
Abstract
The present study describes the kidney transcriptome of Labeo rohita, a freshwater fish, exposed to gradually increased salinity concentrations (2, 4, 6 and 8ppt). A total of 10.25 Gbps data was generated, and a suite of bioinformatics tools, including FEELnc, CPC2 and BLASTn were employed for identification of long non-coding RNAs (lncRNAs) and micro RNAs (miRNAs). Our analysis revealed a total of 170, 118, 99, and 269 differentially expressed lncRNA and 120, 118, 99, and 124 differentially expressed miRNAs in 2, 4, 6 and 8 ppt treatment groups respectively. Two competing endogenous RNA (ceRNA) networks were constructed i.e. A* ceRNA network with up-regulated lncRNAs and mRNAs, down-regulated miRNAs; and B* ceRNA network vice versa. 2ppt group had 131 and 83 lncRNA-miRNA-mRNA pairs in A* and B* networks, respectively. 4ppt group featured 163 pairs in A* network and 191 in B* network, while the 6ppt had 103 and 105 pairs. 8ppt group included 192 and 174 pairs. These networks illuminate the intricate RNA interactions in freshwater fish to varying salinity conditions.
Similar content being viewed by others

Background & Summary
Osmoregulation is a crucial mechanism in fishes to adapt against acute or chronic changes in environmental salinity. Kidney is one of the crucial osmoregulatory organs in fishes to maintain an osmotic balance of body fluids through water influx or efflux1. In freshwater fishes, kidney excrete large volumes of hypotonic urine and reabsorb active ions to maintain ionic homeostasis2. Among three major carp species in India, Labeo rohita (rohu) is the most important freshwater fish. The species have higher consumer demand and economic value3. In recent years, the impact of climate change causes an increase in salinity levels in freshwater resources4, significantly impacting aquatic organisms’ physiology5,6. In a salinity-fluctuating environment, maintaining internal osmotic and ionic homeostasis and adapting to salinity changes involves participating in various enzymes and transporters7,8. The first step toward elucidating molecular mechanisms and core physiological processes behind salinity change is identifying the candidate genes involved9.
The non-coding RNAs, such as miRNAs and lncRNAs, are reported to be regulators of mRNAs at the transcriptional and post-transcriptional levels10,11. The competitive endogenous (ceRNA) hypothesis demonstrates that lncRNAs can act as endogenous sponges to regulate mRNAs expression by negatively mediating miRNAs expression12,13. There were previous reports focused on role of miRNAs in the regulation of osmotic pressure14, salinity stress15, and immune response16, also studies on lncRNA regulation of mRNAs under adverse environmental conditions17. In order to find possible immune response regulators that could be challenged by the pathogenic bacterium Aeromonas salmonicida, ceRNA analysis was conducted in Atlantic salmon16.
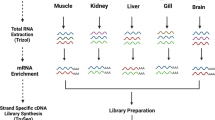
In the present study, L. rohita was treated with 2, 4, 6 and 8ppt salinity concentration and kidney tissue samples were processed for transcriptome sequencing. Differentially expressed mRNAs, miRNAs and lncRNAs were identified. Based on target prediction and correlation analysis ceRNA network was generated. This data will be helpful to the research community in understanding the physiology of fish in hypersaline conditions. The schematic representation of study design and workflow is presented in Fig. 1.

