
Abstract
Beyond transferring genetic information, RNAs are molecules with diverse functions that include catalyzing biochemical reactions and regulating gene expression. Most of these activities depend on RNAs’ specific structures. Therefore, accurately determining RNA structure is integral to advancing our understanding of RNA functions. Here, we summarize the state-of-the-art experimental and computational technologies developed to evaluate RNA secondary and tertiary structures. We also highlight how the rapid increase of experimental data facilitates the integrative modeling approaches for better resolving RNA structures. Finally, we provide our thoughts on the latest advances and challenges in RNA structure determination methods, as well as on future directions for both experimental approaches and artificial intelligence-based computational tools to model RNA structure. Ultimately, we hope the technological advances will deepen our understanding of RNA biology and facilitate RNA structure-based biomedical research such as designing specific RNA structures for therapeutics and deploying RNA-targeting small-molecule drugs.
Similar content being viewed by others

Main
RNA was once conceptualized as a passive passenger for the delivery of genetic information recorded in DNA to the functional products—proteins. However, this view has been changed since the discoveries that RNA can function as catalytic ribozymes, as temperature-sensing and metabolite-sensing riboswitches, and as epigenetically regulatory long noncoding RNAs (lncRNAs), among others1,2,3. These diverse functions, are based on the ability of single-stranded RNA molecules to fold into diverse secondary and tertiary structures4,5. Moreover, it has been reported that mutations disrupting RNA structures can be associated with human diseases such as repeat expansion disorders, retinoblastoma and breast cancer6. The ability to characterize RNA folding and structure is therefore essential to advance our understanding of the diverse functions of RNA.
RNA molecules first fold into secondary structures in a process dominated by canonical Watson–Crick and wobble base pairing, before further folding into tertiary structures, driven by interactions among secondary structural elements (Box 1). It is notable that most structural studies focused on a small number of known functional RNAs, and were conducted in vitro, mainly using X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and more recently cryo-electron microscopy (cryo-EM), small-angle X-ray scattering (SAXS) and gel electrophoresis-based probing methods7.
These RNA structure determination efforts have deepened our understanding of the mechanisms underlying various biological processes. For example, resolving the structure of the translation machine—the ribosome—has revealed that rRNAs both provide a scaffold and form the catalytic core of the ribosome where the nascent peptide synthesis occurs. Moreover, determining the structures of the riboswitches has unveiled fascinating modular architectures and enabled elucidation of the molecular recognition that these biomolecules used to regulate gene expression1. However, the limited scope of known RNA structures obtained so far has led to an incomplete picture of RNA structure and folding in cells.
Efforts over the last decade have developed a new generation of deep sequencing-based RNA structure probing methods with profoundly increased throughput, which have enabled transcriptome-wide structural profiling in vitro8,9 and in vivo10,11,12. These methods have uncovered distinct functions of RNA structures in gene regulation. For instance, global RNA structure maps in Escherichia coli revealed that mRNA translation efficiency is regulated by the unfolding kinetics of mRNA structures overlapping the ribosomal binding site13. During zebrafish development, the structures in the 3′ untranslated region can regulate maternal RNA degradation by modulating microRNA activity14 and RNA-binding protein (RBP) binding15. In cellular innate immunity, circular RNAs with 16–26-bp imperfect RNA duplexes can act as inhibitors of double-stranded RNA (dsRNA)-activated protein kinase (PKR)16. Interestingly, overexpression of the dsRNA-containing circular RNA in T cells can alleviate aberrant PKR activation in the autoimmune disease systemic lupus erythematosus16. The structural organization of the entire HIV-1 RNA genome modulates ribosome elongation to regulate native protein folding17, and alternative RNA structures at splice sites have been shown to affect the abundance of different transcript isoforms18. Recently, several RNA structure probing studies focusing on resolving the structure of the RNA genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have helped uncover functional and structural elements that contribute to the virus’s translation, sub-genome generation and overall infectivity, and have helped identify therapeutic targets and drugs19.
Alongside experimental studies, there is a long tradition of developing computational methods for studying RNA structures20. However, many of these methods are based on assumptions about energy calculations in solution, and do not reflect how RNA molecules fold and function in cells6,21. More recently, methods have been developed to incorporate experimentally determined structural data into computational modeling to support functional analyses of RNAs in their physiologically relevant states; these tools have helped generate alternative structure models for viral RNA genomes18,22 and have supported the discovery of riboSNitches9.
Here, we review recent advances in experimental RNA structure probing methods and computational approaches for RNA structural prediction and modeling; we highlight the advantages of leveraging probing data for structure prediction and analysis. Whenever possible, we discuss the similarities in the methods used for studying RNA structure to the methods used to assess DNA and proteins. Finally, aiming to facilitate efficient communication between RNA experimentalists and computational experts, we consider several directions that deserve additional research efforts to increase the resolution and flexibility of probing methods and better harness machine learning tools for RNA structure research in basic biology and biomedical investigations.

Advances in experimental RNA structure determination
The experimental acquisition of high-resolution RNA structures has a long history (Box 2). X-ray crystallography and NMR have been used successfully to solve RNA structures (starting with the first RNA tertiary structure at atomic resolution in 1974; ref. 23), whereas NMR has remained mainly suitable for assessing small RNAs (typically fewer than 100 nucleotides). RNA crystals are required for X-ray crystallography, yet it is challenging to obtain appropriate RNA crystals owing to the intrinsic structural heterogeneity caused by their flexible backbones and weak long-range interactions7. Moreover, the SAXS method is capable of characterizing the low-resolution, overall shapes of RNA particles in solution (including large RNA molecules). Recent innovations in cryo-EM single-particle technologies have dramatically improved the resolution and capacity to solve macromolecule structures including RNA24. Despite all of these painstaking efforts, there are currently only 6,155 RNA-containing structures in the RCSB Protein Data Bank (PDB), accounting for fewer than 3.2% of the total number of structures (191,869, as of June 2022). And it is also noteworthy that the resolved structures have predominantly been short regulatory and enzymatic RNAs (for example, tRNA, rRNA and ribozyme). Although a few individual structural elements in mRNA and lncRNAs have been solved25, solving the full structure of long RNA molecules remains beyond our current reach.
In addition, these biophysical methods are hard to apply to study structural dynamics in living cells. This, together with the limited applicability of these methods for certain types of RNAs, have led to an incomplete picture of RNA structure and folding. There are now a large variety of RNA structure probing methods that variously combine enzymatic or chemical probes with deep sequencing for high-throughput studies of the RNA ‘structurome’. Broadly, these methods can be categorized into two major groups based on the type of structural information they obtain: footprinting-based methods and proximity ligation-based methods.

Footprinting-based RNA probing methods
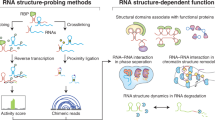
The general principle underlying footprinting-based methods is the use of probes to modify RNA in an RNA structure-specific manner8,10,11,12. These probes leave ‘footprints’ on RNA as a modified base, which can be subsequently captured by reverse transcription (RT) and read out by sequencing and analysis (Fig. 1a). Footprinting does not provide direct base-pairing information, but instead measures the probe reaction intensity with each nucleotide and calculates a reactivity score for each nucleotide (termed a structural score) to represent the probability of forming secondary structure base pairings.

