
Abstract
Ruthenium oxide (RuO2) is the best oxygen evolution reaction (OER) electrocatalyst. Herein, we demonstrated that RuO2 can be also efficiently used as an oxygen reduction reaction (ORR) electrocatalyst, thereby serving as a bifunctional material for rechargeable Zn–air batteries. We found two forms of RuO2 (i.e. hydrous and anhydrous, respectively h-RuO2 and ah-RuO2) to show different ORR and OER electrocatalytic characteristics. Thus, h-RuO2 required large ORR overpotentials, although it completed the ORR via a 4e process. In contrast, h-RuO2 triggered the OER at lower overpotentials at the expense of showing very unstable electrocatalytic activity. To capitalize on the advantages of h-RuO2 while improving its drawbacks, we designed a unique structure (RuO2@C) where h-RuO2 nanoparticles were embedded in a carbon matrix. A double hydrophilic block copolymer-templated ruthenium precursor was transformed into RuO2 nanoparticles upon formation of the carbon matrix via annealing. The carbon matrix allowed overcoming the limitations of h-RuO2 by improving its poor conductivity and protecting the catalyst from dissolution during OER. The bifunctional RuO2@C catalyst demonstrated a very low potential gap (ΔE OER-ORR = ca. 1.0 V) at 20 mA cm−2. The Zn||RuO2@C cell showed an excellent stability (i.e. no overpotential was observed after more than 40 h).
Similar content being viewed by others


Introduction
The demand for high energy-density technologies has gradually shifted the research interests from Li-ion to metal–air batteries1,2,3. The utilization of Zn in metal–air batteries is beneficial because of its low cost, the employment of aqueous electrolytes and the safety characteristics of this metal4, 5. However, a number of challenges lie ahead (i.e. non-uniform Zn dissolution from the anodes, limited solubility of Zn ions in the electrolytes and serious overpotential during charge) before rechargeable Zn–air battery cells are developed. In this sense, the development of efficient and stable bifunctional catalysts for air electrodes is one of the most important issues for rechargeable Zn–air batteries. The efficiency and cyclability of the oxygen reduction and evolution reactions (ORR and OER) should be guaranteed2, 3.
While platinum and ruthenium oxide (RuO2) are known to be the best ORR and OER catalysts, respectively6, 7, these materials exclusively promote these reactions (i.e. the electroactivity of RuO2 towards the ORR is low as compared with that towards OER)8. From a bifunctional viewpoint, the use of ORR-active RuO2 is the best scenario because we are already sure of its superior OER activities. This study was motivated by previous studies on the supercapacitor applications of RuO2 9,10,11. Amorphous hydrous RuO2 (RuO2·xH2O with x > 0, h-RuO2) showed higher capacitances than its crystalline anhydrous counterpart (x = 0 or ah-RuO2)10, 11. This higher capacitance was potentially ascribed to a more active interaction between the space charges on the surface of h-RuO2 and the electrolyte ions, although this phenomenon does not provide a direct measure of the electroactivity. Despite its capacitance characteristics, h-RuO2 has not been proposed as an electrocatalyst yet, and studies describing the effect of the hydration on the electroactivity of this material are lacking in the literature. Therefore, the null hypothesis that partial or complete hydration of RuO2 is ineffective in improving the ORR and/or OER electroactivities is worth testing.
In case the null hypothesis is rejected, there is an additional issue. While ah-RuO2 shows high metallic conductivity (ca. 104 S cm−1) and crystallinity, h-RuO2 has low electric conductivity (ca. 1 S cm−1) and amorphous characteristics12. The poor electric conductivity of the h-RuO2 catalyst particles is likely limiting their electrocatalytic activity. Three conditions are required to achieve high electrocatalytic activities: (1) rapid charge transfer kinetics13 defined by the catalytic active sites; (2) good accessibility of the reactants to the active sites14; and (3) the presence of highly developed electric pathways for the active sites15. Carbon coating of cathode and/or anode materials has been widely used as a key strategy to improve the electric conductivity of electrodes16,17,18. In this sense, electroactive materials can be easily composited with carbon by reducing the carbon precursors under a reductive gas environment at temperatures higher than the thermal decomposition temperature of the precursors and lower than the reduction temperature of the active materials. Therefore, the main concern when fabricating oxide/carbon composites via the thermal method is to prevent oxide reduction. For example, sucrose-coated Fe2O3 was converted to carbon-coated Fe3O4 at 500 °C under argon, and a third of the iron atoms were reduced from Fe3+ to Fe2+ as a result of the thermal treatment19.
