
Abstract
Phenolic acids and tanshinones are two major bioactive components in Salvia miltiorrhiza Bunge. A novel endogenous R2R3-MYB transcription factor, SmMYB36, was identified in this research. This transcript factor can simultaneously influence the content of two types of components in SmMYB36 overexpression hairy roots. SmMYB36 was mainly localized in the nucleus of onion epidermis and it has transactivation activity. The overexpression of SmMYB36 promoted tanshinone accumulation but inhibited phenolic acid and flavonoid biosynthesis in Salvia miltiorrhiza hairy roots. The altered metabolite content was due to changed metabolic flow which was regulated by transcript expression of metabolic pathway genes. The gene transcription levels of the phenylpropanoid general pathway, tyrosine derived pathway, methylerythritol phosphate pathway and downstream tanshinone biosynthetic pathway changed significantly due to the overexpression of SmMYB36. The wide distribution of MYB binding elements (MBS, MRE, MBSI and MBSII) and electrophoretic mobility shift assay results indicated that SmMYB36 may be an effective tool to regulate metabolic flux shifts.
Similar content being viewed by others

Introduction
In the medical model plant Salvia miltiorrhiza Bunge, hydrophilic phenolic acids (rosmarinic acid and salvianolic acid B)1,2,3 and lipophilic tanshinones (dihydrotanshinone I, cryptotanshinone, tanshinone I and tanshinone II A)4,5,6 are major bioactive components for treating cerebrovascular diseases in clinical studies7. However, the supply of S. miltiorrhiza cannot satisfy the production and application demand due to its long growth cycle and degradation of quality. In recent years, much more attention has been paid to the illustration of metabolic pathways and biosynthetic regulation of secondary metabolites using metabolic engineering or fermentation engineering.
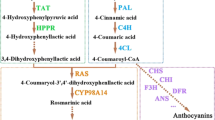
The biosynthetic pathway of phenolic acids is well illuminated in S. miltiorrhiza, which contains a phenylalanine-derived pathway and tyrosine-derived pathway8,9,10,11,12,13,14,15 (Fig. 1). L-phenylalanine is converted to 4-coumaroyl-CoA through phenylpropanoid-derived pathway8,9,10, 13. The 4-hydroxyphenyllactic acid (4-HPLA) is synthesized from L-tyrosine through a tyrosine-derived pathway8,9,10, 13. The 4-coumaroyl-CoA and 4-HPLA are combined to generate rosmarinic acid by rosmarinic acid synthase (RAS) and cytochrome P450-dependent monooxygenase (CYP)9, 10. Salvianolic acid B is thought to be a derivative of rosmarinic acid, but more research is needed to clarify the specific process8, 10. The tanshinone biosynthetic pathway covers the cytoplasmic mevalonate (MVA) pathway and plastidial methylerythritol phosphate (MEP) pathway13, 15,16,17,18 (Fig. 1). Isopentenyl diphosphate (IPP) is a common intermediate of both pathways. Next, IPP is transformed into diterpenoids as catalysed by geranylgeranyl diphosphate synthase (GGPPS), copalyl diphosphate synthase (CPS), kaurene synthase-like (KSL), CYP and other unknown enzymes15, 19, 20. According to the stated pathway, the content of the aimed metabolites was increased through key enzyme gene overexpression of the biosynthetic pathways21,22,23 and gene expression suppression of competitive pathways23, 24 in S. miltiorrhiza hairy roots or plants.

The model for tanshinone and phenolic acid biosynthesis regulation by SmMYB36.
MYB transcription factors are widespread throughout the plant world, regulating development, primary and secondary metabolism and abiotic and biotic stress25, 26. Zhang et al. found that AtMYB12 could regulate primary metabolism to provide an increasing supply of carbon, energy and reducing power for secondary metabolism in tomato fruits, leading to substantial accumulation of novel phenylpropanoids27. In AtMYB12 overexpression tobacco, the enhancement of aromatic amino acids provided more supply for the biosynthesis of phenylpropanoids28.
R2R3-MYB factors are the largest family of MYB factors, with 125 members in Arabidopsis thaliana 29 and 110 members in S. miltiorrhiza 30. Based on the conserved amino acid sequence motifs, R2R3-MYB factors are divided into 22 subgroups in A. thaliana 25, 29 and 37 subgroups in S. miltiorrhiza and A. thaliana 30. Li et al. found that some R2R3-MYBs in the same subgroup showed similar functions to metabolic pathways, while others were species-specialized transcription factors30. The results suggested that the R2R3-MYBs of subgroup 4 (PtMYB14) and subgroup 5 (VvMYB5b) were likely terpenoid biosynthesis regulators and the R2R3-MYBs of subgroup 3, 4, 5, 6, 7, 13 and 21 were potential regulators of the phenylpropanoid-derived pathway30. The overexpression of PtMYB14 and VvMYB5b influenced the accumulation of terpenoids and phenylpropanoids31, 32, which indicated that members of two above subgroups may regulate both terpenoid and phenylpropanoid biosynthetic pathways. SmMYB36 was grouped with those members by phylogenetic analysis (Supplementary Fig. S1) and may regulate the accumulation of two major secondary metabolites in S. miltiorrhiza. Liu et al. summarized the regulating effects of MYB proteins towards plant phenylpropanoid metabolism, combining structural analysis with functional analysis26. For example, AtMYB123 (TT2)33, HvMYB1034, DkMYB235, LjTT2c36, PpMYBPA137, OgMYB139, PtMYB13440 and FaMYB1141 belong to subgroup 5 according to their structures, and all function as activators to promote proanthocyanidin accumulation26. AtMYB75 (also called AtPAP1)11, 42, AtMYB90 (also called AtPAP2)43, PURPLE44, AtMYB11343, GMYB1045, VIMYBA1-346, GmMYB1047, CsRUBY48, PcMYB1049, PyMYB1050, AN251, 52, FaMYB1053, NtAN254 and AtMYB11443, belonging to subgroup 6, could act as activators to regulate anthocyanidin biosynthesis26, 30. Therefore, it is possible to analyse the structure by discussing the function of MYB. It is thought that R2R3-MYB could function individually or cooperate with basic helix-loop-helix (bHLH) and WD-repeat (WDR) transcription factors in regulating metabolite synthesis. For example, the overexpression of either ZmC1 or ZmC1/R promoted tanshinone accumulation in S. miltiorrhiza hairy roots13. The MBW complex (TT2-TT8-TTG1) may cooperatively regulate the production of proanthocyanins55, 56 and flavonoids57, but it is uncertain whether the altered expression of transformed transcription factors led to altered expression of other transcription factors in transgenic lines, which should be studied further.
MYB factors are also applied to manipulate the metabolic process in S. miltiorrhiza due to their transcriptional activation or repression activity on genes of secondary metabolic pathways. The heterologous expression of AtMYB75 (AtPAP1) and snapdragon Rosea1 in S. miltiorrhiza leads to the up-regulation of the expression level of core phenylpropanoid pathway genes and enhanced content of rosmarinic acid and salvianolic acid B58, 59. The overexpression of SmPAP1 promotes the accumulation of rosmarinic acid, salvianolic acid B, total phenolics and total flavonoids in transgenic S. miltiorrhiza Bge.f.alba roots60. Zhang et al. found that SmMYB39 plays a repressor role in gathering rosmarinic acid and salvianolic acid B by inhibiting the gene transcripts of phenolic acid biosynthetic pathway in S. miltiorrhiza 8.
Few studies have been performed on tanshinone metabolic regulation compared to phenolic acid synthetic modulation. The heterogeneous overexpression of VvMYB5b caused an enhancement in carotene and decrease in flavonol and caffeic acid in tomato32. The accumulation of terpene and anthocyanin was stimulated by the heterogeneous overexpression of PtMYB14 in spruce31. The content of phenylpropanoid-derived compounds and terpenoid compounds were enhanced by the overexpression of AtPAP1 in rose flowers61. The heterogeneous overexpression of ZmC1 or ZmC1/R in S. miltiorrhiza hairy roots could simultaneously mediate the increase in tanshinones and the decrease in phenolic acids by changing the transcript levels of pathway genes in S. miltiorrhiza hairy roots13, which may result from the upward transcript levels of most genes in the MEP pathway and the downstream tanshinone biosynthetic pathway. Heterogeneous and the endogenous transcription factors might exhibit different functions due to different genetic backgrounds or induction effects32.