a, The general workflow of footprinting-based probing methods. RNAs are first probed by chemical probes, which modify the nucleotides with structural preferences. The modification footprints then can be converted into RT truncations or RT mutations during RT and can be further read out by sequencing and analyses. Each nucleotide is finally assigned a reactivity score, which represents the frequency of modifications and in turn reflects its structural context. b, The general workflow of proximity ligation-based methods. RNAs are crosslinked at the base-pairing or interacting regions, and are then subjected to fragmentation. Proximity ligation is performed to connect base-pairing or interacting fragments. Usually, an enrichment step for the crosslinked ones is performed before or after proximity ligation. Finally, the information of base pairing or interaction can be recovered from chimeric reads in sequencing libraries.
To conduct footprinting-based RNA probing, users must make careful choices about probing reagents, chemical modification readout methods and the protocol for library construction as these factors strongly influence the structural information obtained (Supplementary Table 1). The base-specific chemical probes target the Hoogsteen and/or the Watson–Crick faces of particular unpaired (or exposed) bases. For example, dimethyl sulfate (DMS) interacts with N1 of adenine and N3 of cytosine and has been used for the development of methods including DMS-seq and Structure-seq10,11. N-Cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide have been used to probe RNA structures by modifying guanine and uracil in vitro26 and in vivo27,28 (Supplementary Table 2). Another category of chemical probes targets the RNA backbone and can thus assess structural information for all types of nucleotides. Among them, selective 2′-hydroxyl acylation detected by primer extension (SHAPE) reagents sense flexibility in the 2′-OH group of the sugar ring12,29 and have been used for the development of SHAPE-seq, SHAPE-MaP and in vivo click (ic)SHAPE12,30,31 (Supplementary Table 2).
SHAPE reagents are able to provide structural information of all four bases and therefore provide an advantage over base-specific probes. However, the reactivity scores obtained from SHAPE reagents rely on the local flexibility of the 2′-OH for each base, which can be affected by base stacking in addition to base pairing32. Moreover, the reactivity of the probing reagents varies when used in different types of cell lines33. Notably, some reported probes (for example, NAI-N3 and N3-kethoxal) have dual functionality, for example, having the ability to couple biotin to help enrich the modified RNAs during library construction, making them attractive to users working with low-abundance RNAs or rare samples (as for example in difficult-to-obtain clinical samples)12.
Moreover, cell membrane permeability and instant RNA kinetic snap capacity are also relevant considerations when selecting appropriate probing reagents34. For example, to support in vivo structural probing, probes should have high cell membrane permeability and long reaction times (for example, DMS, NAI, NAI-N3, 5NIA and 2A3)10,11,12,33,34,35 (Supplementary Table 1).
Chemical modification signals can be read out as RT-truncation or RT-mutation signals10,11,12,13,18,36. In the ‘RT-truncation strategy’, footprints are read out as RT stops (that is, as the reverse transcriptase drops off when encountering the chemical adduct10,11,12). A more recent development is the ‘RT-mutation strategy’, which is based on the tendency of reverse transcriptase to mis-incorporate nucleotides instead of stopping at chemical adduct sites under specific reaction conditions13,18,36. The RT-mutation strategy allows detection of multiple footprints per cDNA molecule, and thus enables studies of RNA structural heterogeneity (that is, multiple conformations of a single RNA molecule) by grouping the reads based on mutation patterns18,22. However, both strategies were found to have bias in detecting DMS modifications: specifically RT mutations tend to occur on modified cytosines, while RT stops favor modified adenosines, and such bias is known to depend on both the reverse transcriptase used and the local structural context37.
For library construction, many protocols have been developed to improve the signal-to-noise ratio and to decrease the material input requirements (Supplementary Table 2). For example, Structure-seq2 uses hairpin adaptors to reduce the ligation bias and introduces biotinylated nucleotides during RT to allow for removal of unwanted by-products and to reduce the number of required PAGE purifications38. SmartSHAPE adds a biotinylated adaptor to cDNA to allow the downstream reactions to be performed in an ‘on beads’ manner, which obviates the need for PAGE purification, and incorporates RNase I digestion to remove the artifact signals of premature RT products. These improvements collectively enable smartSHAPE to investigate samples with very small RNA input concentrations39. The abovementioned methods are all based on short-read sequencing, which precludes us from analyzing structure with its full-length origin. More recently, new methods were developed by combining chemical probing and direct long-read RNA sequencing using Nanopore, such as PORE-cupine40 and nanoSHAPE41; these methods enable us to phase alternative structures for long transcripts.
Proximity ligation-based RNA probing methods
Footprinting-based methods capture only the base-pairing tendencies of a nucleotide; in contrast, proximity ligation-based RNA probing methods can obtain partner information (base-pairing and interaction data) within an RNA (intramolecular RNA structure) or between two RNA molecules (intermolecular RNA–RNA interactions)42,43,44,45,46,47,48. Typically, these methods first crosslink interacting RNA pairs, after which RNAs are fragmented, and interacting RNA pairs are then ligated to form chimeric molecules, which can be identified after sequencing and bioinformatics analyses to represent the interacting RNA fragments (Fig. 1b and Supplementary Table 2).
These methods can be roughly categorized into two groups: base-pairing dependent and protein centric. Base-pairing-dependent methods were developed mainly based on psoralen-mediated or psoralen-derivative-mediated crosslinking of two direct base-paired fragments42,43,44,45. These methods differ in strategies for enriching crosslinked fragments, a step that strongly influences the signal-to-noise ratio. Strategies used to date include two-dimensional (2D) polyacrylamide gel electrophoresis (as in PARIS)43, biotin-psoralen for streptavidin beads selection (SPLASH)42, RNase R (LIGR-seq)44 and antisense oligonucleotides (COMRADES)45. Notably, these methods may suffer from a low proximity ligation rate, and from spurious ligation. The crosslinker psoralen, known to preferentially crosslink staggered uridines and RBPs, can block its crosslinking activity49. These limitations together can lead to noise and severe loss of information in the resulting data, thus limiting their capacity to detect biologically relevant interactions. Indeed, meta-analyses have reported limited overlaps between the interactions detected using SPLASH and PARIS, even from the same cell lines50. Notably, the recently developed reagents trans-bis-isatoic anhydride (TBIA) and dipicolinic acid imidazolide (DPI) have a 2′-hydroxyl acylation crosslinker that can react with two 2′-OH groups of single-stranded nucleotides in proximity51,52. SHAPE-JuMP uses TBIA to capture nucleotide pairing and uses an engineered reverse transcriptase that ‘jumps’ across crosslinked nucleotides to obviate the need for proximity ligation51. SHARC (spatial 2′-hydroxyl acylation reversible crosslinking) drastically improves crosslinking efficiency to >90% using DPI, increases the detection resolution of pairing regions by exonuclease trimming, and enables transcriptome-wide analysis of spatial distances in cells52.