Two points are crucial in our strategy to guarantee simultaneous bifunctional ORR and OER RuO2 electroactivities: (1) the presence of partially hydrated RuO2 as a catalyst and (2) carbon coating of the catalyst particles. We increased the electric conductivity of the catalyst layers by embedding RuO2 (or more precisely h-RuO2) nanoparticles in a carbon matrix phase (RuO2@C). The RuO2@C was synthesized by annealing micelles comprising RuO2 surrounded by double hydrophilic block copolymers of poly(ethylene oxide)-block-poly(acrylic acid) (PEO-b-PAA) as a template9, 20, 21. During the annealing process, the h-RuO2 core was crystallized while the PEO-b-PAA shell was converted to a continuous carbon phase surrounding partially hydrated RuO2. Both ORR and OER electroactivities were significantly improved by incorporating h-RuO2 into the carbon phase. Zn–air cells based on RuO2@C showed the very low potential gaps between the ORR (during discharge) and OER (during charge) processes, thereby confirming the superior reversibility of this material as compared with previously reported Zn–air cells utilizing Pt/C and ah-RuO2 catalysts.
Methods
RuO2@C Synthesis
Poly(ethylene oxide-block-acrylic acid) (PEO-b-PAA; PEO5000-b-PAA6700 where the numbers indicate molecular weights of each block; Polymer Source), ruthenium (III) chloride hydrate (RuCl3·xH2O; Sigma-Aldrich) and hydrazine (N2H4; Sigma-Aldrich) and sodium hydroxide (NaOH; Junsei Chemical) were used as received. Hydrous ruthenium oxide nanoparticles templated by double hydrophilic block copolymer shell (h-RuO2@PEO-b-PAA) were synthesized as reported previously9. Briefly, 25.1 mg PEO-b-PAA (equivalent to 0.20 mmol of carboxylic acid groups) was dissolved in 50.0 mL of deionized water under vigorous stirring until the solution was completely transparent. 0.10 mL of 4.0 M NaOH (0.40 mmol, 2 equivalents to carboxylic acid groups in PAA block) and then 17.8 mg (0.10 mmol) RuCl3·xH2O were introduced to the solution. Subsequently, 0.10 mL of 10.0 M hydrazine (1.0 mmol) was added to the resulting suspensions under vigorous stirring. After a few seconds, the solution color became dark cyan. The resulting solution was dialyzed against deionized water using a dialysis membrane (MWCO 12000–14000; SpectraPore) to remove residuals. The prepared h-RuO2@PEO-b-PAA solution exhibited fairly high colloidal stability, which lasted more than one year without any precipitation. Dry powder of h-RuO2@PEO-b-PAA was obtained by using a rotary evaporator. RuO2@C was obtained by heating the dried h-RuO2@PEO-b-PAA at the rate of 10 °C min−1 to an annealing temperature and then annealing in air at 400 °C for 2 h.
Characterization
The morphology and size of RuO2@C were investigated by using transmission electron microscopy (TEM; JEOL, JEM-2100F; accelerating voltage at 200 kV with Gatan CCD camera). The functional groups of RuO2@C annealed at different temperatures were analysed by X-ray photoelectron spectroscopy (XPS; Thermo Fisher, K-alpha). The crystallography of RuO2 was investigated by high power X-ray diffractometer (XRD; Rigaku, D/MAX 2500 V/PC).
Catalyst inks
Catalyst inks were prepared by mixing 4 mg catalyst composite in a mixture of 50 μl of 0.05% Nafion solution (Sigma-Aldrich) and 450 μl of ethanol by sonication for 30 min. 6 μl of the ink was transferred onto the 4 mm-diameter glassy carbon (GC) disk electrode of Pt/GC ring/disk electrode (ALS) and then dried at ambient temperature. Our RuO2@C was compared with ah-RuO2 (agglomerates of 30~50 nm primary particles, Sigma-Aldrich) as the more anhydrous control and h-RuO2 (Alfa Aesar) as the more hydrous control. Both controls were used as received. The catalyst composites were prepared by mixing the catalysts with 20 wt. % Ketjen Black 600 as a conducting agent. Pt/C (20 wt % loading of Pt on carbon black, Alfa Aesar) was also used as the catalyst composite for comparison.
Electrochemistry
The electrocatalytic activity and stability of the catalysts were measured by using rotating ring disk electrode (RRDE; ALS) and potentiostat (Bio-Logic, VMP3). The catalyst-coated RRDE as a working electrode was immersed in a glass cell containing 0.1 M KOH. Hg/HgO (XR400, Radiometer Analytical) and Pt wire were used as reference and counter electrodes, respectively. All the potentials were reported in VRHE (V versus RHE; RHE = reversible hydrogen electrode) in this work even if the potential values were read from potentiostats in VHg/HgO: VRHE = VHg/HgO + 0.93 V in 0.1 M KOH (aq). The ORR polarization voltammograms at 10 mV s−1 were obtained in the O2-saturated electrolyte between +0.2 VHg/HgO and −0.8 VHg/HgO at various rotation speed (400, 900, 1600 or 2500 rpm). At the same time, +0.4 VHg/HgO was applied to the ring electrode of RRDE to detect peroxide formed from the disk electrode by oxidizing the peroxide completely. Pure faradaic currents were reported in this work by subtracting background capacitive currents obtained in N2-saturated electrolyte from the overall reduction currents from disk electrodes. The collection efficiency (N) was estimated at 0.42 (the same as the theoretical value) in 10 mM potassium ferricyanide (K3Fe(CN)6) in 0.1 M KOH electrolyte under Ar atmosphere. The number of electrons transfer (n) of ORR was calculated by using: n = 4 |I d |/(|I d | + I r /N) where \({I}_{d}\) and \({I}_{r}\) are the disk and ring currents, respectively. The OER polarization voltammograms at 10 mV s−1 were obtained in the N2-saturated electrolyte between +0.35 VHg/HgO and +0.9 VHg/HgO at 1600 rpm. Mass-transfer-corrected currents (iK) were used for Tafel plots: iK = i iL/(iL − i) with iL = limiting current22.