The biosynthesis of secondary metabolites is regulated by a multi-level network62. First-level regulation is achieved by structural genes in the biosynthetic pathways and second-level regulation is performed by transcription factors that can control the expression level of structural genes by binding to their promoter regions62. SmPAP1 is able to increase the promoter activity of SmPAL and SmC4H in transiently-transformed tobacco leaves and interact with SmMYC260. ZmC1 can directly interact with the ZmR or the promoter of SmMDC 13. MYB-responsive elements (MBS, MRE, MBSI and MBSII) and bHLH binding sites (CANNTG) are widely distributed in the promoter regions of phenolic acid and tanshinone biosynthetic pathway genes in S. miltiorrhiza 63, 64. These widespread elements may play crucial roles in regulating metabolic flux shifts.
In this research, we found the endogenous R2R3-MYB transcription factor SmMYB36, which could regulate the accumulation of two major secondary metabolites in S. miltiorrhiza. This transcription factor could be a potential manipulation tool to control metabolic flux flowing to the tanshinone biosynthetic direction in S. miltiorrhiza.
Results
Bioinformatics analysis of SmMYB36
Here, the sequence of SmMYB36 had 98% sequence identity with the sequence in the NCBI database (GenBank Number: KF059390.1). Three nucleotide differences were observed at position 52, 171 and 455 (ACG to TCG, AGA to AGT and AAT to AGT), all of which caused amino acid substitutions (Thr18, Arg56, Asn152 to Ser). Sequence analysis indicated that SmMYB36 contained a complete open reading frame (ORF) and encoded a putative protein of 160 amino acid residues with predicted molecular weight of 18 kDa. Localization prediction results demonstrated that SmMYB36 may localize to the chloroplast, mitochondria and nucleus. The SMART analysis indicated that SmMYB36 contains a complete R2R3 repeat (8 aa to 58 aa and 61 aa to 109 aa) at the N-terminus (Supplementary Fig. S3). The SOPMA analysis and multiple sequence alignment revealed that each repeat is composed of three helices (Supplementary Fig. S3)55, 65.
Phylogenetic analysis reveals that SmMYB36 and the members of subgroup 5 and 15 gathered into a cluster, which differs from other MYBs (SmPAP1 and SmMYB39) that were already reported or analysed in S. miltiorrhiza 8, 60 (Fig. 2 and Supplementary Fig. S5). The bidirectional best BLAST hits and phylogenetic tree analysis indicated that the predicted orthologous genes of SmMYB36 occurred in the Aquilegia coerulea for the first time and that AtMYB23 might be an orthologous gene of SmMYB36. AtMYB114, AtMYB82, AtMYB5, AtMYB8, AtMYB6, ZmC1, SmPAP1 and the members of subgroup 6 (AtMYB75, AtMYB90, AtMB113 and Rosea1), subgroup 5 (AtMYB123), subgroup 15 (AtMYB0, AtMYB23 and AtMYB66) and subgroup 4 (SmMYB39, AtMYB4, AtMYB32, AtMYB7 and AtMYB3) were selected for further motif analysis, which aims to discover specific motifs (Supplementary Table S2) in various subgroups based on previous research8, 25, 26, 29, 55, 66,67,68,69,70,71,72. SmMYB36 contains complete R2 and R3 domains, and each domain has a helix-helix-turn-helix motif. The primary structure (-W-(X19)-W-(X19)-W-……-F/I/L/M-(X18)-W-(X18)-W-) of the R2 domain and R3 domain were in accordance with previous reports25. Motif analysis indicated that SmMYB36 has only the DNEI motif, which can be found in subgroup 5 and subgroup 4 (Supplementary Fig. S3)66. The specific motifs of subgroup 5 (Sg5 motif and motif 5), subgroup 4 (C1 motif and C3 motif) and subgroup 15 (WVxxDxFELSxL motif) could not be found (Supplementary Table S2)67, 68. Moreover, SmMYB36 harbours the [D/E]Lx2[R/K]x3Lx6Lx3R motif (Supplementary Fig. S3) in the R3 domain which is reported to be responsible for interacting with bHLH protein70, 73. Members of subgroup 5 and subgroup 4 contain the DNEI motif, which is a conserved element related to regulation of proanthocyanidin synthesis66. This suggests that SmMYB36 may also regulate the synthesis of proanthocyanidin, similar to AtMYB12333. However, the absence of other motifs specific to subgroup 4, 5 or 15 indicated that SmMYB36 may be a novel member in evolution.

The phylogenetic tree of R2R3-MYB transcription factors. The phylogenetic tree was constructed by maximum likelihood method of MEGA 6.06 based on the multiple sequence alignment using MUSCLE method.
Subcellular localization of SmMYB36
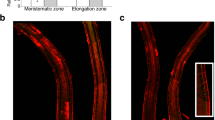
To reveal the potential function of SmMYB36 in the transcriptional regulation system, the subcellular localization of SmMYB36-GFP was performed in onion epidermis. The GFP fluorescence of the control existed in the nucleus and cytoplasm. The GFP fluorescence of SmMYB36 was intensive in the nucleus and dispersive in the cytoplasm (Fig. 3). These results indicate that SmMYB36 may play a role as a transcription factor in the transcriptional regulation system.

Subcellular localization of SmMYB36 protein. The pA7-GFP (upper lane) and pA7-GFP-SmMYB36 (bottom lane) plasmids were transiently expressed in onion epidermal cells. Fluorescence was observed using a confocal laser scanning microscope at 24 h after incubation. The pictures show bright field, green fluorescent field, DAPI and overlay of three fields from left to right.
Transactivation analysis of SmMYB36
The yeast containing the pDEST-GBKT7 or pDEST-GBKT7-SmMYB36 was able to survive on SD/-Trp medium regardless of the concentration change in triazol-3-amine (3AT) (from 0 mM to 20 mM) (Fig. 4). However, on SD/-Trp/-His/-Ade medium, only the yeast with the pDEST-GBKT7-SmMYB36 plasmid grew normally in the absence of 3AT (Fig. 4a). With increasing concentration of 3AT (from 0 mM to 20 mM), the growth state of the yeast with recombinant plasmid increasingly worsened (Fig. 4b). The results imply that SmMYB36 has transactivation activity.

The transactivation analysis of SmMYB36. (a) The yeast AH109 containing pDEST-GBKT7 or pDEST-GBKT7-SmMYB36 construct could survive on SD/-Trp medium regardless of the concentration change of 3AT (from 0 mM to 20 mM). (b) While on SD/-Trp/-His/-Ade medium, only the yeast with pDEST-GBKT7-SmMYB36 plasmid grew normally in the absence of 3AT.
Identification and selection of hairy roots
In this study, seventeen independent SmMYB36-overexpressing lines were identified. Twelve SmMYB36-overexpressing lines were used in a preliminary experiment to detect the metabolite content (Supplementary Fig. S6). According to the preliminary results, four SmMYB36-overexpressing lines (3610, 3611, 3613 and 3615, which were renamed as 36-1, 36-2, 36-3 and 36-4, respectively) with efficient expression of SmMYB36 and one empty-vector line (EV8, renamed to EV) were chosen for QPCR analysis. Line EV, line 36-1, line 36-2, line 36-3 and line 36-4 emitted red fluorescence, but no red fluorescence signal was observed in line WT (Supplementary Fig. S2a). PCR analysis of rolB, rolC, neomycin phosphotransferase II gene (NPT) and SmMYB36 was used to detect the gene integration status of hairy roots (Supplementary Fig. S2b). Specifically, rolB and rolC are Agrobacterium rhizogene-derived genes, which were diagnostic among all hairy roots. In addition, NPT was identified in EV and SmMYB36 transgenic lines as it is an element of pK7WG2R. The existence of exogenous SmMYB36 was only observed in SmMYB36 transgenic lines.