The protein-centric methods aim to detect RNA interactions mediated by proteins. These methods can be further classified into two categories: methods that assess interactions with one or several proteins (using analyte-specific antibodies to purify proteins and associated RNAs, such as CLASH, hiCLIP and RIPPLiT46,47,48) and methods that attempt to reveal global interaction maps of all proteins (such as RPL, MARIO and RNA in situ conformation sequencing (RIC-seq))53,54,55. Notably, proximity ligation is usually a rate-limiting step due to its low efficiency, and a variety of improvement approaches have been invented. For example, RIC-seq uses in situ proximity ligation and increases the reaction time to increase the yield of the ligated products and to reduce spurious ligation55.
Footprinting-based methods only obtain a structural score of base-pairing probability for each nucleotide; and proximity ligation-based RNA probing methods only generate information for interacting RNA fragments. Each of these methods provides only partial information so computational methods (which we address below) are typically required to generate full models of RNA secondary structures.
Computational approaches for RNA structure prediction and modeling
RNA secondary structure modeling methods
In parallel to experimental methods for RNA structure probing, computational methods have also been developed to predict RNA secondary structures over the past decades. Herein, we classify these computational methods into knowledge-based methods and learning-based methods. The details of representative methods are shown in Table 1.
Knowledge-based methods
Experimental work to characterize RNA structures has generated data from which researchers have gleaned principles about how RNA molecules fold into their intricate structures. These principles have in turn formed the basis for developing computational RNA secondary structure prediction methods; these knowledge-based prediction methods can be further categorized into energy-based methods and covariation-based methods.
Energy-based methods
Energy-based methods search for the thermodynamically most stable secondary structure of an analyte RNA molecule by minimizing free energy using dynamic programming algorithms (Fig. 2a). The calculation of the free energy is based on the experimentally determined parameters, synthesized into the ‘Tuner rules’, about how RNA folds20. Examples in this category include Mfold20, RNAstructure56, MC-fold57, RNAfold58, and so on. Generally speaking, energy-based methods have been at the forefront of RNA secondary structure prediction, and remained the most widely used methods to date. The main limitations of these methods are their increasing inaccuracy (owing to error accumulation in energy calculations) and computational complexity as the length of the analyte RNA increases, as well as their tendency to ‘overfold’ RNA structures and their inability to take into account key determinants of RNA folding in the context of living cells, such as the co-transcriptional nature of folding, protein binding or RNA modifications21,59,60. Concerning RNA modifications, we note that secondary structure prediction for RNA sequences containing N6-methyladenosine has been made possible61. So far, energy-based methods remain recommended for prediction of secondary structures of small RNA molecules or fragments (for example, <200 nucleotides), but caution is strongly warranted for longer RNA molecules.

a, The energy-based secondary structure modeling methods assume the native structure is the most thermodynamically stable structure and search the structure with minimum free energy using dynamic programming algorithms. b, The covariation-based secondary structure modeling methods fold the target sequence into a secondary RNA structure based on the assumption that base-pairing nucleotides tend to have coevolution, which could be identified as covarying positions from the alignment of multiple homologous RNA sequences. c, The learning-based secondary structure modeling methods typically use graph models (as deep neural networks in recent deep learning-based methods) to represent RNA structures.
Covariation-based methods
Covariation-based methods have been developed based on the understanding that the structurally and functionally relevant base pairings in RNA secondary structures tend to coevolve in sequence to maintain the consistency of an RNA’s structure (Fig. 2b). Examples include Dynalign II62, R-scape63, CaCofold64, and so on; these methods start by identifying covariations from an alignment of multiple homologous RNA sequences, and then fold the target sequence into a secondary RNA structure constrained with results from covariation analysis. Among them, R-scape and CaCofold are notable for their rigidity in evolutionary analyses and the evaluation of statistical significance for covariations. In general, covariation-based methods avoid the inaccuracies in energy calculation and are suitable for predicting functionally relevant RNA structures. The accuracy of covariation-based methods is heavily dependent on the quality of the multiple sequence alignment65,66; accordingly, several semiautomated approaches67,68 take advantage of the Infernal package69 to facilitate multiple sequence alignment construction.
As approaches based only on energy calculation or evolutionary analysis have their own limitations, integrative methods have been proposed to combine the strength of both. For example, RNAalifold70 and TurboFold II71 estimate RNA folding by considering both thermodynamic parameters and coevolution information from homologous sequences. These integrative methods frequently achieve higher prediction performance for a broad range of RNAs.
Learning-based methods
With the increase of RNA secondary structure data and the rapid development of artificial intelligence, learning-based strategies are gaining popularity in RNA secondary structure prediction (Supplementary Table 2). In general, learning-based methods use a model to represent the RNA secondary structures, with the ability to learn model parameters from the experimentally determined RNA structure data and, for a given input sequence, to predict RNA secondary structure based on the maximum probabilities (Fig. 2c).
Traditional machine learning-based methods
Traditional machine learning-based methods include ContextFold72, Pfold73, CONTRAfold74, TORNADO75, and so on (Fig. 2c). While models in early years only used a limited number of parameters, new methods have proposed feature-rich (~70,000 free parameters for ContextFold) scoring functions. These feature-rich models partially avoid the problem of error accumulation, and have achieved considerable success59,76. This trend toward ever-richer feature scope has been boosted by recently developed deep neural networks.
Deep learning-based methods
Deep learning-based methods are similar to traditional machine learning-based methods but use more complex neural networks. These methods can be traced back about a decade, and started with a multilayer perceptron approach77; however, this did not receive widespread attention, owing to its insufficient generalization ability. Notably, while most reported methods tend to be based on one type of neural network (for example, convolutional neural network (CNN), recurrent neural network, Transformer and U-Net) for structure predictions, as with CDPfold78, DMfold79, E2Efold80 and Ufold81 (Fig. 2c and Table 1), there are also now methods that combine technologies to improve their prediction accuracy. For example, SPOT-RNA82 trains an ensemble model comprising both residual neural networks (ResNets) and long short-term memory (LSTM) networks to help to capture the flexibility of RNA structures. SPOT-RNA and SPOT-RNA2 both use transfer learning to pretrain models based on a large dataset82,83, and refines the models with small, high-quality datasets; their developers reported that this refinement is particularly useful in avoiding the concern of overfitting complex deep neural networks onto the currently sparse data of high-quality RNA structures. In addition to transfer learning, MXfold2 (ref. 84) also used a strategy based on integrating thermodynamic parameters with RNA folding scores learnt from deep neural networks, an approach used previously in MXfold85 and SimFold86.
To date, knowledge-based methods have remained the mainstay for exploration of RNA structure through computational prediction, but learning-based methods are gaining popularity for their seemingly excellent performance in terms of prediction accuracy and computational efficiency (with Ufold, SPOT-RNA2 and MXfold2 as the best performers)81,83,84. However, in contrast to knowledge-based methods, where the energy terms or parameters used are estimated from experiments or evolution, learning-based methods learn model parameters from a small set of known structures, for example, PDB, Archive II87, RNAstralign71 and bpRNA88. The inevitable bias toward certain RNA types in the small training set could potentially cause overfitting of model parameters; and such parameters often lack biophysical or evolutionary meaning, making it difficult to generalize across different RNA families89. Moreover, it should be noted that the assessments were typically performed by the research groups that developed those prediction methods; our opinion is that third-party assessments, as in CompaRNA90 and RNA-Puzzles91, are essential for bias-free evaluations to support the best practice guidelines.