Zn–air battery
Zn–air cells were constructed in a previously reported configuration23 based on: Zn plate (Alfa Aesar) as an anode, carbon on nickel mesh (MEET, Korea) as a gas diffusion layer (GDL) of an cathode, microporous membrane (Celgard 3501) as a separator and Ni mesh as current collectors with 6 M KOH aqueous electrolyte. 100 μl catalyst ink was loaded on a carbon GDL electrode (geometric area = 2.834 cm2) and the catalyst-loaded electrode was dried at 80 °C for >1 h. Zn-air cells were galvanostatically discharged and charged at various currents by a potentiostat (Bio-Logic, VMP3).
Results and Discussion
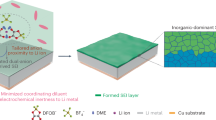
The ruthenium precursors templated by PEO-b-PAA were converted to RuO2 nanoparticles (4–5 nm in size) embedded in a continuous carbon matrix phase forming RuO2@C nanoclusters (Figs. 1 and S1). The electrostatic interaction between the ruthenium precursor cations and the anionic PAA blocks in the double hydrophilic block copolymer PEO-b-PAA afforded RuO2 spherical nanoparticles. The additional hydrophilic PEO blocks decorating the surface of the nanoparticles and exposed to solution stabilized the nanoparticles in solution, preventing them from aggregation.

RuO2@C nanoclusters. (a) Schematic. (b and c) TEM images.
Completely anhydrous form of RuO2 (ah-RuO2) has high crystallinity while its completely hydrous form (h-RuO2) is amorphous with no characteristic XRD patterns (Fig. S2). Partially hydrous RuO2’s (x-RuO2) show the in-between patterns depending on their hydration degree. RuO2@C showed a well-defined X-ray diffraction (XRD) RuO2 pattern at annealing temperatures higher than 350 °C. The amorphous characteristics of RuO2@PEO-b-PAA decreased with increasing temperature as its hydrated RuO2 was dehydrated with x decreasing in RuO2·xH2O. After annealing at 400 °C (i.e. the temperature used for preparing RuO2@C), the apparent crystallographic peaks of RuO2 were identified. The crystallite size calculated by Scherrer equation increased with the annealing temperature (i.e. 12 nm at 350 °C; <17 nm at 400 °C (RuO2@C); and <20 nm at 450 °C). The crystallite size of x-RuO2, obtained upon heating h-RuO2 at 400 °C, was 23 nm, which was larger than that of RuO2@C obtained by heating RuO2@PEO-b-PAA at 400 °C. ah-RuO2 showed the largest crystallite size among the materials studied (27 nm).
X-ray photoelectron spectra (XPS) measurements confirmed that the RuO2 crystallites in RuO2@C as well as the amorphous RuO2 phase in RuO2@PEO-b-PAA were both hydrated (Fig. S3a–d). Significant amounts of surface-adsorbed H2O and OH− species were identified in the O1s spectra of both samples. Thus, surface water was dominant in RuO2@PEO-b-PAA, while OH− prevailed in RuO2@C. In addition to lattice Ru(IV), Ru(III) species originating from hydrous Ru(III)–OH was found in the Ru3d spectra of both samples, although the relative amount of Ru(III) to Ru(IV) significantly decreased after thermal annealing24. When compared with h-RuO2 and ah-RuO2, the material prepared herein showed intermediate properties (Fig. S3e and f). Thus, the main peak in the O1s spectra of RuO2@C was placed between the surface OH−-characteristic peak of h-RuO2 and the lattice O2−-characteristic peak of ah-RuO2. The OH− to O2− or Ru(III) to Ru(IV) area ratios of RuO2@C (1.1 or 1.8, respectively) were between those of ah-RuO2 (0.4 or 1.3, respectively) and h-RuO2 (2.4 or 2.6, respectively). The relative hydration degree (x) of RuO2@C was estimated to be 0.27 (i.e. a quarter hydrous) by interpolating the peak ratio data as a measure of the hydration and assuming x values of 0.0 and 1.0 respectively for ah-RuO2 and h-RuO2 (Fig. 2). The x values of RuO2@PEO-b-PAA and x-RuO2 were 0.81 (i.e. highly hydrated) and 0.08 (nearly anhydrous), respectively. Therefore, we can consider RuO2@C to be formed by a mixture of hydrous and anhydrous RuO2 phases25,26,27. The anhydrous characteristics were dominant in the bulk properties (e.g., crystallography), whereas the hydrous characteristics were relevant when considering the surface properties (e.g., XPS spectra).