Study design of the kidney transcriptome profile under hypersaline environment.
Methods and Results
Ethical approval
All the experimental protocols were approved by Institute biosafety committee of PGIFER (Postgraduate Institute of Fisheries Education and Research), Kamdhenu University, Gandhinagar, Gujarat. The guidelines of the CPCSEA (Committee for the Purpose of Control and Supervision of Experiments on Animals, Ministry of Environment and Forests (Animal Welfare Division) on care and use of animals and ARRIVE2.0 (Animal Research: Reporting of In Vivo Experiments) in scientific research were followed during the experiment.
Sample collection and library preparation
The salinity stress experiment was conducted at Postgraduate Institute of Fisheries Education and Research (PGIFER), Kamdhenu University, Himmatnagar, Gujarat. Fingerlings (>10 g) were acquired from the State Fisheries Department Fish Hatchery, Gujarat. They were kept in 150-liter tanks with continuous aeration at 27 ± 5 °C. The fish were fed at 5% of the body weight till the end of the experiment, and 25% water was replaced each day, along with feces, to keep the tanks clean. The fingerlings were randomly split into control and salinity treatment groups. The control group was constantly maintained at 0ppt whereas in the treatment group the salinity was gradually raised (1ppt/day) to 2, 4, 6 and 8 ppt salinity by adding (55 ppt) of Red Sea Coral Pro Salt (Red Sea, USA). Each week the fish were gradually transferred to increased salinity and 3 fish were randomly euthanized and tissue samples were collected from the control and treatment groups. The samples were stored at −80 °C in RNAlater® until further use. The total RNA was extracted from the kidney tissues using RNeasy Plus Mini Kit (Qiagen, Germany). The integrity and quality of RNA were assessed with Agilent 2100 Bioanalyzer system (Agilent technologies, Ca) and Qubit 4 Fluorometer (Thermo Fisher Scientific, United States). The cDNA libraries were prepared by TruSeq Stranded Total RNA Library Prep Kit (Illumina, Ca) after removing ribosomal RNA with RiboMinus™ Eukaryote System v2 (Thermo Fisher, Ma). The samples were sequenced on an Illumina MiSeq and NovaSeq 6000 platform with paired-end forward and reverse reads.
Data processing and expression analysis
A total of 10.25 Gbps data was generated and processed for a quality check using FastQC (v0.11.9). The reads were aligned with the NCBI reference genome Rohu (Labeo rohita) (GenBank assembly accession GCA_004120215.1 v1) using segemehl (v0.2.0-418), and expression levels of mRNAs were computed with featureCounts (v2.0.1). The expression matrix of mRNA genes from individual salinities was used in the DESeq 2 package for differential expression analysis. The significant DEGs were considered with p-value ≤ 0.05 | log2FoldChange ≥0.5 for the enrichment and pathway analysis. The data was visualized using the ggplot2 package for each salinity-treated group. The detailed results of expression profile of transcriptome of kidney and significant mRNAs can be found in our previously published study18.
Prediction of putative lncRNAs
For identification of lncRNAs, transcripts were de-novo assembled with Cufflinks version (v2.2.1) using aligned bam files from individual samples. Cuffmerge was used to obtain a combined assembly, which was then processed through FEELnc pipeline (v.0.2.1) (https://github.com/tderrien/FEELnc)19. FEELncfilter was initially utilized to filter out transcripts less than 200 bp, including single-exon transcripts. Next, FEELnccodpot was used to evaluate the coding potential of each transcript based on the length of ORF, sequence bias, and transcript length to differentiate lncRNA from mRNA. Of 37,462 transcripts, 4,170 potential candidate lncRNAs were identified from the FEELnc program. Subsequently, FEELncclassifier was used to classify the identified lncRNA into genic, intergenic, containing, same strand, convergent, divergent, overlapping, and nested categories (Fig. 2). Finally, CPC2 (v0.1) (http://cpc2.gao-lab.org/) was utilized as an additional assessment method for the identification of the coding potential of transcripts, which uses a support vector machine (svm)20, 1,447 non-coding lncRNA were finalized, and an input matrix was prepared with featureCounts using GTF file of lncRNA for differential expression analysis using DESeq2 package in R software (v 4.2.3). In the 2, 4, 6, and 8ppt salinity groups, 170, 118, 99, and 269 differentially expressed lncRNA with p-value ≤ 0.05 & Log2FoldChange ≥0.5, respectively (Figs. 3, 4 and Figshare Dataset 121).

Bar plot representing various classes of lncRNAs predicted in kidney transcriptome of Labeo rohita.

Bar plot depicting no. of differentially expressed mRNAs, lncRNAs and miRNAs identified in 2, 4, 6, and 8ppt salinity treated groups.