RNA tertiary structure modeling methods
As noted above, due to the intrinsic flexibility of RNA structures, knowledge about how RNA folds in 3D space is very limited (relative to solved protein tertiary structures). As a consequence, the development of prediction tools for RNA tertiary structures lags far behind that for protein structures. Nevertheless, there exists several representative methods, which could be classified into three categories (so as to methods for protein tertiary structure prediction), and the details of representative methods can be found in Table 1.
Ab initio folding methods
Ab initio folding methods calculate the most stable tertiary structures from the unfolded conformation of an RNA molecule based on knowledge-based energy functions derived from known RNA structures (Fig. 3a). Examples include iFold92 and SimRNA93. Briefly, these methods use a coarse-grained representation of each residue while preserving the physical and chemical properties of RNA molecules. Unlike iFold, which simulates RNA folding based on discrete molecular dynamics and replica exchange molecular dynamics separately, SimRNA instead uses a replica exchange Monte Carlo scheme, which simulates potential folding of RNA. Although these approaches (especially SimRNA) have been shown to perform well in solving RNA tertiary structures for certain RNAs68,94, the oversimplified representation of RNA molecules does not consider high-resolution, atomic-level structural information.

a, The ab initio folding methods often use a coarse-grained representation of each nucleotide and then perform sampling or molecular dynamics to generate the optimal RNA conformation. b, The fragment assembly methods search for possible structures for a fragment from a library constructed from known structures and then assemble them into full structural models. c, A geometric deep learning-based scoring function of RNA tertiary structure by using atomic coordinates information; r.m.s.d., root mean square deviation.
Fragment assembly methods
Fragment assembly methods build RNA structural models by assembling structural fragments in a template library (Fig. 3b). Example methods that use this strategy include FARNA95, MC-Sym57, RNAComposer96, FARFAR2 (ref. 97) and so on. In general, these methods sample fragments from a structure library and then use energy minimization to assemble them into a full structural model. Currently, fragment assembly methods are, by far, the largest category for prediction of RNA tertiary structures, but these methods inherently have the same problem (and potential bias) noted above: they rely on the number of experimentally solved RNA structures.
Deep learning-based methods
Exploitation of deep learning-based methods remains limited for RNA tertiary structure modeling, again owing to the paucity of available RNA structural data. A scoring function based on a geometric deep neural network named Atomic Rotationally Equivariant Scorer (ARES)98 was recently developed to identify the best conformation generated by FARFAR2 (Fig. 3c). Notably, ARES learns the 3D coordinates and chemical element type of each atom, rather than each residue. Although ARES remains a scoring function without the ability to adequately sample RNA structural space, its development should be understood as a landmark achievement for artificial intelligence-based RNA tertiary structure prediction, and will likely inspire future research into RNA tertiary structure prediction using cutting-edge deep learning techniques.
Given the distinctions between the chemical composition and folding mechanism between RNAs and proteins, we anticipate that the phenomenal success of Alphafold2 (ref. 99) will be difficult to directly reproduce in the RNA structure prediction field. Having said that, there are certain informative similarities between the higher-order structures of RNA and protein100. And the differences between nucleotides and amino acids are further narrowed when operating at the atomic level, suggesting that the fundamental knowledge underlying the success of protein structure prediction tools do have the capacity to be transferred to RNA tertiary structure prediction in the near future.
Integrative RNA structural modeling based on experimental probing data
Although it appears that methods discussed above have achieved high accuracy, it cannot be overemphasized that these tools were developed based on energy terms and parameters derived from RNA structures obtained in vitro and are also evaluated using RNA structures obtained in vitro. While the functional structures of RNA molecules are known to be strongly impacted by specific interactions that occur in specific cell types and circumstances101,102, it is a nontrivial problem that these prediction methods do not reflect RNA structures under biological context. Excitingly, the aforementioned development of the RNA structure probing technologies has enabled the acquisition of large amounts of experimental probing data. We are therefore at an opportune moment, as this probing data can be incorporated into RNA structure modeling (that is, can be harnessed in model training, and for data mining, by computational specialists) to both improve prediction accuracy and to yield structure models that reliably represent the RNA structures that perform specific functions in particular cells.
Modeling assisted by footprinting RNA probing data
There are now methods that have started to make use of the increasingly rich resource of in vivo probing data for modeling RNA structure in biological context103. For example, RNAstructure56, RME104 and RNAprob105 explicitly convert probing data (for example, SHAPE reactivity scores) into ‘pseudoenergy terms’ and applies them for energy or statistical models by penalizing base-pairing nucleotides (Fig. 4a). Among them, RNAstructure is the most widely used tool for RNA structure studies. To date, it has been used to study diverse RNA classes, including small RNAs, lncRNAs, mRNAs and viral RNA genomes13,14,17,19,106. In contrast, SeqFold107 uses a ‘sample and select’ approach to sample an ensemble of RNA structures, and then select the one(s) that agree with experimental reactivity scores (Fig. 4b). It can be used to study the differential effects of RNA secondary structure on gene regulation at the transcriptome scale.

a, Methods that optimize the predicted secondary structure using a pseudoenergy term generated by probing data. b, Methods that select the optimal secondary structure conformations from a large structural ensemble generated using various sampling strategies. c, Methods that model multiple structures by grouping reads based on their mutational patterns. d, Methods that leverage proximity ligation-based probing data for segmentation of structural or topological domains and for modeling of RNA secondary structures. EM, expectation maximization.
While the aforementioned methods typically report only one (optimal) structural model for one RNA molecule, there are also tools, including SLEQ108 and Rsample109, that consider multiple structural conformations. Distinct from Rsample, SLEQ selects the structure ensembles that best explain the observed read patterns instead of reactivity scores. SLEQ has also been shown as useful for studying the structural heterogeneity of riboSNitches108.
Methods have also been developed that exploit the linked structural information for simultaneous mutations present in multiple nucleotides in one RNA molecule; these can be used to directly detect heterogeneous conformations based on grouping of sequencing reads by mutational patterns (Fig. 4c). For example, the RNA interaction groups by mutational profiling (RING-MaP) method110 uses spectral clustering to group reads from the same putative structural conformation; this has been used to identify two conformations of the thiamine pyrophosphate riboswitch. Moreover, a tool for the detection of RNA folding ensembles named DREEM18, which adopts an expectation–maximization algorithm to assign reads generated by DMS-based mutational profiling and sequencing (DMS-MaPseq) to heterogeneous different structural conformations, has been used to investigate alternative conformations at the splice sites of the HIV-1 RNA. Recently, the deconvolution of coexisting RNA conformations from mutational profiling (DRACO) method22 was developed based on a combination of spectral clustering and fuzzy clustering of reads, and was applied to analyze the SARS-CoV-2 RNA genome structure.