Hydration degree (x). The x values of ah-RuO2 and h-RuO2 were assumed to be 0.0 and 1.0, respectively. The OH− to O2− or Ru3+ to Ru4+ peak area ratios were used as a measure of the hydration degree. The x values of other RuO2 samples were estimated from the ratios obtained by interpolating two pre-fixed points of the XPS peak ratio versus x.
When used as an electrocatalyst, the structure of RuO2@C is expected to be beneficial owing to several reasons. First, the nanosized catalyst provided a large electrocatalytic surface area per mass. This high surface area was achieved by using the PEO-b-PAA template, which restricted the growth of RuO2 primary particles during the synthesis. Second, the electrons effectively reached the catalyst surface through the continuous carbon phase of the RuO2@C nanoclusters. Third, non-carbon PEO-b-PAA residues generated voids and pore space after carbonization, thereby allowing reactants to be readily transferred through the porous carbon matrix. Fourth, the carbon matrix surrounding RuO2 possibly prevented the catalyst from dissolution.
While ORR electroactivity of RuO2 has been rarely reported, its OER and hydrogen evolution reaction (HER) activities have been widely investigated28. Poor electroactivities (i.e. low ORR currents and high overpotentials) have been reported for RuO2 (Table S1, ESI†)3, 29,30,31. As an example, RuO2 showed a potential at half of the limiting current (i.e. E 2/L at i L/2) of +0.56 VRHE at −1.2 mA cm−2 (cf. +0.9 VRHE at −3 mA cm−2 for Pt/C)29. This high overpotential of RuO2 was indicative of very sluggish ORR kinetics. More seriously, the electron transfer number (n) was estimated to be ca. 2 (4 is the preferred value for n; discussed below). Interestingly, significantly higher ORR electroactivities were obtained herein even for a commercially available ah-RuO2 control sample (Fig. 3). The RuO2@C nanoclusters prepared herein, mixed at 20 wt% with carbon black (CB) as a conducting agent (RuO2@C in Fig. 3 and RuO2@C + CB in Fig. S4), showed the most rapid kinetics (E 2/L = +0.7 VRHE at −3 mA cm−2) among the RuO2 ORR catalysts tested. When CB was not used, the onset potential was significantly shifted to negative potentials (RuO2@C in Fig. S4).

ORR in O2-saturated 0.1 M KOH (aq). (a) ORR polarization curves at 1,600 rpm and 10 mV s−1. (b) Electron transfer number (n). (c) Tafel plots. Mass transfer-corrected currents (i K) are used. Tafel slopes (b) are indicated in mV dec−1. (d) Chronoamperometric stability of ORR at +0.4 VRHE. The initial currents are indicated next to the names of the catalysts.
When compared with h-RuO2 and ah-RuO2, RuO2@C combined the advantages of both forms of RuO2: the RuO2@C was anhydrous-like in terms of smaller ORR onset overpotential; and hydrous-like due to its higher number of electron transfer. In the conductive environments realized by 20 wt. % CB, the presence of ah-RuO2 was beneficial in terms of the onset potential for ORR polarization (Fig. 3a). On the other hand, h-RuO2 was beneficial in terms of the number of electron transferred (n), especially at low overpotentials (Fig. 3b). Annealing at 400 °C (x-RuO2 in Fig. 3) allowed reducing the high overpotential of h-RuO2 while shifting the onset potential to that of ah-RuO2. However, the annealing promoted dehydration, decreasing the n values in x-RuO2 and reaching those of ah-RuO2. RuO2@C showed lower overpotentials as compared with the two extreme control samples (i.e. h-RuO2 and ah-RuO2) with n values comparable to those of h-RuO2. The hydration degree (x in RuO2·xH2O) of RuO2@C was higher than that of x-RuO2 (as revealed via XRD and XPS data in Figs. S2 and S3, respectively) such that high n values (>3.9) were obtained. The n values measured from disk and ring RRDE currents coincided with the values estimated from the Koutecky–Levich plots (Fig. S5). The 4e ORR showed by RuO2@C revealed complete reduction of oxygen without producing hydrogen peroxide (i.e. the 2e ORR intermediate product). RuO2@C showed a Tafel slope at low overpotentials of 69 mV dec−1, close to that of Pt/C (Fig. 3c). Therefore, we concluded that the carbon matrix compensated the poor conductivity of h-RuO2, preventing the Ohmic potential shift (i.e. low overpotential as in the case of conductive ah-RuO2) and enabling efficient utilization of the active mass (i.e. high n values as in the case of h-RuO2).