SmMYB36 inhibits phenolic acids or flavonoids biosynthesis and promotes tanshinone biosynthesis in S. miltiorrhiza hairy roots
The preliminary experiment results for twelve lines (without replicates) show a decreasing trend of rosmarinic acid, salvianolic acid B, total phenolics and total flavonoids and the increasing tendency of dihydrotanshinone I, cryptotanshinone, tanshinone I and tanshinone II A (Supplementary Fig. S6). The content of four major tanshinones varied to different extents in different SmMYB36 transgenic lines (Supplementary Fig. S6 and Fig. 5g). Four major tanshinone contents increased markedly in line 36-3 and 36-4, compared to the WT and EV lines. However, only the content of tanshinone I and tanshinone II A increased observably in line 36-1 and 36-2 while the content of dihydrotanshinone and cryptotanshinone did not change obviously. Because samples would be washed with distilled water three times before harvest, tanshinone content suffered some losses. The SmMYB36-overexpressing hairy roots and their extracts appeared much redder than the WT and EV lines (Fig. 5a and 5b). Due to the correlation between colour difference and the total tanshinone content74, the SmMYB36-overexpressing lines may contain more tanshinones, as was also proved by the total content of dihydrotanshinone I, cryptotanshinone, tanshinone I and tanshinone II A (Fig. 5c). The content of rosmarinic acid, salvianolic acid B, total phenolics and total flavonoids was significantly reduced in SmMYB36 transgenic hairy roots (Supplementary Fig. S6, Fig. 5f, 5d and 5e). The correlation analysis indicated a negative correlation between the content of rosmarinic acid, salvianolic acid B, total phenolics and total flavonoids and the transcription level of SmMYB36. The results demonstrate that SmMYB36 could promote tanshinone accumulation but inhibit phenolic acid and flavonoid biosynthesis processes.

The phenotypes of hairy roots. (a) Hairy roots of S. miltiorrhiza induced by A. rhizogenes strain ATCC15834. Hairy roots were cultured in 6,7-V liquid medium for 18 days before being photographed. (b) The colour differences between different extracts of dry hairy roots. (c) The content of total tanshinones in hairy roots. (d) The content of total phenolics in hairy roots. (e) The content of total flavonoids in hairy roots. The pictures show WT (infected by A. rhizogenes strain ATCC15834), EV (infected by A. rhizogenes strain ATCC15834 containing plasmid pK7WG2R-EV), line 36-1, line 36-2, line 36-3 and line 36-4 (infected by A. rhizogenes strain ATCC15834 containing plasmid pK7WG2R-SmMYB36) hairy roots from left to right. (f) The content of dihydrotanshinone I, cryptotanshinone, tanshinone I and tanshinone II A in transgenic and control hairy roots of S. miltiorrhiza. The x represents the values calculated by standard curves when the peak area is zero (18 days). (g) The content of rosmarinic acid and salvianolic acid B in transgenic and control hairy roots of S. miltiorrhiza. The physiological and HPLC analysis both have three biological repeats of transgenic lines and each biological repeat has three technological repeats. The metabolite contents were shown by their means ± SD.
SmMYB36 regulates fatty acid content
Compared to EV lines, the content of total fatty acids showed an increasing trend in SmMYB36 transgenic lines (Supplementary Fig. S7a). Among the five major fatty acids in hairy roots, the relative content of oleic acid (C18:1) decreased markedly, while linoleic acid (C18:2) increased significantly in SmMYB36 transgenic lines (Supplementary Fig. S7b).
SmMYB36 down-regulates expression of phenolic acid biosynthesis pathway genes and up-regulates expression of tanshinone pathway genes
To further uncover the transcription regulatory function of SmMYB36, the key gene expression level of the phenolic acid biosynthesis pathway and tanshinone biosynthetic pathway was detected using quantitative RT-PCR (Fig. 6 and Fig. 7). Most candidate genes of the phenylpropanoid pathway (PAL1, C4H1, 4CL2) and tyrosine pathway (TAT1, HPPR1) were down-regulated, except for 4CL1. However, the expression level of RAS1 and CYP98A14 did not change significantly. The expression pattern of phenolic acid biosynthesis pathway genes was consistent with the change in phenolic acid content. The transcript level of methylerythritol phosphate pathway genes (DXS1, DXS2, DXR, MCT, MDS, HDS, CMK, HDR1) was enhanced substantially, while no obvious expression differences were found in mevalonate pathway genes (AACT1, AACT2, HMGS, MK, PMK, MDC), except for HMGR2. The tanshinone biosynthetic downstream pathway genes (GGPPS1, CPS1, KSL1, CYP76AHI) represented increased expression. The variation in tanshinone biosynthetic pathway gene transcription and tanshinone content was generally consistent. These results show that the main secondary metabolism pathways of S. miltiorrhiza could be regulated by SmMYB36.

Relative expression level of phenolic acid biosynthesis pathway genes in transgenic hairy roots. Each line has two biological repeats and each biological repeat has three technological repeats. All values are expressed as means ± SD.

Relative expression level of tanshinone biosynthesis pathway genes in transgenic hairy roots. Each line has two biological repeats and each biological repeat has three technological repeats. All values are expressed as means ± SD.
SmMYB36 binds to the predicted MYB-binding elements
To further reveal whether SmMYB36 could directly interact with the promoters of pathway genes, the promoter sequences and MYB-related elements of pathway genes were predicted and are shown in Supplementary Table S3. Several pathway genes (PAL1, C4H1, 4CL1, 4CL2, TAT1, HPPR1, DXS1, DXS2, DXR, MCT, MDS, HDS, CMK, HDR1, GGPPS1, CPS1, KSL1, CYP76AHI, HMGR2) displayed different patterns between SmMYB36-overexpressed and control lines. Among these genes, C4H1, 4CL2, HPPR1, DXR, MCT and GGPPS1 were selected to conduct electrophoretic mobility shift assay (EMSA) due to their promoters contain MYB-binding elements. All of the specific MYB-related probes for these gene promoters (Supplementary Table S1) could interact with SmMYB36; the control probes could not interact with SmMYB36 (Fig. 8). The specific MYB-related probes of IPPI and HMGS1, with unchanged expression, could also interact with SmMYB36. To determine whether the combination ability of SmMYB36 is highly specific to only some gene promoters, the probes for the MYB-binding core elements (MBS1, CAACTG; MBS2, CGGTCA; MBS3, TAACTG; MRE, AACCTAA; MBSI, AAAAAAC(C/G)GTTA; MBSII, AAAAGTTAGTTA) were synthesized and used for EMSA. Interestingly, SmMYB36 could interact with most of these MYB-related core elements (MBS1, CAACTG; MBSI, AAAAAAC(C/G)GTTA; MBSII, AAAAGTTAGTTA) (Fig. 8).

Analysis of SmMYB36 binding to the predicted promoter probes of secondary metabolism pathway genes in S.miltiorrhiza. Arrows indicates the combination between SmMYB36 and probes.
Discussion
The members of subgroups 4, 5, 6 and 15 were selected to perform motif analysis; these members have relatively close relationships to SmMYB36 in phylogenetic trees (Fig. 2 and Supplementary Fig. S5). Motif analysis indicates that SmMYB36 contains a DNEI motif that is widely present in subgroup 4 and 555, 66. The DNEI motif is highly conserved and specific to the proanthocyanidin accumulation55, 66. However, no other motif (Supplementary Table S2) specific to subgroups 4, 5, 6 and 15 was found in the C-terminal region of SmMYB36, which demonstrates that SmMYB36 might be a novel member in evolution. The predicted orthologous genes of SmMYB36 occurred in Aquilegia coerulea for the first time; this organism is a model plant used to study the evolutionary relationships of eudicot and monocot plants (Supplementary Fig. S4 and Table S4). The evolutionary distances of R2R3-MYBs in monocots (maize)75 and dicots (A. thaliana)29 provided evidence for the expansion hypothesis of R2R3-MYBs. It is hypothesized that rapid evolution of R2R3-MYBs occurred during the last 400 million years, after evolutionary divergence from bryophytes to tracheophytes75. The amplification of R2R3-MYBs was used to satisfy the specific cellular functions of new plants76, thus adjusting plant metabolism and development through plasticity77. Phylogenetic tree analysis indicates that SmMYB36 is in a close relationship with the AtMYB0, AtMYB23 and AtMYB66 of subgroup 15; AtMYB23 might be an orthologous gene of SmMYB36 (Fig. 2 and Supplementary Fig. S5). AtMYB23 plays a crucial role in controlling trichome development, including initiation and branching78, 79. However, no reports have suggested that AtMYB23 can regulate metabolism. Trichome and artemisinin regulator 1, an AP2/ERF transcription factor, acts as an important regulator in both the development of trichomes and the biosynthesis of artemisinin in Artemisia annua 80, which is an example of a transcription factor regulating development and metabolism meanwhile. It was shown that tanshinones primarily accumulate in the periderm of S. miltiorrhiza roots81. Root epidermis (mainly trichomes) and periderm are both specific plant tissues, where secondary metabolites accumulate, indicating the possible regulatory roles of AtMYB23 and SmMYB36 in the development and metabolism of plants. We deduced that SmMYB36 might not only regulate secondary metabolism but also influence plant development; more experimental evidence is needed to clarify.