Volcano plot representing differentially expressed (A) long non-coding RNAs (lncRNAs) (B) micro RNAs (miRNAs) at 2, 4, 6, and 8ppt salinity treated groups. The red and blue colour indicates up and down regulated genes respectively, whereas the grey colour indicates non-significant genes.
Prediction of putative miRNAs
To identify miRNAs, fasta file was prepared from raw fastq using the fastx toolkit (https://github.com/agordon/fastx_toolkit). The collapsed reads function from the mirdeep2 package was implemented to identify miRNA sequence whose length varies between (16 to 24 bp) which is shorter than the sequence read length. The standalone BLASTn tool was implemented for the identification of putative mature miRNA sequences obtained from the miRbase database (https://www.mirbase.org), for teleostei species with E-value (1E-1) and percent identity ≥ 95 as a cut-off. The Differential expression analysis of miRNAs was performed using EdgeR package from Bioconductor (v.3.40.2)22. A total of 120, 118, 99, and 124 differentially expressed miRNAs with p-value ≤ 0.05 and log2FoldChange ≥0.5 were considered (Figs. 3, 4 and Figshare Dataset 221).
Identification of target mRNAs for lncRNA and miRNA
LncRNAs competitively bind microRNAs to alter the expression of specific mRNAs23. The targeted mRNAs were predicted for miRNAs and lncRNAs using miRanda (v3.3a) (http://www.microrna.org/microrna/home.do)24, which uses scoring matrix for the individual alignment for detection of potential target sites in coding sequences, with score cutoff ≥ 145 and energy ≤ −1025 to predict lncRNA-miRNA pairs and miRNA-lncRNA pairs. A total of 953, 863, 494 and 1983 lncRNA-miRNA pairs and 766, 869, 532, and 1226 miRNA-mRNA pairs were identified in 2, 4, 6, and 8ppt salinity treated groups, respectively (Figshare Dataset 321). Correlation between lncRNA and miRNA was calculated using corr.test() function by R software. LncRNA-miRNA pairs using Pearson correlation coefficients (PCC) with | r | ≥ 0.94 and p-value ≤ 0.05 were selected. A total of 10,999; 20,341; 7,575; 36,919 significant lncRNA-mRNA pairs were identified in 2, 4, 6, and 8ppt groups respectively. These lncRNA-mRNA pairs include 159 lncRNAs and 152 mRNAs, 118 lncRNAs and 351 mRNAs, 99 lncRNAs and 155 mRNAs, and 268 lncRNAs and 279 mRNAs in 2, 4, 6, and 8ppt groups respectively (Figshare Dataset 421).
Construction of ceRNA network
Among the predicted, lncRNA-miRNA and miRNA-mRNA pairs, under the stipulation that both lncRNA and mRNA are concurrently targeted by the same miRNA and display a negative co-expression. Those pairs were considered to construct lncRNA-miRNA-mRNA network, and the network topology was graphically depicted using the Cytoscape software (v3.9.1) for visualization and subsequent analysis. According to the ceRNA hypothesis, ceRNAs (lncRNA and mRNA) have positive correlation expression by competing for the same miRNA, which is negatively correlated. Thus, two different ceRNA networks were constructed for each treatment group consisting of, i.e., (1.) up-regulated lncRNAs and mRNAs, and down-regulated miRNAs (A* ceRNA network) and (2.) down-regulated lncRNAs and mRNAs, and up-regulated miRNAs (B* ceRNA network). Both positive and negative correlation pairs were identified based on log2FoldChange values. In the 2ppt treatment group, the A* integrated network contains 131 lncRNA-miRNA-mRNA pairs which include 64 lncRNAs, 36 miRNAs, and 31mRNAs and the B* integrated network contains 83 lncRNA-miRNA-mRNA pairs, including 41 lncRNAs 16 miRNAs and 26mRNAs (Fig. 5 and Figshare Dataset 521).

ceRNA (lncRNA-miRNA-mRNA) network of 2ppt salinity treated group. (A) The A* ceRNA network. Blue, orange, and green colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 424 edges, and 83 nodes consists of 64, 36 and 31 lncRNAs, miRNAs, and mRNAs respectively. (B) The B* ceRNA network. Pink, blue, and yellow colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 880 edges, and 131 nodes consists of 41, 16 and 26 lncRNAs, miRNAs and mRNAs respectively.
In 4ppt, A* network contains 163 lncRNA-miRNA-mRNA pairs, including 43 lncRNAs, 40 miRNAs, and 80 mRNAs. B* network includes 191 lncRNA-miRNA-mRNA pairs which include 53 lncRNAs, 60 miRNAs, and 78 mRNAs (Fig. 6 and Figshare Dataset 5 25). In 6ppt, A* network includes 103 lncRNA-miRNA-mRNA pairs which include 43 lncRNAs, 38 miRNAs, and 22 mRNAs. B* network contains 105 lncRNA-miRNA-mRNA pairs with 42 lncRNAs, 33 miRNAs, and 30 mRNAs (Fig. 7 and Figshare Dataset 5 25). In 8ppt, A* network contains 192 lncRNA-miRNA-mRNA pairs which include 103 lncRNAs, 23 miRNAs, and 66 mRNAs. B* network contains 174 lncRNA-miRNA-mRNA pairs which include 111 lncRNAs, 24 miRNAs, and 39 mRNAs (Fig. 8 and Figshare Dataset 5 25).