The chronoamperometric ORR stability of RuO2 was also improved by the carbon shell in RuO2@C (Fig. 3d). The currents of ah-RuO2 and Pt/C at +0.4 VRHE significantly decreased (to 80% of the initial values) after 30 h. In contrast, RuO2@C showed excellent stability without current decay. Although Pt/C is the best ORR catalyst from the kinetic standpoint, this material is well known to suffer from instability as a result of Pt aggregation via surface diffusion and dissolution/re-precipitation processes.
The OER electroactivities of RuO2@C were investigated with the full knowledge that RuO2 is one of the best OER catalysts32. RuO2@C, regardless of CB presence, showed remarkably higher current densities and clearly reduced overpotentials as compared to its noncarbon-matrix counterpart (ah-RuO2; Figs. 4a and S6; current at 1.8 VRHE = 54 mA cm−2 (RuO2@C with CB, >31 mA cm−2 (RuO2@C without CB), >13 mA cm−2 (ah-RuO2 with and without CB) and >5.4 mA cm−2 (Pt/C); potential at 10 mA cm−2 (E 10) = 1.52 VRHE (RuO2@C with CB), <1.59 VRHE (RuO2@C without CB), <1.75 VRHE (RuO2 with and without CB) and <1.87 VRHE (Pt/C)). To the best of our knowledge, the RuO2@C material prepared herein showed better OER current and onset potential values than any previously reported RuO2 catalyst (Table S1). Note that all polarization data obtained herein were not IR-compensated unless specified. Since IR compensation correction seriously affected the OER polarization (not the ORR), the percentage of compensation (f) should be carefully selected (Fig. S7): R c = f R u, where R c is the resistance for correction and R u is the uncompensated resistance between the working and reference electrodes. The E 10 of RuO2@C significantly decreased from 1.55 VRHE at f = 0% to 1.48 VRHE at f = 85% and 1.47 VRHE at f = 100%. The nanosized particles of the catalyst well connected to electric and ionic pathways would be partly responsible for the improved OER electroactivity of RuO2@C. The hydrated nature of RuO2 can also account for the improved results. Thus, the OER onset potentials decreased with increasing hydrous character of RuO2 in the catalysts (Fig. 4a, inset) as follows: ah-RuO2 < x-RuO2 < RuO2@C < h-RuO2.

OER in N2-saturated 0.1 M KOH (aq). (a) OER polarization curves at 1,600 rpm for the first potential sweep cycles at 10 mV s−1. (b) OER current retention at 1.83 VRHE during repeated cyclic voltammograms. (c) Chronoamperometric stability of OER at 1.73 VRHE for 60 min in 0.1 M KOH. The initial currents are indicated next to the names of the catalysts. (d) Chronopotentiometric stability of OER at 5 mA cm−2 for 40 h.
Despite its good onset potential characteristics, h-RuO2 showed important stability issues (Fig. 4b). This OER instability has been previously ascribed to RuO2 dissolution issues during OER, especially in the case of h-RuO2 7, 33. A broad anodic peak was found at 1.6 VRHE for h-RuO2 during the initial anodic scan of the potential (Fig. 4a). The stability of h-RuO2 was improved by the carbon matrix in RuO2@C. Thus, the specific current density of RuO2@C reached 54 mA cm−2 at 1.83 VRHE and slightly decreased (to 93% of the initial value) after repeating the potential sweep between 1.3 VRHE and 1.83 VRHE 100 times at 10 mV s−1 (Fig. 4b). The carbon matrix of RuO2@C was believed to protect its partially hydrated RuO2 from dissolution. RuO2@C also showed high stability under different chronoamperometric conditions (1.73 VRHE for 60 min, 0.1 M KOH) (Fig. 4c). The noise-like fluctuation in the current during OER was produced by O2 bubbles generated on the electrode surface. The RuO2 particle size in the carbon matrix of RuO2@C did not significantly change after the chronoamperometric test, in contrast with Pt/C that showed particle agglomeration under identical conditions (Fig. S9).
Carbon corrosion is one of the most serious issues of air electrodes in Zn–air batteries during rechargeable operations. The loss of solid carbon via corrosion leads to catalyst loss and electrode leakage, thereby resulting in performance decay34. Carbon corrosion processes can be described as follows35, 36:
When considering the reduction potentials, carbon corrosion is thermodynamically inevitable in the OER potential range. The only way of mitigating carbon corrosion is the reduction of the OER overpotential. The overpotential advantage of RuO2@C was clearly reflected in the OER stability (i.e. the potential remained stable below 1.6 VRHE at 5 mA cm−2 over 40 h, Fig. 4d). In contrast, the rest of RuO2 samples significantly increased their overpotential values with time at the same current density.