Localization results demonstrated that SmMYB36 is localized in the cytoplasm and nucleus. The function of transcription factors is closely related to their localization82. SmMYB36 localized to the nucleus and has transactivation activity, suggesting that it can regulate the transcription of target genes by itself in the nucleus. Some transcription factors function outside the nucleus82,83,84. Here, the fluorescence of SmMYB36-GFP is dispersive in the cytoplasm. Cytoplasm is where many physiological processes run. SmMYB36 may be involved in some processes such as transcription in plastids. The function or localization of one transcription factor may be influenced by other transcription factors83. For example, AtMYC1 localized in the cytoplasm and can interact with GL1, leading to the relocalization of GL1 from the nucleus to the cytoplasm and further increasing the number of trichomes83. There may be bHLHs that cooperate with SmMYB36 to regulate the physiological processes in the cytoplasm and nucleus. More research is needed to illustrate the accurate localization results of SmMYB36 using Arabidopsis and tobacco protoplasts.
Over-expression of SmMYB36 can inhibit the biosynthesis of phenolic acids in S. miltiorrhiza hairy roots. A negative correlation was found between the content of phenolic acids and the transcription level of SmMYB36. The changes in metabolite content were in accordance with transcript-level changes in metabolic pathway genes. Specifically, the decrease in rosmarinic acid, salvianolic acid B and total phenolic content in transgenic lines were correlated with the reduced transcript expression of PAL1, 4CL2 and TAT1, which suggested the limitation of enzyme activity and substrate content, potentially leading to reduced phenolic acid content. In addition, down-regulation of C4H1 transcription in transgenic lines was also involved in the reduced alteration of total phenolic content. Different 4CLs can direct carbon flux into various phenylpropanoid branch pathways85, 86; 4CL2 is more important than 4CL1 in the phenolic acid biosynthesis of S. miltiorrhiza 87, 88. Hence, it is reasonable that 4CL2 was inhibited, but 4CL1 was not affected, in SmMYB36 overexpressed lines. Total flavonoid content of SmMYB36 overexpressed lines decreased remarkably, coordinated with the transcript levels of general phenylpropanoid pathway genes. Overall, the transcript levels of most genes in the upstream phenylpropanoid and tyrosine derived pathways exhibit a decreased tendency compared with control (EV), which might contribute to reduced phenolic compounds in SmMYB36-overexpressed lines. Comprehensively, because of the reduction of metabolic flow to the phenylpropanoid derived pathway and tyrosine derived pathway, the content of phenolic acid was reduced in SmMYB36 overexpressed lines.
The R2R3-MYBs of subgroup 4 and R3-MYBs are two types of repressors for the phenylpropanoid metabolite pathway26. The R2R3-MYBs of subgroup 4 usually function as repressors participating in phenylpropanoid-derived metabolite accumulation26 and contain the C1 and C3 (EAR or ERF) motif 89. The C3 motif was conserved at the C-terminus of subgroup 4 members and is required for repression activity8, 89. R2R3-MYB, bHLH and WD normally form a functional MBW complex to work. R3-MYBs are the other type of repressors, which can replace R2R3-MYB and competitively bind bHLH proteins to perform repressor activity90, 91. However, SmMYB36 is an R2R3-MYB rather than R3-MYB, and the C3 motif was absent in SmMYB36, which indicates that the inhibition mechanism of SmMYB36 may differ from the above two types repressors and specialize in phenolic acid accumulation. More evidence is needed to determine the inhibition mechanism of SmMYB36.
Overexpression of SmMYB36 can promote the biosynthesis of tanshinones in S. miltiorrhiza hairy roots. Changes in metabolite content and changes of pathway gene transcript level were identical. It is generally thought that the MEP pathway, compared to the MVA pathway, plays a larger role in diterpenoid synthesis92. A similar phenomenon was observed in this research: gene expression of the MVA pathway was not obviously altered. However, the contents of dihydrotanshinone I and cryptotanshinone varied from the independent lines (Fig. 5g), showing an elevated trend. This is likely because the sampling method (washed with distilled water three times) caused different degrees of tanshinone losses in the harvesting of SmMYB36-overexpressed hairy roots. As far as we know, this is the first report that endogenous R2R3-MYB can regulate the biosynthesis of tanshinones in S. miltiorrhiza hairy roots. Many studies have found that terpenoid metabolism is regulated by AP2/ERF, WRKY, bHLH and basic leucine zipper (bZIP) transcription factors93. However, most studies concern the function of R2R3-MYBs in phenylpropanoid metabolism, rather than terpenoid biosynthesis regulation26. This may be because some phenylpropanoid metabolites are closely related to the colour of organisms (e.g. anthocyanin), making them much easier to study94. Second, the regulation of active metabolite biosynthesis is complicated; for example, the complexity of protein complexes (such as MBW complexes and MYB/bHLH complexes95) and the complexity of regulatory networks are difficult to illuminate94, 95.
SmMYB36 can simultaneously regulate the biosynthesis of phenolic acids and tanshinones in S. miltiorrhiza hairy roots. We found that the phenolic acid content was negatively correlated with tanshinone content in SmMYB36-hairy roots. The altered transcription of SmMYB36 leads to changed expression of biosynthetic pathway genes and modulated the metabolic flux shift from phenolic acid accumulation to tanshinone accumulation. The regulation of metabolic flux has been achieved by the altered expression of secondary metabolic pathway genes21,22,23,24 and transcription factors8, 13, 58,59,60 in S. miltiorrhiza. The regulation of transcription factors is thought to function by interacting with cis-elements of secondary metabolic pathway genes. The predicted responsive elements of MYB and bHLH are widely distributed in the gene promoter regions of known phenolic acid and tanshinone biosynthetic pathways63, 64. Our EMSA results indicated that SmMYB36 could interact with most of the predicted core MYB-related elements, providing more evidence for clarifying the metabolic flux shift. These predicted responsive elements and transcription factors may be effective tools to regulate secondary metabolism, which should be studied further. Since AtMYB23 and SmPAP1 can interact with bHLHs (subgroup III f or d + e)60, 79, 96 and SmMYB36 contains a bHLH-binding motif, there may be SmbHLHs that interact with SmMYB36. The illumination of these SmbHLHs will lead to a better understanding of the regulation mechanism of SmMYB36. In addition, transcription factors can regulate primary metabolism. According to our analysis, fatty acid profiles were altered significantly in SmMYB36-hairy roots compared to the control (Supplementary Fig. S7). Taken together, SmMYB36 not only regulated secondary metabolism but also influenced primary metabolism and may be potential tools to manipulate metabolic flux in S. miltiorrhiza hairy roots, suggesting complicated and comprehensive roles for SmMYB36.
Based on the results and analysis, we proposed a model for tanshinone and phenolic acid biosynthesis regulation by SmMYB36 (Fig. 1). SmMYB36 inhibited the biosynthesis of phenolic acids and promoted the accumulation of tanshinones in S. miltiorrhiza hairy roots. Some bHLHs may exist in S. miltiorrhiza that can interact with SmMYB36 to participate in primary and secondary metabolism. In secondary metabolism, the biosynthesis of tanshinones could be promoted by SmMYB36 or SmMYB36-bHLH complexes while the biosynthesis of phenylpropanoids could be inhibited. High throughput techniques (transcriptome-, proteome-, metabolome- and ChIP-sequencing) can be applied to further illustrate the complicated mechanism. SmMYB36 was the only transcription factor found that could regulate the accumulation of two major secondary metabolites at the same time in S. miltiorrhiza. This regulating effect is quite novel, and SmMYB36 is a double-edged sword for the quality of S. miltiorrhiza. Higher expression levels of SmMYB36 promote the accumulation of tanshinones but not phenolic acids. Thus, it is important to control the expression level of SmMYB36 when accumulating the two types of bioactive compounds.