Modeling assisted by proximity ligation-based RNA probing data
Analyses of proximity ligation-based probing data have also yielded many insights into RNA structure modeling and functional RNA structural elements. For example, visualization of both PARIS data and RIC-seq data generated Hi-C-like connectivity maps for distinct RNAs, which were termed ‘structural domains’106 or ‘topological domains’55 in different studies (Fig. 4d). For example, Li et al. implemented an algorithm to search for an optimal hierarchical division of large RNAs iteratively based on PARIS data, and successfully chopped the Zika virus RNA into dozens of structural domains, notably reporting similar domain boundaries as two different Zika virus strains106. Note that studies of mutually exclusive interactions have collectively indicated that the coexistence of multiple conformations (that is, alternative structures) occurs ubiquitously in cells43,45.
There are much fewer tools utilizing proximity ligation-based probing data. Recently, IRIS111 was developed to include the long-range interaction information in PARIS data in its modeling (Fig. 4d). By converting PARIS data into supporting scores that represent pairing probabilities between nucleotides, IRIS is thus able to use information of interaction fragments from PARIS data to output representative secondary structural models.
Modeling aided by cryo-electron microscopy and small-angle X-ray scattering RNA structure data
In addition to integrating probing data to model RNA secondary structures in vivo, tools have also been built to integrate other types of data to model RNA tertiary structures. Researchers have started to assess RNA tertiary structures using cryo-EM; a recent development is the use of low-resolution density maps to computationally model RNA tertiary structures112 (Fig. 5a). Specifically, RNA structure probing experiments are first conducted to obtain RNA secondary structural information, which is then used to constrain the prediction of secondary structural models. Then, these secondary structural models are combined with cryo-EM density maps representing the overall architecture of the analyte RNA, to construct all-atom models of RNA tertiary structure with auto-DRRAFTER113. These efforts have established that cryo-EM can routinely resolve maps of RNA-only systems and shown that cryo-EM maps enable coordinate estimation when complemented with multidimensional RNA structure mapping and auto-DRRAFTER computational modeling.

a, Methods that combine low-resolution density maps generated by cryo-EM and secondary structure models inferred from probing data to model RNA tertiary structures. b, Methods that combine SAXS scattering information and predicted secondary structure based on probing data to model RNA tertiary structures.
SAXS can also be used to characterize tertiary structures of RNA molecules (Fig. 5b). For example, RS3D is a program that adopts hierarchical moves and simulated annealing for 3D RNA structure resolving114. It incorporates RNA secondary structures and SAXS data to generate tertiary RNA structural models, and the results from RS3D can be further refined using suitable force-field information.
Conclusion and future directions
As discussed before, RNA occupies a conceptual middle ground between DNA and proteins; and the methods used to study RNA structure share informative similarities with the sequencing, biophysical and computational technologies used to analyze DNA and proteins (Box 2). At the same time, we show how the intrinsic structural heterogeneity of RNA molecules and the sensitivity of their functional structures to cellular context make RNA structure determination a uniquely challenging research area.
Remarkably, there have been profound advances in RNA structural probing methods, for example increasing in throughput (from studying single transcripts to the transcriptome-wide scale), moving from in vitro to in vivo, and achieving ever-increasing gains in resolution and scope by incorporating innovative chemical probes and sequencing technologies. Nonetheless, it is obvious that there is much room for further improvement of these methods.
For example, the regulation of RNAs is known to be strongly tied to their localization; we know that where a given RNA localizes in cells can determine whether it is translated, stored or degraded. One direction for RNA structure probing technology improvement is therefore to increase spatial resolution, seeking to reveal more fine-grained subcellular structural maps and spatial structural maps in cells, which should broaden our knowledge about posttranscriptional regulation from a structural view. The well-established traditional cell compartment purification methods, such as using centrifugation and/or further immunoprecipitation, have successfully enriched the membrane-bound organelles (nucleus, mitochondria, and so on) and membraneless assemblies (P-bodies, stress granules and so on)102. Recently reported technologies like APEX-seq, which uses the peroxidase enzyme APEX2 for direct proximity labeling of RNA, can greatly expand the scope of experimentally accessible subcellular compartments115. These methods may be combined with current RNA structure probing technologies for RNA spatial structurome investigations.
Recent breakthroughs in single-cell experimental technologies offer a potential solution to resolve the RNA structures at the single-cell level, which should provide an opportunity to study the heterogeneity of RNA structure at the cellular (and thus tissue) levels during, for example, the pathological development of diseases. However, hurdles need to be conquered to increase the signal-to-noise ratio to sufficiently recover RNA structural information.
Beyond experimental structure determination methods, computational modeling methods have also made rapid advances. One continuing challenge, however, is that all learning-based methods (and especially those based on deep neural networks) likely suffer from overfitting, an issue acknowledged by many researchers in the field. The overfitting problem may be attributed to the incompatibility between the complexity of the models and the limited number of known RNA structures. Although several methods have used certain techniques like transfer learning and integration with thermodynamic energy terms to address this challenge, innovations from small sample learning are highly desired and will likely yield substantial improvements in prediction accuracy. On the other hand, the training datasets used as input by these models to date include mainly structures of tRNA and rRNA, and predominately with the data obtained in vitro. Thus, given the known variability/flexibility of RNA structures, we can assume that predictions will have difficulty in reflecting the structures as they actually occur in diverse cellular contexts. Emerging computational methods integrating structure probing data are likely going to radically bolster RNA structure studies; however, much remains to be done. Importantly, structure prediction should also consider the multiple conformations of an RNA, rather than the optimal one, especially for those tools that use only sequence as input, because an RNA can adopt multiple conformations.
Second, current deep learning-based RNA structure predictions have been limited to secondary structure predictions, owing largely to the insufficient quantity of experimentally validated RNA tertiary structures. However, there is a strong desire to model RNA tertiary structures with coordinate information98,113. Although deep learning-based RNA tertiary structure predictions lag far behind the state-of-the-art methods for protein tertiary structure prediction—which is certainly understandable given the very limited number of native RNA structures that have been reported—the historic advance presented by Alphafold2 (ref. 99) for protein tertiary structure prediction and the remarkable breakthrough of ARES98 for RNA structural conformation scoring seem very likely to inspire the development of innovative computational methods for predicting RNA tertiary structures in the near future.
RNA structures have been applied in studies of RNA functions and regulation, for example, for predicting RBP binding101 and RNA modification sites12. Specific RNA structures are known to prevent the degradation of RNA25 and to increase the half-life, which can aid the design of stable mRNA vaccines. As our understanding of how RNA structures form, interact and function in cells improves, it seems obvious that researchers will begin to engineer RNAs with desired functions. Ideally, the same principles underlying endogenous RNA behavior will inform the design of de novo RNA molecules. It will also be exciting to see whether the RNA structure modeling tools will perform well as we expand into RNA design and engineering. Moreover, analogous to protein structure-guided drug screening and design, structured RNA molecules can be targeted by small molecules with high selectivity and strong affinity. RNA structural modeling can help to find potential drugs for treating human disease, with the particularly attractive prospect of targeting the mRNA molecules encoding ‘undruggable’ target proteins. In short, accurate RNA structural determination will be a prerequisite for RNA biotechnology and biomedical applications.