Reversible operation of Zn–air batteries (Fig. S10a) was achieved by using the bifunctional RuO2@C material (Fig. 5). RuO2@C, Pt/C or a carbon electrode were used as air electrodes, while zinc was selected as the metal electrode. The ORR and OER kinetics were investigated from the discharge and charge rate capabilities under fixed slow charge (Fig. 5a) and discharge (Fig. 5b) conditions, respectively. Pt/C showed the lowest ORR overpotential, although it developed a severe OER overpotential (even larger than the non-catalytic carbon electrode) especially at high current densities (>200 mA). RuO2@C showed a very good ORR polarization behaviour despite presenting larger overpotentials than Pt/C. Interestingly, the overpotential difference between RuO2@C and Pt/C in the Zn–air battery cells (ca. 0.1 V) was lower than the half wave potential difference (EL/2) between them in the linear sweep voltammograms (ca. 0.2 V, Fig. 3a). The RuO2@C-based battery was successfully charged at a lower potential as compared to Pt/C- and carbon electrode-based batteries, especially at high currents (Fig. 5b). Stable potential profiles were obtained in the presence of RuO2@C, up to fast charges of 200 mA. The potential difference between charge and discharge (ΔE OER-ORR) of RuO2@C (1.1 V) was significantly enhanced as compared to those of Pt/C and carbon air electrodes (1.6–1.7 V, Fig. S10b) at 100 mA (1.65 V for RuO2@C versus ca. 5 V for the rest of electrodes at 200 mA). The lower ΔE OER-ORR value of RuO2@C was indicative of the higher ORR–OER reversibility of this material.

Zn–air batteries. 20 wt% carbon black used for RuO2@C. Pt loading of Pt/C is 20 wt%. (a) Discharge rate capability at 20 mA cm−2 charge. ORR proceeds on air electrodes. The currents used for discharge are indicated in mA (geometric electrode area = 2.8 cm2). (b) Charge rate capability at 20 mA cm−2 discharge. OER proceeds on air electrodes. The currents used for charge are indicated in mA (geometric electrode area = 2.8 cm2). (c and d) Galvanostatic charge/discharge cycles at 20 mA cm−2.
To further confirm the rechargeability of the RuO2@C-based Zn–air batteries, the cells were repeatedly discharged and charged in the galvanostatic manner at 20 mA cm−2 following a 20-min cycle period for 50 h (Fig. 5c and d). The potentials of RuO2@C remained stable at 1.04 V during discharge and at 2.11 V during charge for over 40 h or 120 cycles (Fig. S10c). The observed decrease in the ORR potential during discharge after 40 h was not caused by catalyst deterioration. Instead, this issue was caused by an electrolyte leakage through the gas diffusion layer of the air electrode, thereby hindering the oxygen supply. No problematic deterioration was found in the Zn–RuO2@C cell during OER operation. Unlike RuO2@C, Pt/C- or carbon-based Zn–air cells showed loss of performance during OER as a result of catalyst failure. Serious OER overpotentials were developed from the initial charge. When compared with the cell operation data previously reported, the RuO2@C-based cell demonstrated the most reversible behaviour among the published Zn–air cells23, 37,38,39,40. This cell showed a ΔE OER-ORR value of 1.0 V, which is the lowest value among the air batteries reported (Fig. S10d and Table S2). The potential gap decreased to 0.85 V when operating the cell under a 100% oxygen stationary atmosphere instead of air. The reversibility of our Zn–oxygen cell was among the best reported so far, although lower ΔE OER-ORR values have been reported in the literature (Fig. S11 and Table S2)41,42,43,44,45, although in these cells oxygen was forcibly introduced through the cells or electrolytes.
Conclusions
The advantages of h-RuO2 and ah-RuO2 were combined by embedding partially hydrated RuO2 nanoparticles in a carbon matrix (RuO2@C). RuO2@C showed an x value of 0.27 (i.e. quarter hydrous). RuO2@C demonstrated both anhydrous- (i.e. lower ORR onset overpotential and superior OER stability) and hydrous-like (i.e. higher number of electron transfer during ORR and lower OER onset overpotential behaviour. The superior characteristics of RuO2@C were demonstrated by operating Zn–air battery cells. These cells showed the smallest overpotentials reported so far, thereby guarantying the efficient operation of rechargeable Zn–air batteries.
References
Lee, J. S. et al. Metal-air batteries with high energy density: Li-air versus Zn-air. Adv. Energy Mater. 1, 34–50 (2011).
Rahman, M. A., Wang, X. & Wen, C. High Energy Density Metal-Air Batteries: A Review. J. Electrochem. Soc. 160, A1759–A1771 (2013).
Zhang, J., Zhao, Z., Xia, Z. & Dai, L. A metal-free bifunctional electrocatalyst for oxygen reduction and oxygen evolution reactions. Nat. Nanotechnol. 10, 444–452 (2015).
Li, Y. & Dai, H. Recent advances in zinc-air batteries. Chem. Soc. Rev. 43, 5257–5275 (2014).