Materials and Methods
Plasmid construction
The plasmids pDONR207, pK7WG2R, pK7WG2R-EV and pDEST-GBKT7 were provided by Prof. Cathie Martin (John Innes Centre, UK). The constructs pA7-GFP and pET32a (+) were kept in our own laboratory. Total RNA was extracted from two-week-old sterile plantlets of S. miltiorrhiza according to the instructions of the RNAprep Pure Plant Kit (TIANGEN, China). The total RNA was reverse transcribed into cDNA using the instructions of the PrimeScript RT Reagent Kit (Takara, Japan). The whole CDS sequences of SmMYB36 were amplified with primers (Supplementary Table S1) using EasyPfu DNA Polymerase (Transgen, China). The PCR products were recombined into the pDONR207 entry vector using a BP reaction and introduced into the destination vector pK7WG2R or pDEST-GBKT7 using an LR reaction. To generate subcellular localization vectors and a prokaryotic expression vector, the cDNA fragments with digestion sites for SmMYB36 were separately double-digested and cloned into the pA7-GFP vector and pET32a (+) vector. The inserted sequences in the vectors were identified by sequencing (Shanghai Sangon, China).
Phylogenetic tree construction and bioinformatics analysis
BLAST was used to determine differences between the SmMYB36 sequences we cloned and the NCBI database. ExPASy, SMART and SOPMA software were employed to predict the molecular weight, domains and secondary structures. Potential positioning prediction was confirmed by cNLS Mapper, Plant-Ploc and TargetP. The amino acid sequence of SmMYB36 was submitted to the R2R3-MYB protein family of A. thaliana, Oryza sativa L., Brachypodium distachyon and Lotus japonicas in the IT3F website (http://jicbio.nbi.ac.uk/IT3F/) to construct the phylogenetic tree (Supplementary Fig. S5). Rosea1 (ABB83826.1), ZmC1 (P10290.1), SmPAP1 (ACZ48688.2), SmMYB39 (AGS48990.1), SmMYB36 and other 125 R2R3-MYB factors of A. thaliana 25 in the NCBI database were used to construct the phylogenetic tree (Fig. 2) using the maximum likelihood method of MEGA 6.06 based on the multiple sequence alignment using a MUSCLE method. The homologous analysis was based on the Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html) and BLAST tools from NCBI and Phytozome (Supplementary Table S4 and Fig. S3). The orthologous genes of SmMYB36 were predicted by the bidirectional best BLAST hits and phylogenetic tree analysis (Supplementary Table S4). The species phylogenetic tree was from the Phytozome database (https://phytozome.jgi.doe.gov/pz/portal.html) and the species containing the predicted orthologous genes of SmMYB36 (Supplementary Fig. S4) were emphasized.
Hairy root culture
The hairy roots were derived from S. miltiorrhiza sterile leaves infected by A. rhizogenes strain ATCC15834 containing the plasmid pK7WG2R-SmMYB36 or pK7WG2R-EV97. The wild-type hairy roots were from leaves infested with empty A. rhizogenes ATCC15834. The transgenic hairy roots were confirmed by fluorescence of DsRed protein and PCR using rolB, rolC, NPT and SmMYB36 specific primers (Supplementary Table S1). One wild-type line, one empty-vector line and four transgenic lines of hairy roots were selected. Each line used three repeats for further analysis. Each 100-mL conical flask contained 50 mL 6,7-V liquid medium inoculated with 0.3 g fresh hairy roots to propagate in a constant-temperature shaking incubator (25 °C,120 rev.min−1). The hairy roots were harvested after 18 days and used for real-time quantitative PCR analysis, HPLC analysis and a physiological assay.
Real-time quantitative PCR analysis
The total RNA of hairy roots was extracted according to the instructions of the RNAprep Pure Plant Kit (TIANGEN, China). The total RNA was reverse transcribed into cDNA based on the instructions of the PrimeScript RT Reagent Kit (Takara, Japan). Real-time quantitative PCR was performed following the instructions of the SYBR Premix Ex Taq II Kit (Takara, Japan). The actin gene with constitutive expression was used as the internal control. The transcript levels of the following genes were quantitated: phenylalanine ammonia-lyase (PAL), cinnamic acid 4-hydroxylase (C4H), 4CL, tyrosine amino transferase (TAT), hydroxyphenylpyruvic acid reductase (HPPR), RAS, CYP98A14, CYP76AH1, acetoacetyl-CoA thiolase(AACT), 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS), 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR), mevalonate kinase (MK), 5-phosphomevalonate kinase (PMK), mevalonate pyrophosphate decarboxylase (MDC), 1-deoxy-D-xylulose 5-phosphate synthase (DXS), 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR), 2C-methyl-D-erythritol 4-phosphate cytidylyl-transferase (MCT), 4-diphosphocytidyl-2C-methyl-erythritol kinase (CMK), 2C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (MDS), 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate synthase (HDS), 1-hydroxy-2-methyl-2-(E)-butenyl 4-diphosphate reductase (HDR), GGPPS, CPS and KSL. The primers used in this experiment were used according to previous reports63, 18. Gene expression level was calculated by the ΔΔCT method and represented by their means ± SD. Real-time PCR employed the following protocol: 95 °C for 30 s, 1 cycle; 95 °C for 5 s, 60 °C for 30 s, 40 cycles. The relative expression level of SmMYB36 was shown in Supplementary Fig. S2c.
Subcellular localization
The plasmids pA7-GFP-SmMYB36 and pA7-GFP were transiently transformed into onion epidermis using a gene gun (Bio-Rad, Hercules, CA, USA). After incubation for 24 hours, the onion epidermis was stained with DAPI (Solarbio, Beijing, China) for 20 minutes and washed twice with PBS buffer (pH 7.2). GFP fluorescence, DAPI fluorescence and bright field of onion epidermis were observed under a confocal laser scanning microscope (Nikon A1, Tokyo, Japan).
Transactivation assay
To determine whether SmMYB36 has transactivation function, the pDEST-GBKT7-SmMYB36 and pDEST-GBKT7 plasmids were transformed into the yeast strain AH109. The 3AT was selected to inhibit transactivation activity (from 0 mM to 20 mM). The transformed yeast cells were first screened on synthetic dropout (SD) medium lacking tryptophan (SD/-Trp/+3AT) and selected on SD medium without tryptophan, histidine and adenine (SD/-Trp/-His/-Ade/+3AT).
HPLC and GC analysis
The hairy roots were dried at 45 °C to a constant weight in an oven. The contents of tanshinones and phenolic acids in hairy roots were determined using HPLC method described by Liang et al.98 and Zhang et al.8. All the tanshinones and phenolic acids were detected at 270 nm and 288 nm, respectively. All components were determined using a standard curve from Peng et al.99. The same extract of hairy roots used here was exploited for further physiological assays.
The contents of total fatty acids, palmitic acid (C16:0), stearic acid (18:0), oleic acid (C18:1), linoleic acid (C18:2), and linolenic acid (C18:3) were determined by GC analysis following the protocol of Li et al.100.
Physiological assay
Photos of hairy roots and their extracts were recorded to display the correlation between colour difference and the total tanshinone content74, 101. The sum of dihydrotanshinone I, cryptotanshinone, tanshinone I and tanshinone II A was calculated as total tanshinones in this research. Total phenolics content was determined using the Folin-Ciocalteu method102, with minor modification. Specifically, 0.04 mL extracts, 1 mL distilled water and 1 mL Folin-Ciocaleu reagent were thoroughly mixed. Afterwards, 1.6 mL Na2CO3 (7.5 g/100 mL) was added, and the mixture was incubated in a water bath (30 °C, dark) for 1.5 hours. The absorbance of samples was measured at 765 nm. Gallic acid was used to construct a calibration curve to determine the total phenolics content. The phenolic acids represent rosmarinic acid and salvianolic acid B. Total flavonoids were detected according to Jia’s method103. The absorption peak was estimated at 506 nm. Using rutin as a standard, the calibration curve was established to determine the total flavonoid content.