ceRNA (lncRNA-miRNA- mRNA) network of 4ppt salinity treated group. (A) The A* ceRNA network. Blue, orange, and green colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 2023 edges, and 191 nodes consists of 43, 40 and 80 lncRNAs, miRNAs, and mRNAs respectively. (B) The B* ceRNA network. Pink, blue, and yellow colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 1139 edges, and 163 nodes consists of 53, 60 and 78 lncRNAs, miRNAs and mRNAs respectively.

ceRNA (lncRNA-miRNA- mRNA) network of 6ppt salinity treated group. (A) The A* ceRNA network. Blue, orange, and green colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 592 edges, and 105 nodes consists of 43, 38 and 22 lncRNAs, miRNAs, and mRNAs respectively. (B) The B* ceRNA network. Pink, blue, and yellow colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 477 edges, and 103 nodes consists of 42, 33 and 30 lncRNAs, miRNAs and mRNAs respectively.

ceRNA (lncRNA-miRNA- mRNA) network of 8ppt salinity treated group. (A) The A* ceRNA network. Blue, orange, and green colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 1746 edges, and 174 nodes consists of 103, 23 and 66 lncRNAs, miRNAs, and mRNAs respectively. (B) The B* ceRNA network. Pink, blue, and yellow colour circles represent lncRNA, miRNA and mRNA respectively. Network includes 3236 edges, and 192 nodes consists of 111, 24 and 39 lncRNAs, miRNAs and mRNAs respectively.
Functional enrichment of the ceRNA network
The functional enrichment and pathway analysis was performed using DAVID (https://david.ncifcrf.gov/). The significantly enriched terms classified in BP, CC, MF, and KEGG pathways were considered for identifying differentially expressed genes involved in salinity stress (Fig. 9 and Figshare Dataset 621).