References
Serganov, A. & Patel, D. J. Ribozymes, riboswitches and beyond: regulation of gene expression without proteins. Nat. Rev. Genet. 8, 776–790 (2007).
Pyle, A. M. Ribozymes: a distinct class of metalloenzymes. Science 261, 709–714 (1993).
Rinn, J. L. & Chang, H. Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166 (2012).
Mortimer, S. A., Kidwell, M. A. & Doudna, J. A. Insights into RNA structure and function from genome-wide studies. Nat. Rev. Genet. 15, 469–479 (2014).
Ganser, L. R., Kelly, M. L., Herschlag, D. & Al-Hashimi, H. M. The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol. 20, 474–489 (2019).
Wan, Y., Kertesz, M., Spitale, R. C., Segal, E. & Chang, H. Y. Understanding the transcriptome through RNA structure. Nat. Rev. Genet. 12, 641–655 (2011).
Ma, H., Jia, X., Zhang, K. & Su, Z. Cryo-EM advances in RNA structure determination. Signal Transduct. Target Ther. 7, 58 (2022).
Kertesz, M. et al. Genome-wide measurement of RNA secondary structure in yeast. Nature 467, 103–107 (2010).
Wan, Y. et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505, 706–709 (2014).
Ding, Y. et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700 (2014). This paper presents structure-seq, an in vivo genome-wide RNA structure probing method at nucleotide resolution and illustrates the use of structure-seq to resolve the RNA structurome of Arabidopsis thaliana.
Rouskin, S., Zubradt, M., Washietl, S., Kellis, M. & Weissman, J. S. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–705 (2014). This paper presents DMS-seq, an in vivo genome-wide RNA structure probing method at nucleotide resolution and shows how DMS-seq was used to resolve yeast and human structuromes. The study also found that RNA tends to be more structured in vitro than in vivo.
Spitale, R. C. et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490 (2015). This paper presents the icSHAPE method and an RNA structurome in mouse cells for all four nucleotides. The paper also reports a correlation between RNA modification and RNA structure.
Mustoe, A. M. et al. Pervasive regulatory functions of mRNA structure revealed by high-resolution SHAPE probing. Cell 173, 181–195 (2018).
Beaudoin, J. D. et al. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat. Struct. Mol. Biol. 25, 677–686 (2018).
Shi, B. et al. RNA structural dynamics regulate early embryogenesis through controlling transcriptome fate and function. Genome Biol. 21, 120 (2020).
Liu, C. X. et al. Structure and degradation of circular RNAs regulate PKR activation in innate immunity. Cell 177, 865–880 (2019).
Watts, J. M. et al. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460, 711–716 (2009). This study characterized the structure of the entire HIV-1 genome at single-nucleotide resolution using SHAPE and the paper reports a correlation between high levels of RNA structure and sequences that encode inter-domain loops in HIV proteins.
Tomezsko, P. J. et al. Determination of RNA structural diversity and its role in HIV-1 RNA splicing. Nature 582, 438–442 (2020).
Sun, L. et al. In vivo structural characterization of the SARS-CoV-2 RNA genome identifies host proteins vulnerable to repurposed drugs. Cell 184, 1865–1883 (2021). This paper reports the in vivo and in vitro structure maps of the SARS-CoV-2 RNA genome, and the study used the obtained RNA structure data to predict host proteins that bind to the SARS-CoV-2 genome.
Zuker, M. On finding all suboptimal foldings of an RNA molecule. Science 244, 48–52 (1989).
Leamy, K. A., Assmann, S. M., Mathews, D. H. & Bevilacqua, P. C. Bridging the gap between in vitro and in vivo RNA folding. Q. Rev. Biophys. 49, e10 (2016).
Morandi, E. et al. Genome-scale deconvolution of RNA structure ensembles. Nat. Methods 18, 249–252 (2021).
Kim, S. H. et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science 185, 435–440 (1974).
Liu, Z. et al. Cryo-EM structure of human dicer and its complexes with a pre-miRNA substrate. Cell 173, 1191–1203 (2018).
Akiyama, B. M. et al. Zika virus produces noncoding RNAs using a multi-pseudoknot structure that confounds a cellular exonuclease. Science 354, 1148–1152 (2016).
Incarnato, D., Neri, F., Anselmi, F. & Oliviero, S. Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome. Genome Biol. 15, 491 (2014).
Wang, P. Y., Sexton, A. N., Culligan, W. J. & Simon, M. D. Carbodiimide reagents for the chemical probing of RNA structure in cells. RNA 25, 135–146 (2019).
Mitchell, D. 3rd et al. In vivo RNA structural probing of uracil and guanine base-pairing by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC). RNA 25, 147–157 (2019).
Deigan, K. E., Li, T. W., Mathews, D. H. & Weeks, K. M. Accurate SHAPE-directed RNA structure determination. Proc. Natl Acad. Sci. USA 106, 97–102 (2009).
Siegfried, N. A., Busan, S., Rice, G. M., Nelson, J. A. & Weeks, K. M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 11, 959–965 (2014).
Lucks, J. B. et al. Multiplexed RNA structure characterization with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-seq). Proc. Natl Acad. Sci. USA 108, 11063–11068 (2011).
Bindewald, E. et al. Correlating SHAPE signatures with three-dimensional RNA structures. RNA 17, 1688–1696 (2011).
Busan, S., Weidmann, C. A., Sengupta, A. & Weeks, K. M. Guidelines for SHAPE reagent choice and detection strategy for RNA structure probing studies. Biochemistry 58, 2655–2664 (2019).
Spitale, R. C. et al. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 9, 18–20 (2013).
Marinus, T., Fessler, A. B., Ogle, C. A. & Incarnato, D. A novel SHAPE reagent enables the analysis of RNA structure in living cells with unprecedented accuracy. Nucleic Acids Res. 49, e34 (2021).
Zubradt, M. et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 75–82 (2017).
Sexton, A. N., Wang, P. Y., Rutenberg-Schoenberg, M. & Simon, M. D. Interpreting reverse transcriptase termination and mutation events for greater insight into the chemical probing of RNA. Biochemistry 56, 4713–4721 (2017).
Ritchey, L. E. et al. Structure-seq2: sensitive and accurate genome-wide profiling of RNA structure in vivo. Nucleic Acids Res. 45, e135 (2017).
Piao, M. et al. An ultra low-input method for global RNA structure probing uncovers Regnase-1-mediated regulation in macrophages. Fundamental Res. 2, 2–13 (2022).
Aw, J. G. A. et al. Determination of isoform-specific RNA structure with nanopore long reads. Nat. Biotechnol. 39, 336–346 (2021).
Stephenson, W. et al. Direct detection of RNA modifications and structure using single-molecule nanopore sequencing. Cell Genom. 2, 100097 (2022).
Aw, J. G. et al. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell 62, 603–617 (2016).
Lu, Z. et al. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 165, 1267–1279 (2016). This paper presents PARIS, a method based on reversible psoralen crosslinking for global mapping of RNA duplexes with near base-pair resolution in mouse cells. The study discovered many long-range as well as alternative RNA–RNA interactions.