Lee, J.-S., Lee, T., Song, H.-K., Cho, J. & Kim, B.-S. Ionic liquid modified graphene nanosheets anchoring manganese oxide nanoparticles as efficient electrocatalysts for Zn-air batteries. Energy Environ. Sci. 4, 4148–4154 (2011).
Nie, Y., Li, L. & Wei, Z. Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 44, 2168–2201 (2015).
Cherevko, S. et al. Oxygen and hydrogen evolution reactions on Ru, RuO2, Ir, and IrO2 thin film electrodes in acidic and alkaline electrolytes: A comparative study on activity and stability. Catal. Today 262, 170–180 (2015).
Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Chem Soc Rev hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 44, 2060–2086 (2015).
Seo, E., Lee, T., Lee, K. T., Song, H.-K. & Kim, B.-S. Versatile double hydrophilic block copolymer: dual role as synthetic nanoreactor and ionic and electronic conduction layer for ruthenium oxide nanoparticle supercapacitors. J. Mater. Chem. 22, 11598–11598 (2012).
Kim, I.-H. & Kim, K.-B. Electrochemical Characterization of Hydrous Ruthenium Oxide Thin-Film Electrodes for Electrochemical Capacitor Applications. J. Electrochem. Soc. 153, A383–A383 (2006).
Zheng, J. P. Hydrous Ruthenium Oxide as an Electrode Material for Electrochemical Capacitors. J. Electrochem. Soc. 142, 2699–2699 (1995).
Adeyemo, A., Hunter, G. & Dutta, P. K. Interaction of CO with hydrous ruthenium oxide and development of a chemoresistive ambient CO sensor. Sens. Actuators, B 152, 307–315 (2011).
Jahan, M., Liu, Z. & Loh, K. P. A Graphene Oxide and Copper-Centered Metal Organic Framework Composite as a Tri-Functional Catalyst for HER, OER, and ORR. Adv. Funct. Mater. 23, 5363–5372 (2013).
Wu, G. et al. Synthesis–structure–performance correlation for polyaniline–Me–C non-precious metal cathode catalysts for oxygen reduction in fuel cells. J. Mater. Chem. 21, 11392 (2011).
Chung, D. Y. et al. Highly Durable and Active PtFe Nanocatalyst for Electrochemical Oxygen Reduction Reaction. Journal of the American Chemical Society 137, 15478–15485 (2015).
Lee, S., Cho, Y., Song, H.-K., Lee, K. T. & Cho, J. Carbon-Coated Single-Crystal LiMn2O4 Nanoparticle Clusters as Cathode Material for High-Energy and High-Power Lithium-Ion Batteries. Angew. Chem. Int. Ed. 51, 8748–8752 (2012).
Park, H.-S., Kim, T.-H., Lee, M.-H. & Song, H.-K. Catalytic carbonization of an uncarbonizable precursor by transition metals in olivine cathode materials of lithium ion batteries. J. Mater. Chem. 22, 20305 (2012).
Kim, T.-H., Park, H.-S., Lee, M.-H., Lee, S.-Y. & Song, H.-K. Restricted growth of LiMnPO4 nanoparticles evolved from a precursor seed. J. Power Sources 210, 1–6 (2012).
Kim, T.-H. & Song, H.-K. Hollow versus nonhollow: The electrochemical preference in a case study of the conversion reaction of Fe3O4. Electrochim. Acta 105, 47–52 (2013).
Leong, W. L. et al. Non-volatile organic memory applications enabled by in situ synthesis of gold nanoparticles in a self-assembled block copolymer. Adv. Mater. 20, 2325–2331 (2008).
Sun, Y., Wu, J., Tian, J., Jin, C. & Yang, R. Sulfur-doped carbon spheres as efficient metal-free electrocatalysts for oxygen reduction reaction. Electrochim. Acta 178, 806–812 (2015).
Park, S. et al. Electrochemical Reduction of Oxygen at Platinum Electrodes in KOH Solutions‐Temperature. Journal of The Electrochemical Society 133, 1641–1649 (1986).
Chen, Z. et al. Highly Active and Durable Core–Corona Structured Bifunctional Catalyst for Rechargeable Metal–Air Battery Application. Nano Lett. 12, 1946–1952 (2012).
Zhang, J. et al. Template Synthesis of Tubular Ruthenium Oxides for Supercapacitor Applications. J. Phys. Chem. C 114, 13608–13613 (2010).
Dmowski, W., Egami, T., Swider-Lyons, K. E., Love, C. T. & Rolison, D. R. Local Atomic Structure and Conduction Mechanism of Nanocrystalline Hydrous RuO2 from X-ray Scattering. J. Phys. Chem. B 106, 12677–12683 (2002).
Sugimoto, W., Iwata, H., Yokoshima, K., Murakami, Y. & Takasu, Y. Proton and electron conductivity in hydrous ruthenium oxides evaluated by electrochemical impedance spectroscopy: The origin of large capacitance. J. Phys. Chem. B 109, 7330–7338 (2005).