Electrophoretic mobility shift assays (EMSA)
The plasmids pET32a-SmMYB36 and pET32a were transformed and expressed in E. coli BL21. HIS-labelled protein was purified out using Ni-NTA Resin (Solarbio, Beijing, China). The elution buffer (pH 8.0) contains 50 mM NaH2PO4·2H2O, 300 mM NaCl and 250 mM imidazole. The promoter fragments were predicted based on the genome sequence of S. miltiorrhiza (http://www.ndctcm.org/shujukujieshao/2015-04-23/27.html) and PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/) databases. The MBS, MRE, MBSI and MBSII specific or core element sequences of promoter fragments were used as probes and the sequences of the same length as the above probes of the SmMYB36 open reading frame were used as control probes. (Supplementary Table S1). The EMSA assay was conducted according to the instructions of the Electrophoretic Mobility Shift Assay (EMSA) Kit (Invitrogen). The mass ratio of probe and protein was 1:15 in each reaction mixture (10 µL).
Statistical analysis
Significance analysis of gene expression and metabolite content was performed by means of Analysis of Variance (ANOVA), Least Significance Difference (LSD) and Student-Newman-Keuls (S-N-K). Correlation analysis was performed between the different gene expressions or between gene expression and metabolite content. Pearson test and Spearman test were used to calculate statistically significant correlations (with P value less than 0.05). IBM SPSS Statistics was used for various computations.
References
Chen, H., Feng, C., Chiu, F. C. K. & Lo, C. M. Y. The effect of yeast elicitor on the growth and secondary metabolism of hairy root cultures of Salvia miltiorrhiza. Enzyme & Microbial Technology 28, 100–105 (2001).
Liu, A. H. et al. Simultaneous quantification of six major phenolic acids in the roots of Salvia miltiorrhiza and four related traditional Chinese medicinal preparations by HPLC–DAD method. Journal of Pharmaceutical & Biomedical Analysis 41, 48–56 (2006).
Petersen, M. & Simmonds, M. S. J. Molecules of Interest Rosmarinic Acid. Phytochemistry 62, 121–125 (2003).
Li, L. N. Biologically active components from traditional Chinese medicines: Pure and Applied Chemistry. Pure & Applied Chemistry 70, 547–554 (1998).
Don, M. J., Shen, C. C., Syu, W. J., Ding, Y. H. & Sun, C. M. Cytotoxic and aromatic constituents from Salvia miltiorrhiza. Phytochemistry 67, 497–503 (2006).
Shi, Z., He, J., Yao, T., Chang, W. & Zhao, M. Simultaneous determination of cryptotanshinone, tanshinone I and tanshinone IIA in traditional Chinese medicinal preparations containing Radix salvia miltiorrhiza by HPLC. Journal of Pharmaceutical & Biomedical Analysis 37, 481–486 (2005).
Li, C. J. et al. Bis-sesquiterpenes and diterpenes from Chloranthushenryi. Phytochemistry 69, 2867–2874 (2008).
Zhang, S. et al. Cloning and characterization of a putative R2R3 MYB transcriptional repressor of the rosmarinic acid biosynthetic pathway from Salvia miltiorrhiza. PloS One 8, e73259 (2013).
Petersen, M. et al. Evolution of rosmarinic acid biosynthesis. Phytochemistry 70, 1663–1679 (2009).
Petersen, M. Rosmarinic acid: new aspects. Phytochemistry Reviews 12, 207–227 (2013).
Di, P. et al. 13C tracer reveals phenolic acids biosynthesis in hairy root cultures of Salvia miltiorrhiza. Acs Chemical Biology 8, 1537–1548 (2013).
Ma, P., Liu, J., Zhang, C. & Liang, Z. Regulation of Water-Soluble Phenolic Acid Biosynthesis in Salvia miltiorrhiza Bunge. Applied Biochemistry & Biotechnology 170, 1253–1262 (2013).
Zhao, S., Zhang, J., Tan, R., Yang, L. & Zheng, X. Enhancing diterpenoid concentration in Salvia miltiorrhiza hairy roots through pathway engineering with maize C1 transcription factor. Journal of Experimental Botany 66, 7211–7226 (2015).
Bo, W. et al. Genome-wide identification of phenolic acid biosynthetic genes in Salvia miltiorrhiza. Planta 241, 711–725 (2014).
Ma, X. H. et al. The Biosynthetic Pathways of Tanshinones and Phenolic Acids in Salvia miltiorrhiza. Molecules 20, 16235–16254 (2015).
Rohmer, M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Natural Product Reports 16, 565–574 (1999).
Hemmerlin, A., Harwood, J. L. & Bach, T. J. A raison d’être for two distinct pathways in the early steps of plant isoprenoid biosynthesis? Progress in Lipid Research 51, 95–148 (2012).
Ma, Y. et al. Genome-wide identification and characterization of novel genes involved in terpenoid biosynthesis in Salvia miltiorrhiza. Journal of Experimental Botany 63(7), 2809–2823 (2012).
Pan, L., Hemmerlin, A., Bach, T. J. & Chye, M. L. The potential of the mevalonate pathway for enhanced isoprenoid production. Biotechnology Advances 34, 697–713 (2016).
Guo, J. et al. Cytochrome P450 promiscuity leads to a bifurcating biosynthetic pathway for tanshinones. New Phytologist 210, 525–531 (2015).
Kai, G. et al. Metabolic engineering tanshinone biosynthetic pathway in Salvia miltiorrhiza hairy root cultures. Metabolic Engineering 13, 319–327 (2011).
Dai, Z., Cui, G., Zhou, S. F., Zhang, X. & Huang, L. Cloning and characterization of a novel 3-hydroxy-3-methylglutaryl coenzyme A reductase gene from Salvia miltiorrhiza involved in diterpenoid tanshinone accumulation. Journal of Plant Physiology 168, 148–157 (2011).
Ying, X. et al. The c4h, tat, hppr and hppd Genes Prompted Engineering of Rosmarinic Acid Biosynthetic Pathway in Salvia miltiorrhiza Hairy Root Cultures. PloS One 6, e29713 (2011).
Zhang, Y. et al. Pathway engineering for phenolic acid accumulations in Salvia miltiorrhiza by combinational genetic manipulation. Metabolic Engineering 21, 71–80 (2014).
Dubos, C. et al. MYB transcription factors in Arabidopsis. Trends in Plant Science 15, 573–581 (2010).
Liu, J., Osbourn, A. & Ma, P. MYB Transcription Factors as Regulators of Phenylpropanoid Metabolism in Plants. Molecular Plant 8, 689–708 (2015).
Zhang, Y. Multi-level engineering facilitates the production of phenylpropanoid compounds in tomato. Nature Communications 6, 219–246 (2015).
Misra, P. et al. Modulation of transcriptome and metabolome of tobacco by Arabidopsis transcription factor, AtMYB12, leads to insect resistance. Plant Physiology 152, 2258–2268 (2010).
Stracke, R., Werber, M. & Weisshaar, B. The R2R3-MYB gene family in Arabidopsis thaliana. Current Opinion in Plant Biology 4, 447–456 (2001).
Li, C. & Lu, S. Genome-wide characterization and comparative analysis of R2R3-MYB transcription factors shows the complexity of MYB-associated regulatory networks in Salvia miltiorrhiza. BMC Genomics 15, 1–12 (2014).
Bedon, F. et al. Subgroup 4 R2R3-MYBs in conifer trees: gene family expansion and contribution to the isoprenoid- and flavonoid-oriented responses. Journal of Experimental Botany 61, 3847–3864 (2010).
Mahjoub, A. et al. Overexpression of a grapevine R2R3-MYB factor in tomato affects vegetative development, flower morphology and flavonoid and terpenoid metabolism. Plant Physiology & Biochemistry 47, 551–561 (2009).
Sharma, S. B. & Dixon, R. A. Metabolic engineering of proanthocyanidins by ectopic expression of transcription factors in Arabidopsis thaliana. Plant Journal 44, 62–75 (2005).
Himi, E. et al. Ant28 gene for proanthocyanidin synthesis encoding the R2R3 MYB domain protein (Hvmyb10) highly affects grain dormancy in barley. Euphytica 188, 141–151 (2012).