Sharma, E., Sterne-Weiler, T., O’Hanlon, D. & Blencowe, B. J. Global mapping of human RNA–RNA interactions. Mol. Cell 62, 618–626 (2016).
Ziv, O. et al. COMRADES determines in vivo RNA structures and interactions. Nat. Methods 15, 785–788 (2018).
Helwak, A., Kudla, G., Dudnakova, T. & Tollervey, D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665 (2013).
Sugimoto, Y. et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494 (2015).
Metkar, M. et al. Higher-order organization principles of pre-translational mRNPs. Mol. Cell 72, 715–726 (2018).
Lu, Z. & Chang, H. Y. The RNA base-pairing problem and base-pairing solutions. Cold Spring Harb. Perspect. Biol. 10, a034926 (2018).
Gong, J. et al. RISE: a database of RNA interactome from sequencing experiments. Nucleic Acids Res. 46, D194–D201 (2018).
Christy, T. W. et al. Direct mapping of higher-order RNA interactions by SHAPE-JuMP. Biochemistry 60, 1971–1982 (2021).
Van Damme, R. et al. Chemical reversible crosslinking enables measurement of RNA 3D distances and alternative conformations in cells. Nat. Commun. 13, 911 (2022).
Ramani, V., Qiu, R. & Shendure, J. High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol. 33, 980–984 (2015).
Nguyen, T. C. et al. Mapping RNA–RNA interactome and RNA structure in vivo by MARIO. Nat. Commun. 7, 12023 (2016).
Cai, Z. et al. RIC-seq for global in situ profiling of RNA–RNA spatial interactions. Nature 582, 432–437 (2020). This paper reports RIC-seq, a technology to profile the transcriptome-wide in intramolecular and intermolecular RNA–RNA interactions mediated by proteins. The study also revealed many RNA-based enhancer and promoter interactions.
Reuter, J. S. & Mathews, D. H. RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129 (2010). This paper presents RNAstructure, a software package for RNA secondary structure prediction and analysis based on minimum free-energy calculation. RNAstructure can incorporate experimental probing data to improve prediction performance.
Parisien, M. & Major, F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 452, 51–55 (2008).
Lorenz, R. et al. ViennaRNA Package 2.0. Algorithms Mol. Biol. 6, 26 (2011). This paper presents the ViennaRNA package, a toolkit for RNA secondary structure prediction, comparison and visualization.
Lorenz, R., Wolfinger, M. T., Tanzer, A. & Hofacker, I. L. Predicting RNA secondary structures from sequence and probing data. Methods 103, 86–98 (2016).
Simmonds, P. Pervasive RNA secondary structure in the genomes of SARS-CoV-2 and other coronaviruses. mBio 11, e01661-20 (2020).
Kierzek, E. et al. Secondary structure prediction for RNA sequences including N6-methyladenosine. Nat. Commun. 13, 1271 (2022).
Fu, Y., Sharma, G. & Mathews, D. H. Dynalign II: common secondary structure prediction for RNA homologs with domain insertions. Nucleic Acids Res. 42, 13939–13948 (2014).
Rivas, E., Clements, J. & Eddy, S. R. A statistical test for conserved RNA structure shows lack of evidence for structure in lncRNAs. Nat. Methods 14, 45–48 (2017).
Rivas, E. RNA structure prediction using positive and negative evolutionary information. PLoS Comput. Biol. 16, e1008387 (2020).
Rivas, E., Clements, J. & Eddy, S. R. Estimating the power of sequence covariation for detecting conserved RNA structure. Bioinformatics 36, 3072–3076 (2020).
Rivas, E. Evolutionary conservation of RNA sequence and structure. Wiley Interdiscip. Rev. RNA 12, e1649 (2021).
Gao, W., Jones, T. A. & Rivas, E. Discovery of 17 conserved structural RNAs in fungi. Nucleic Acids Res. 49, 6128–6143 (2021).
Manfredonia, I. et al. Genome-wide mapping of SARS-CoV-2 RNA structures identifies therapeutically relevant elements. Nucleic Acids Res. 48, 12436–12452 (2020).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Bernhart, S. H., Hofacker, I. L., Will, S., Gruber, A. R. & Stadler, P. F. RNAalifold: improved consensus structure prediction for RNA alignments. BMC Bioinformatics 9, 474 (2008).
Tan, Z., Fu, Y., Sharma, G. & Mathews, D. H. TurboFold II: RNA structural alignment and secondary structure prediction informed by multiple homologs. Nucleic Acids Res. 45, 11570–11581 (2017).
Zakov, S., Goldberg, Y., Elhadad, M. & Ziv-Ukelson, M. Rich parameterization improves RNA structure prediction. J. Comput. Biol. 18, 1525–1542 (2011).
Knudsen, B. & Hein, J. Pfold: RNA secondary structure prediction using stochastic context-free grammars. Nucleic Acids Res. 31, 3423–3428 (2003).
Do, C. B., Woods, D. A. & Batzoglou, S. CONTRAfold: RNA secondary structure prediction without physics-based models. Bioinformatics 22, e90–e98 (2006).
Rivas, E., Lang, R. & Eddy, S. R. A range of complex probabilistic models for RNA secondary structure prediction that includes the nearest-neighbor model and more. RNA 18, 193–212 (2012).
Rivas, E. The four ingredients of single-sequence RNA secondary structure prediction. A unifying perspective. RNA Biol. 10, 1185–1196 (2013).
Koessler, D. R., Knisley, D. J., Knisley, J. & Haynes, T. A predictive model for secondary RNA structure using graph theory and a neural network. BMC Bioinformatics 11, S21 (2010).
Zhang, H. et al. A new method of RNA secondary structure prediction based on convolutional neural network and dynamic programming. Front. Genet. 10, 467 (2019).
Wang, L. et al. DMfold: a novel method to predict RNA secondary structure with pseudoknots based on deep learning and improved base-pair maximization principle. Front. Genet. 10, 143 (2019).
Chen, X., Li, Y., Umarov, R., Gao, X. & Song, L. RNA secondary structure prediction by learning unrolled algorithms. Preprint at https://arxiv.org/abs/2002.05810 (2020).
Fu, L. et al. UFold: fast and accurate RNA secondary structure prediction with deep learning. Nucleic Acids Res. 50, e14 (2022).
Singh, J., Hanson, J., Paliwal, K. & Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 10, 5407 (2019).
Singh, J. et al. Improved RNA secondary structure and tertiary base-pairing prediction using evolutionary profile, mutational coupling and two-dimensional transfer learning. Bioinformatics btab165 (2021).
Sato, K., Akiyama, M. & Sakakibara, Y. RNA secondary structure prediction using deep learning with thermodynamic integration. Nat. Commun. 12, 941 (2021).
Akiyama, M., Sato, K. & Sakakibara, Y. A max-margin training of RNA secondary structure prediction integrated with the thermodynamic model. J. Bioinform. Comput. Biol. 16, 1840025 (2018).
Andronescu, M., Condon, A., Hoos, H. H., Mathews, D. H. & Murphy, K. P. Computational approaches for RNA energy parameter estimation. RNA 16, 2304–2318 (2010).
