both hepatitis C virus persistence and hepatitis C virus F protein generation in the Chinese Han population. International journal of molecular sciences 15, 9826–9843 (2014).
Wu, M.-p et al. Genetic variations in vitamin D receptor were associated with the outcomes of hepatitis C virus infection among Chinese population. Journal of human genetics 61, 129–135 (2016).
Tang, S. et al. Association of genetic variants in estrogen receptor α with HCV infection susceptibility and viral clearance in a high-risk Chinese population. European journal of clinical microbiology & infectious diseases 33, 999–1010 (2014).
Cheng, C.-W. et al. Effects of NFKB1 and NFKBIA gene polymorphisms on hepatocellular carcinoma susceptibility and clinicopathological features. PLoS One 8, e56130 (2013).
Yu, Y. et al. The joint association of REST and NFKB1 polymorphisms on the risk of colorectal cancer. Annals of human genetics 76, 269–276 (2012).
Fakhir, F.-Z. et al. The-94Ins/DelATTG polymorphism in NFκB1 promoter modulates chronic hepatitis C and liver disease progression. Infection, Genetics and Evolution 39, 141–146 (2016).
Bowes, J. et al. Comprehensive assessment of rheumatoid arthritis susceptibility loci in a large psoriatic arthritis cohort. Annals of the rheumatic diseases 71, 1350–1354 (2012).
Daley, D. et al. Associations and interactions of genetic polymorphisms in innate immunity genes with early viral infections and susceptibility to asthma and asthma-related phenotypes. Journal of Allergy and Clinical Immunology 130, 1284–1293 (2012).
Chorev, M. & Carmel, L. The function of introns. Front genet 3, 1–15 (2012).
Damgaard, C. K. et al. A 5′ splice site enhances the recruitment of basal transcription initiation factors in vivo. Molecular cell 29, 271–278 (2008).
Seeff, L. B. Natural history of chronic hepatitis C. Hepatology 36, S35–S46 (2002).
Fish, E. N. The X-files in immunity: sex-based differences predispose immune responses. Nature reviews. Immunology 8, 737–744 (2008).
Cui, Y. & Jia, J. Update on epidemiology of hepatitis B and C in China. Journal of gastroenterology and hepatology 28, 7–10 (2013).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81502853, 81773499, 81703273, 81473029), the Natural Science Foundation of Jiangsu Province of China (No. BK20151026, BK20171054), Jiangsu Program for Young Medical Talents (QNRC2016616) and the Medical Innovation Team Project of Jiangsu Province (CXTDA2017023). We would like to thank Russell Miller for English editing assistance of the manuscript.
Author information
Authors and Affiliations
Contributions
M.Y., Y.Z. participated in the design of the study. T.T., J.W., H.Z.F. and Y.P. H. carried out the surveys and experiments. M.Y. T.T., X.S.X., P.H. performed the statistical analysis. M.Y., Y.Z., R.B.Y. and J.L. contributed materials and analytical tools. M.Y. and T.T. wrote the manuscript and tables. M.Y. prepared the figures. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, T., Wang, J., Huang, P. et al. Genetic variations in NF-κB were associated with the susceptibility to hepatitis C virus infection among Chinese high-risk population. Sci Rep 8, 104 (2018). https://doi.org/10.1038/s41598-017-18463-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18463-y
This article is cited by
-
Genetic mutations in NF-κB pathway genes were associated with the protection from hepatitis C virus infection among Chinese Han population
Scientific Reports (2019)
-
Impact of CYP2R1, CYP27A1 and CYP27B1 genetic polymorphisms controlling vitamin D metabolism on susceptibility to hepatitis C virus infection in a high-risk Chinese population
Archives of Virology (2019)
-
Genetic markers of lipid metabolism genes associated with low susceptibility to HCV infection
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