Akagi, T., Ikegami, A. & Yonemori, K. DkMyb2 wound-induced transcription factor of persimmon (Diospyros kaki Thunb.), contributes to proanthocyanidin regulation. Planta 232, 1045–1059 (2010).
Yoshida, K. et al. Functional differentiation of Lotus japonicus TT2s, R2R3-MYB transcription factors comprising a multigene family. Plant & Cell Physiology 49, 157–169 (2008).
Ravaglia, D. et al. Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biology 13, 68 (2013).
Chiou, C. Y. & Yeh, K. W. Differential expression of MYB gene (OgMYB1) determines color patterning in floral tissue of Oncidium Gower Ramsey. Plant Molecular Biology 66, 379–388 (2008).
Mellway, R. D., Tran, L. T., Prouse, M. B., Campbell, M. M. & Constabel, C. P. The wound-, pathogen-, and ultraviolet B-responsive MYB134 gene encodes an R2R3 MYB transcription factor that regulates proanthocyanidin synthesis in poplar. Plant Physiology 150, 924–941 (2009).
Schaart, J. G. et al. Identification and characterization of MYB‐bHLH‐WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria × ananassa) fruits. New Phytologist 197, 454–467 (2013).
Dianalucia, Z. et al. Arabidopsis thaliana MYB75/PAP1 transcription factor induces anthocyanin production in transgenic tomato plants. Functional Plant Biology 35, 606–618 (2008).
Teng, S. & Smeekens, S. Sucrose-specific induction of anthocyanin biosynthesis in Arabidopsis requires the MYB75/PAP1 gene. Plant Physiology 139, 1840–1852 (2005).
Gonzalez, A., Zhao, M., Leavitt, J. M. & Lloyd, A. M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant Journal 53, 814–827 (2008).
Chiu, L. W. et al. The purple cauliflower arises from activation of a MYB transcription factor. Plant Physiology 154, 1470–1480 (2010).
Elomaa, P. et al. Activation of anthocyanin biosynthesis in Gerbera hybrida (Asteraceae) suggests conserved protein-protein and protein-promoter interactions between the anciently diverged monocots and eudicots. Plant Physiology 133, 1831–1842 (2003).
Azuma, A. et al. Genomic and genetic analysis of Myb-related genes that regulate anthocyanin biosynthesis in grape berry skin. Theoretical and Applied Genetics 117, 1009–1019 (2008).
Palapol, Y., Ketsa, S., Lin-Wang, K., Ferguson, I. B. & Allan, A. C. A. MYB transcription factor regulates anthocyanin biosynthesis in mangosteen (Garcinia mangostana L.) fruit during ripening. Planta 229, 1323–1334 (2009).
Butelli, E. et al. Retrotransposons control fruit-specific, cold-dependent accumulation of anthocyanins in blood oranges. Plant Cell 24, 1242–1255 (2012).
Pierantoni, L. et al. Mapping of an anthocyanin-regulating MYB transcription factor and its expression in red and green pear, Pyrus communis. Plant Physiology & Biochemistry 48, 1020–1026 (2010).
Feng, S., Wang, Y., Yang, S., Xu, Y. & Chen, X. Anthocyanin biosynthesis in pears is regulated by a R2R3-MYB transcription factor PyMYB10. Planta 232, 245–255 (2010).
Quattrocchio, F., Wing, J. F., Van, dW. K., Mol, J. N. & Koes, R. Analysis of bHLH and MYB domain proteins: species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. Plant Journal 13, 475–488 (1998).
Quattrocchio, F. & Koes, R. Molecular analysis of the anthocyanin2 gene of Petunia and its role in the evolution of flower color. Plant Cell 11, 1433–1444 (1999).
Laitinen, R. A. E., Ainasoja, M., Broholm, S. K., Teeri, T. H. & Elomaa, P. Identification of target genes for a MYB-type anthocyanin regulator in Gerbera hybrida. Journal of Experimental Botany 59, 3691–3703 (2008).
Böhler, J., Schollmeyer, P., Dressel, B., Dobos, G. & Hörl, W. H. Isolation and functional characterization of a floral tissue-specific R2R3 MYB regulator from tobacco. Planta 231, 1061–1076 (2010).
Nesi, N., Jond, C., Debeaujon, I., Caboche, M. & Lepiniec, L. The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell 13, 2099–2114 (2001).
Baudry, A. et al. TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant Journal for Cell & Molecular Biology 39, 366–380 (2004).
Baudry, A., Caboche, M. & Lepiniec, L. TT8 controls its own expression in a feedback regulation involving TTG1 and homologous MYB and bHLH factors, allowing a strong and cell-specific accumulation of flavonoids in Arabidopsis thaliana. Plant Journal 46, 768–779 (2006).
Yuan, Z., Yan, Y. P. & Wang, Z. Z. The Arabidopsis PAP1 transcription factor plays an important role in the enrichment of phenolic acids in Salvia miltiorrhiza. Journal of Agricultural & Food Chemistry 58, 12168–12175 (2010).
Wang, D. et al. Metabolic pools of phenolic acids in Salvia miltiorrhiza are enhanced by co-expression of Antirrhinum majus Delila and Rosea1 transcription factors. Biochemical Engineering Journal 74, 115–120 (2013).
Hao, G. et al. Cloning, molecular characterization and functional analysis of a putative R2R3-MYB transcription factor of the phenolic acid biosynthetic pathway in S. miltiorrhiza Bge. f. alba. Plant Cell Tissue & Organ Culture 124, 151–168 (2016).
Zvi, M. M. B. et al. PAP1 transcription factor enhances production of phenylpropanoid and terpenoid scent compounds in rose flowers. New Phytologist 195, 335–345 (2012).
Zhong, R., Lee, C. & Ye, Z. H. Evolutionary conservation of the transcriptional network regulating secondary cell wall biosynthesis. Trends in Plant Science 15, 625–632 (2010).
Wang, B. et al. Genome-wide identification of phenolic acid biosynthetic genes in Salvia miltiorrhiza. Planta 241, 711–725 (2015).
Zhang, X. et al. Genome-wide characterisation and analysis of bHLH transcription factors related to tanshinone biosynthesis in Salvia miltiorrhiza. Scientific Reports 5, 11244 (2014).
Zhou, H. et al. Peach MYB7 activates transcription of the proanthocyanidin pathway gene encoding leucoanthocyanidin reductase, but not anthocyanidin reductase. Frontiers in Plant Science 6, 908 (2015).
Heppel, S. C. et al. Identification of key amino acids for the evolution of promoter target specificity of anthocyanin and proanthocyanidin regulating MYB factors. Plant Molecular Biology 82, 457–471 (2013).
Takos, A. M. et al. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiology 142, 1216–1232 (2006).
Gesell, A., Yoshida, K., Lan, T. T. & Constabel, C. P. Characterization of an apple TT2-type R2R3 MYB transcription factor functionally similar to the poplar proanthocyanidin regulator PtMYB134. Planta 240, 497–511 (2014).
Kranz, H. D. et al. Towards functional characterisation of the members of the R2R3-MYB gene family from Arabidopsis thaliana. Plant Journal for Cell & Molecular Biology 16, 263–276 (1998).
Deluc, L. et al. The transcription factor VvMYB5b contributes to the regulation of anthocyanin and proanthocyanidin biosynthesis in developing grape berries. Plant Physiology 147, 2041–2053 (2008).
Czemmel, S. et al. The grapevine R2R3-MYB transcription factor VvMYBF1 regulates flavonol synthesis in developing grape berries. Plant Physiology 151, 1513–1530 (2009).
Lin-Wang, K. et al. An R2R3 MYB transcription factor associated with regulation of the anthocyanin biosynthetic pathway in Rosaceae. BMC Plant Biology 10, 1–17 (2010).
Bogs, J., Jaffé, F. W., Takos, A. M., Walker, A. R. & Robinson, S. P. The grapevine transcription factor VvMYBPA1 regulates proanthocyanidin synthesis during fruit development. Plant Physiology 143, 1347–1361 (2007).
Wang Hai, Y. Z., Shen Yuxiang, H. E., Dongmei, L. A. N. & Ying, W. A. N. Deguang. Correlation of Color Feature with Effective Constituents of Radix Salviae Miltiorrhizae. Traditional Chinese Drug Research & Clinical Pharmacology 25, 333–338 (2014).