David enrichment plot for differentially expressed genes involved in ceRNA network for 2, 4, 6, and 8ppt treatment groups.
Data Records
The raw FASTQ files were submitted to NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP384125 (2022)26. The files of differentially expressed mRNAs are published18. The tables representing the information of predicted putative lncRNAs and miRNAs, identified lncRNA-miRNA, miRNA-mRNA, lncRNA-miRNA pairs, lncRNA-miRNA-mRNA pairs, dataset used for RNA network and enrichment analysis are deposited on Figshare21. The Labeo rohita reference genome assembly and annotation used in this study are available on NCBI (https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_004120215.1) (2019)27.
Technical Validation
RNA quality and integrity assessment
RNA quality was assessed using QIAxpert instrument (QIAGEN, Germany). A260/A280 ratio was ranged from 1.93–2.09, which is acceptable range. The quantity and integrity of the RNA were assessed with the Qubit 4 Fluorometer (Thermo Fisher Scientific, United States) and Agilent 2100 Bioanalyzer system (Agilent technologies, California, United States), respectively.
RNA-seq data quality assessment
The raw fastq files were assessed for per base sequence quality, Phred sore, GC content and sequence duplication levels using FASTQC tool (v0.11.9).
Code availability
The following software’s and tools were used in this manuscript. No custom code was utilized during the analysis of the study.
fastx toolkit (v0.0.14) https://github.com/agordon/fastx_toolkit
segemehl (v0.2.0–418) http://legacy.bioinf.uni-leipzig.de/Software/segemehl
STAR (v2.7.4a) https://github.com/alexdobin/STAR
miRbase database https://www.mirbase.org
miRanda (v3.3a) (http://www.microrna.org/microrna/home.do)
FEELnc pipeline (v.0.2.1) https://github.com/tderrien/FEELnc
CPC2 (v0.1) http://cpc2.gao-lab.org/
DESeq 2 (v.1.38.3) https://bioconductor.org/packages/release/bioc/html/DESeq2.html
EdgeR (v.3.40.2) https://bioconductor.org/packages/release/bioc/html/edgeR.html
featureCounts (v2.0.1) https://github.com/ShiLab-Bioinformatics/subread
psych package (corr.test() for Pearson correlation coefficient)
References
Baldisserotto, B. Fish osmoregulation. (CRC Press, 2019).
Greenwell, M. G., Sherrill, J. & Clayton, L. A. Osmoregulation in fish: mechanisms and clinical implications. Vet. Clin. Exot. Anim. Pract. 6, 169–189 (2003).
Prasad, U., Satanand, P., Prasad, P. D. & Amitabh, P. Length weight relationship and condition factor of Labeo rohita in Govindgarh Lake, Rewa (MP). Indian J. Res 1, 185–187 (2012).
Haque, R., Parr, N. & Muhidin, S. Climate-related displacement, impoverishment and healthcare accessibility in mainland Bangladesh. Asian Popul. Stud. 16, 220–239 (2020).
Chong-Robles, J. et al. Osmoregulation pattern and salinity tolerance of the white shrimp Litopenaeus vannamei (Boone, 1931) during post-embryonic development. Aquaculture 422, 261–267 (2014).
Yang, W.-K., Chung, C.-H., Cheng, H. C., Tang, C.-H. & Lee, T.-H. Different expression patterns of renal Na+/K+-ATPase $α$-isoform-like proteins between tilapia and milkfish following salinity challenges. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 202, 23–30 (2016).
Hwang, P. P. & Lee, T. H. New insights into fish ion regulation and mitochondrion-rich cells. Comp. Biochem. Physiol. - A Mol. Integr. Physiol. 148, 479–497 (2007).
Tseng, Y.-C. & Hwang, P.-P. Some insights into energy metabolism for osmoregulation in fish. Comp. Biochem. Physiol. Part C Toxicol. \& Pharmacol. 148, 419–429 (2008).
Zhang, X. et al. RNA-Seq analysis of salinity stress–responsive transcriptome in the liver of spotted sea bass (Lateolabrax maculatus). PLoS One 12, e0173238 (2017).
Qu, K. et al. MicroRNAs: key regulators of endothelial progenitor cell functions. Clin. Chim. acta 448, 65–73 (2015).
Nelson, B. R. et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science (80-.). 351, 271–275 (2016).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146, 353–358 (2011).
Guo, L.-L. et al. Competing endogenous RNA networks and gastric cancer. World J. Gastroenterol. 21, 11680 (2015).
Yan, B., Zhao, L.-H., Guo, J.-T. & Zhao, J.-L. miR-429 regulation of osmotic stress transcription factor 1 (OSTF1) in tilapia during osmotic stress. Biochem. Biophys. Res. Commun. 426, 294–298 (2012).
Tian, Y., Shang, Y., Guo, R., Chang, Y. & Jiang, Y. Salinity stress-induced differentially expressed miRNAs and target genes in sea cucumbers Apostichopus japonicus. Cell Stress Chaperones 24, 719–733 (2019).
Xia, Y. et al. Genome-wide integrated analysis reveals functions of lncRNA-miRNA-mRNA interactions in Atlantic salmon challenged by Aeromonas salmonicida. Genomics 114, 328–339 (2022).
Huo, D. et al. The regulation mechanism of lncRNAs and mRNAs in sea cucumbers under global climate changes: Defense against thermal and hypoxic stresses. Sci. Total Environ. 709, 136045 (2020).
Harshini, V. et al. Kidney transcriptome response to salinity adaptation in Labeo rohita. Front. Physiol. 2183 (2022).
Wucher, V. et al. FEELnc: a tool for long non-coding RNA annotation and its application to the dog transcriptome. Nucleic Acids Res. 45, e57–e57 (2017).
Kang, Y.-J. et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 45, W12–W16 (2017).
Shukla, N. Dataset: lncRNA-miRNA-mRNA network in kidney transcriptome of Labeo rohita under hypersaline environment. Figshare https://doi.org/10.6084/m9.figshare.24770961 (2023).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. bioinformatics 26, 139–140 (2010).
Ai, L.-Y. et al. Integrated Analysis of lncRNA and mRNA Expression Profiles Indicates Age-Related Changes in Meniscus. Front. Cell Dev. Biol. 10, (2022).
Wong, N. & Wang, X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 43, D146–D152 (2015).
Zhang, Y. & Verbeek, F. J. Comparison and integration of target prediction algorithms for microRNA studies. J. Integr. Bioinform. 7, 169–181 (2010).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP384125 (2022).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_004120215.1 (2019).
Acknowledgements
Not applicable. The project was funded by the Government of Gujarat-Department of Science and Technology (GOG-DST), project reference no: MB-18.
Author information
Authors and Affiliations
Contributions
N.S.–primarily involved in the wet laboratory work, data analysis and writing of the manuscript; V.H.–wet laboratory work, literature review, writing of the manuscript; I.R.–troubleshooting in the wet laboratory work, guidance in manuscript writing, and review; A.P.–experimental design, troubleshooting in the wet laboratory work, and manuscript proof-reading; C.J.–experimental design, guidance in the data analysis, and manuscript proof-reading.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shukla, N., Harshini, V., Raval, I. et al. lncRNA-miRNA-mRNA network in kidney transcriptome of Labeo rohita under hypersaline environment. Sci Data 11, 226 (2024). https://doi.org/10.1038/s41597-024-03056-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03056-y