Sloma, M. F. & Mathews, D. H. Exact calculation of loop formation probability identifies folding motifs in RNA secondary structures. RNA 22, 1808–1818 (2016).
Danaee, P. et al. bpRNA: large-scale automated annotation and analysis of RNA secondary structure. Nucleic Acids Res. 46, 5381–5394 (2018).
Szikszai, M., Wise, M., Datta, A., Ward, M. & Mathews, D.H. Deep learning models for RNA secondary structure prediction (probably) do not generalise across families. Bioinformatics 38, 3892–3899 (2022).
Puton, T., Kozlowski, L. P., Rother, K. M. & Bujnicki, J. M. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 41, 4307–4323 (2013).
Magnus, M. et al. RNA-Puzzles toolkit: a computational resource of RNA 3D structure benchmark datasets, structure manipulation and evaluation tools. Nucleic Acids Res. 48, 576–588 (2020).
Ding, F. et al. Ab initio RNA folding by discrete molecular dynamics: from structure prediction to folding mechanisms. RNA 14, 1164–1173 (2008).
Boniecki, M. J. et al. SimRNA: a coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 44, e63 (2016). This paper presents SimRNA, a method for computational RNA tertiary structure prediction. SimRNA invents a coarse-grained representation for RNA molecules and integrates an energy function and Monte Carlo sampling for structure prediction.
Johnson, P. Z., Kasprzak, W. K., Shapiro, B. A. & Simon, A. E. Structural characterization of a new subclass of panicum mosaic virus-like 3’ cap-independent translation enhancer. Nucleic Acids Res. 50, 1601–1619 (2022).
Das, R. & Baker, D. Automated de novo prediction of native-like RNA tertiary structures. Proc. Natl Acad. Sci. USA 104, 14664–14669 (2007).
Biesiada, M., Pachulska-Wieczorek, K., Adamiak, R. W. & Purzycka, K. J. RNAComposer and RNA 3D structure prediction for nanotechnology. Methods 103, 120–127 (2016).
Watkins, A. M., Rangan, R. & Das, R. FARFAR2: improved de novo rosetta prediction of complex global RNA folds. Structure 28, 963–976 (2020). This paper presents FARFAR2, a method for computational RNA tertiary structure prediction based on a fragment assembly strategy and an all-atom scoring function. The RNA structural fragments used by FARFAR2 are collected from a nonredundant crystallographic database.
Townshend, R. J. L. et al. Geometric deep learning of RNA structure. Science 373, 1047–1051 (2021). This paper presents ARES, a scoring function to assess RNA tertiary structure. ARES uses geometric deep learning to develop a scoring function based on studying the type and the atomic 3D coordinates of 18 known RNA structures.
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Rother, K., Rother, M., Boniecki, M., Puton, T. & Bujnicki, J. M. RNA and protein 3D structure modeling: similarities and differences. J. Mol. Model. 17, 2325–2336 (2011).
Sun, L. et al. Predicting dynamic cellular protein–RNA interactions by deep learning using in vivo RNA structures. Cell Res. 31, 495–516 (2021).
Sun, L. et al. RNA structure maps across mammalian cellular compartments. Nat. Struct. Mol. Biol. 26, 322–330 (2019).
Li, P., Zhou, X., Xu, K. & Zhang, Q. C. RASP: an atlas of transcriptome-wide RNA secondary structure probing data. Nucleic Acids Res. 49, D183–D191 (2021).
Wu, Y. et al. Improved prediction of RNA secondary structure by integrating the free-energy model with restraints derived from experimental probing data. Nucleic Acids Res. 43, 7247–7259 (2015).
Deng, F., Ledda, M., Vaziri, S. & Aviran, S. Data-directed RNA secondary structure prediction using probabilistic modeling. RNA 22, 1109–1119 (2016).
Li, P. et al. Integrative analysis of Zika virus genome RNA structure reveals critical determinants of viral infectivity. Cell Host Microbe 24, 875–886 (2018).
Ouyang, Z., Snyder, M. P. & Chang, H. Y. SeqFold: genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data. Genome Res. 23, 377–387 (2013).
Li, H. & Aviran, S. Statistical modeling of RNA structure profiling experiments enables parsimonious reconstruction of structure landscapes. Nat. Commun. 9, 606 (2018).
Spasic, A., Assmann, S. M., Bevilacqua, P. C. & Mathews, D. H. Modeling RNA secondary structure folding ensembles using SHAPE mapping data. Nucleic Acids Res. 46, 314–323 (2018).
Homan, P. J. et al. Single-molecule correlated chemical probing of RNA. Proc. Natl Acad. Sci. USA 111, 13858–13863 (2014).
Zhou, J. et al. IRIS: a method for predicting in vivo RNA secondary structures using PARIS data. Quant. Biol. 8, 369–381 (2020).
Kappel, K. et al. Accelerated cryo-EM-guided determination of three-dimensional RNA-only structures. Nat. Methods 17, 699–707 (2020).
Kappel, K. et al. De novo computational RNA modeling into cryo-EM maps of large ribonucleoprotein complexes. Nat. Methods 15, 947–954 (2018).
Bhandari, Y. R. et al. Topological structure determination of RNA using small-angle X-ray scattering. J. Mol. Biol. 429, 3635–3649 (2017).
Fazal, F. M. et al. Atlas of subcellular RNA localization revealed by APEX-seq. Cell 178, 473–490 e426 (2019).
Umeyama, T. & Ito, T. DMS-seq for in vivo genome-wide mapping of protein–DNA interactions and nucleosome centers. Cell Rep. 21, 289–300 (2017).
Wu, T., Lyu, R., You, Q. & He, C. Kethoxal-assisted single-stranded DNA sequencing captures global transcription dynamics and enhancer activity in situ. Nat. Methods 17, 515–523 (2020).
Klemm, S. L., Shipony, Z. & Greenleaf, W. J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 20, 207–220 (2019).
Jerkovic, I. & Cavalli, G. Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 22, 511–528 (2021).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants nos. 32125007 and 91940306 to Q.C.Z., and 32100504 to Y.F.), the Postdoctoral Science Foundation of China (2021M691811 to Y.F., and 2021M690091 and 2021T140380 to L.S.) and the Postdoctoral Foundation of Tsinghua-Peking Center for Life Sciences (J.Z., Y.F. and L.S.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Methods thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Lei Tang, in collaboration with the Nature Methods team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Tables 1 and 2
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, J., Fei, Y., Sun, L. et al. Advances and opportunities in RNA structure experimental determination and computational modeling. Nat Methods 19, 1193–1207 (2022). https://doi.org/10.1038/s41592-022-01623-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41592-022-01623-y
This article is cited by
-
A deep learning-based method for modeling of RNA structures from cryo-EM maps
Nature Biotechnology (2024)
-
Liver fibrosis pathologies and potentials of RNA based therapeutics modalities
Drug Delivery and Translational Research (2024)
-
RNA contact prediction by data efficient deep learning
Communications Biology (2023)
-
trRosettaRNA: automated prediction of RNA 3D structure with transformer network
Nature Communications (2023)