McKeown, D. A. et al. Structure of Hydrous Ruthenium Oxides: Implications for Charge Storage. J. Phys. Chem. B 103, 4825–4832 (1999).
Over, H. Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: From fundamental to applied research. Chem. Rev. 112, 3356–3426 (2012).
Sunarso, J. et al. Bi-Functional Water/Oxygen Electrocatalyst Based on PdO-RuO2 Composites. J. Electrochem. Soc. 160, H74–H79 (2012).
Masa, J. et al. MnxOy/NC and CoxOy/NC nanoparticles embedded in a nitrogen-doped carbon matrix for high-performance bifunctional oxygen electrodes. Angew. Chem. Int. Ed. 53, 8508–8512 (2014).
Jiang, H. et al. Iron Carbide Nanoparticles Encapsulated in Mesoporous Fe-N-Doped Graphene-Like Carbon Hybrids as Efficient Bifunctional Oxygen Electrocatalysts. ACS Appl. Mater. Interfaces 7, 21511–21520 (2015).
Man, I. C. et al. Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces. ChemCatChem 3, 1159–1165 (2011).
Kötz, R. XPS Studies of Oxygen Evolution on Ru and RuO2 Anodes. J. Electrochem. Soc. 130, 825–825 (1983).
Pei, P., Wang, K. & Ma, Z. Technologies for extending zinc-air battery’s cyclelife: A review. Appl. Energy 128, 315–324 (2014).
Maass, S., Finsterwalder, F., Frank, G., Hartmann, R. & Merten, C. Carbon support oxidation in PEM fuel cell cathodes. J. Power Sources 176, 444–451 (2008).
Gallagher, K. G., Darling, R. M. & Fuller, T. F. In Handbook of Fuel Cells (eds Vielstich, W., Gasteiger, H. A., Lamm, A. & Yokokawa, H.) (John Wiley & Sons, 2010).
Lee, D. U. et al. Highly Active and Durable Nanocrystal-Decorated Bifunctional Electrocatalyst for Rechargeable Zinc-Air Batteries. Chemsuschem 8, 3129–3138 (2015).
Han, L.-N. et al. Ultra-durable two-electrode Zn-air secondary batteries based on bifunctional titania nanocatalysts: a Co2+ dopant boosts the electrochemical activity. J. Mater. Chem. A 4, 7841–7847 (2016).
Du, G. et al. Co3O4 nanoparticle-modified MnO2 nanotube bifunctional oxygen cathode catalysts for rechargeable zinc-air batteries. Nanoscale 5, 4657–4661 (2013).
Chen, Z. et al. Manganese dioxide nanotube and nitrogen-doped carbon nanotube based composite bifunctional catalyst for rechargeable zinc-air battery. Electrochim. Acta 69, 295–300 (2012).
Wang, Z. et al. Co(II)1–xCo(0)x/3Mn(III)2x/3S Nanoparticles Supported on B/N-Codoped Mesoporous Nanocarbon as a Bifunctional Electrocatalyst of Oxygen Reduction/Evolution for High-Performance Zinc-Air Batteries. ACS Appl. Mater. Interfaces 8, 13348–13359 (2016).
Song, J. et al. Optimization of cobalt/nitrogen embedded carbon nanotubes as an efficient bifunctional oxygen electrode for rechargeable zinc-air batteries. J. Mater. Chem. A 4, 4864–4870 (2016).
Prabu, M., Ramakrishnan, P. & Shanmugam, S. CoMn2O4 nanoparticles anchored on nitrogen-doped graphene nanosheets as bifunctional electrocatalyst for rechargeable zinc–air battery. Electrochem. Commun. 41, 59–63 (2014).
Prabu, M., Ketpang, K. & Shanmugam, S. Hierarchical nanostructured NiCo2O4 as an efficient bifunctional non-precious metal catalyst for rechargeable zinc-air batteries. Nanoscale 6, 3173–3181 (2014).
Li, Y. et al. Advanced zinc-air batteries based on high-performance hybrid electrocatalysts. Nat. Commun. 4, 1805 (2013).
Acknowledgements
This work was supported by MOTIE (Materials & components (KEIT): 10062092) and NRF (2016H1A2A1909427, 2015R1A2A2A04003160), Korea.
Author information
Authors and Affiliations
Contributions
H.-S.P. and H.-K.S. conceived the idea and designed experiments. H.-S.P., J.Y. and Y.L. conducted electrochemical characterization and analysis. E.S. and B.-S.K. performed material preparation. H.-K.S. conducted data analysis, planned the project and wrote this paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, HS., Seo, E., Yang, J. et al. Bifunctional hydrous RuO2 nanocluster electrocatalyst embedded in carbon matrix for efficient and durable operation of rechargeable zinc–air batteries. Sci Rep 7, 7150 (2017). https://doi.org/10.1038/s41598-017-07259-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07259-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