Rabinowicz, P. D., Braun, E. L., Wolfe, A. D., Bowen, B. & Grotewold, E. Maize R2R3 Myb genes: Sequence analysis reveals amplification in the higher plants. Genetics 153, 427–444 (1999).
Martin, C. & Pazares, J. MYB transcription factors in plants. Trends in Genetics 13, 67–73 (1997).
Romero, I. et al. More than 80 R2R3-MYB regulatory genes in the genome of Arabidopsis thaliana. Plant Journal 14, 273–284 (1998).
Yeon Hee, K. et al. The MYB23 Gene Provides a Positive Feedback Loop for Cell Fate Specification in the Arabidopsis Root Epidermis. Plant Cell 21, 1080–1094 (2009).
Kirik, V. et al. Functional diversification of MYB23 and GL1 genes in trichome morphogenesis and initiation. Development 132, 1477–1485 (2005).
Hexin et al. Trichome and Artemisinin Regulator 1 Is Required for Trichome Development and Artemisinin Biosynthesis in Artemisia annua. Molecular Plant 8, 1396–1411 (2015).
Xu Cui-hong, S. Z.-M., Wang, Y. & Miao Fang, ZhouL. The Accumulation Rule of the Main Medicinal Components in Different Organs of Salvia miltiorrhiza Bunge. and Salvia miltiorrhiza Bunge. f. alba. Lishizhen Medicine And Materia Medica Research 21, 2129–2132 (2010).
Millar, A. H., Carrie, C., Pogson, B. & Whelan, J. Exploring the function-location nexus: using multiple lines of evidence in defining the subcellular location of plant proteins. Plant Cell 21, 1625–1631 (2009).
Pesch, M., Schultheiß, I., Digiuni, S., Uhrig, J. F. & Hülskamp, M. Mutual control of intracellular localisation of the patterning proteins AtMYC1, GL1 and TRY/CPC in Arabidopsis. Development 140, 3456–3467 (2013).
Yin, Y. et al. Functional analysis of CsCBF3 transcription factor in tea plant (Camellia sinensis) under cold stress. Plant Growth Regulation 80, 335–343 (2016).
Knobloch, K. H. & Hahlbrock, K. Isoenzymes of p-Coumarate: CoA Ligase from Cell Suspension Cultures of Glycine max. European Journal of Biochemistry 52, 311–320 (1975).
Grand, C., Boudet, A. & Boudet, A. M. Isoenzymes of hydroxycinnamate: CoA ligase from poplar stems properties and tissue distribution. Planta 158, 225–229 (1983).
Jin, X. Q., Chen, Z. W., Tan, R. H., Zhao, S. J. & Hu, Z. B. Isolation and functional analysis of 4-coumarate:coenzyme A ligase gene promoters from Salvia miltiorrhiza. Biologia Plantarum 56, 261–268 (2011).
Shu-JuanZhao, Z.-B. H., DiLiu & Leung, F. C. Two Divergent Members of 4-Coumarate: Coenzyme A Ligase from Salvia miltiorrhiza Bunge: cDNA Cloning and Functional Study. Journal of Integrative Plant Biology 48, 1355–1364 (2006).
Ohta, M., Matsui, K., Hiratsu, K., Shinshi, H. & Ohme-Takagi, M. Repression domains of class II ERF transcriptional repressors share an essential motif for active repression. Plant Cell 13, 1959–1968 (2001).
Albert, N. W. & Schwinn, K. E. A conserved network of transcriptional activators and repressors regulates anthocyanin pigmentation in eudicots. Plant Cell 26, 962–980 (2014).
Nemiefeyissa, D., Olafsdottir, S. M., Heidari, B. & Lillo, C. Nitrogen depletion and small R3-MYB transcription factors affecting anthocyanin accumulation in Arabidopsis leaves. Phytochemistry 98, 34–40 (2014).
Skorupinska-Tudek, K. et al. Contribution of the mevalonate and methylerythritol phosphate pathways to the biosynthesis of dolichols in plants. Journal of Biological Chemistry 283, 21024–21035 (2008).
Lu, X., Tang, K. & Li, P. Plant Metabolic Engineering Strategies for the Production of Pharmaceutical Terpenoids. Frontiers in Plant Science 7, 1647 (2016).
Zhang, X. et al. Research progress of the regulation on active compound biosynthesis by the bHLH transcription factors in plants. Yao xue xue bao=Acta pharmaceutica Sinica 49, 435–442 (2014).
Pireyre, M. & Burow, M. Regulation of MYB and bHLH transcription factors: a glance at the protein level. Molecular Plant 8, 378–388 (2015).
Zimmermann, I. M., Heim, M. A., Weisshaar, B. & Uhrig, J. F. Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. Plant Journal 40, 22–34 (2004).
Chen, H., Chen, F., Zhang, Y. L. & Song, J. Y. Production of lithospermic acid B and rosmarinic acid in hairy root cultures of Salvia miltiorrhiza. Journal of Industrial Microbiology & Biotechnology 22, 133–138 (1999).
Liang, Z.-S. et al. Roles of reactive oxygen species in methyl jasmonate and nitric oxide-induced tanshinone production in Salvia miltiorrhiza hairy roots. Plant Cell Reports 31, 873–883 (2012).
Peng, L. et al. Spaceflight environment-induced variation in root yield and active constituents of Salvia miltiorrhiza. Planta Medica 80, 1029–1035 (2014).
Li, C. et al. Investigation of Plant Species with Identified Seed Oil Fatty Acids in Chinese Literature and Analysis of Five Unsurveyed Chinese Endemic Species. Frontiers in Plant Science 8, 224 (2017).
Shi, M. et al. Increased accumulation of the cardio-cerebrovascular disease treatment drug tanshinone in Salvia miltiorrhiza hairy roots by the enzymes 3-hydroxy-3-methylglutaryl CoA reductase and 1-deoxy-Dxylulose 5-phosphate reductoisomerase. Functional & Integrative Genomics 14, 603–615 (2014).
Nerantzaki, A. A., Tsiafoulis, C. G., Charisiadis, P., Kontogianni, V. G. & Gerothanassis, I. P. Novel determination of the total phenolic content in crude plant extracts by the use of 1H NMR of the –OH spectral region. Analytica Chimica Acta 688, 54–60 (2011).
Jia, Z., Tang, M. & Wu, J. The determination of flavonoid contents in mulberry and their scavenging effects on superoxide radicals. Food Chemistry 64, 555–559 (1999).
Acknowledgements
The authors would like to thank Prof. Cathie Martin (John Innes Centre, UK) for providing vectors. This work was funded by the National Natural Science Foundation of China (81373908 and 31670295) and the Chinese Universities Scientific Fund (2452017159).
Author information
Authors and Affiliations
Contributions
P.M. and Z.L. designed and supervised this study; K.D., T.P., Z.B. and Y.J. performed the experiments; K.D. and T.P. performed the data analysis; K.D. wrote this manuscript; P.M. and Z.L. revised the manuscript; all authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ding, K., Pei, T., Bai, Z. et al. SmMYB36, a Novel R2R3-MYB Transcription Factor, Enhances Tanshinone Accumulation and Decreases Phenolic Acid Content in Salvia miltiorrhiza Hairy Roots. Sci Rep 7, 5104 (2017). https://doi.org/10.1038/s41598-017-04909-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-04909-w
This article is cited by
-
Comparative transcriptome analysis and identification of candidate R2R3-MYB genes involved in anthraquinone biosynthesis in Rheum palmatum L.
Chinese Medicine (2024)
-
Contemporary understanding of transcription factor regulation of terpenoid biosynthesis in plants
Planta (2024)
-
Establishment and validation of a callus tissue transformation system for German chamomile (Matricaria chamomilla L.)
BMC Plant Biology (2023)
-
Biological Function and Stress Response Mechanism of MYB Transcription Factor Family Genes
Journal of Plant Growth Regulation (2023)
-
The diurnal emission of floral scent in Oncidium hybrid orchid is controlled by CIRCADIAN CLOCK ASSOCIATED 1 (CCA1) through the direct regulation on terpene synthase
BMC Plant Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
