
Abstract
The advent of chimeric antigen receptor (CAR) T cell therapy has resulted in unprecedented long-term clearance of relapse/refractory hematological malignancies in both pediatric and adult patients. However, severe toxicities, such as cytokine release syndrome and neurotoxicity, associated with CAR T cells affect therapeutic utility; and treatment efficacies for solid tumors are still not impressive. As a result, engineering strategies that modify other immune cell types, especially natural killer (NK) cells have arisen. Owing to both CAR-dependent and CAR-independent (innate immune-mediated) antitumor killing capacity, major histocompatibility complex-independent cytotoxicity, reduced risk of alloreactivity and lack of major CAR T cell toxicities, CAR NK cells constitute one of the promising next-generation CAR immune cells that are also amenable as ‘off-the-shelf’ therapeutics. In this Review, we compare CAR T and CAR NK cell therapies, with particular focus on immunological synapses, engineering strategies and challenges.
Similar content being viewed by others

Main
CAR T cell therapies are based on engineering T cells with recombinant antigen-specific CAR molecules that bestow CAR T cells with target-specific cytotoxicity, independent of the T cell antigen receptor (TCR). Compared to regular cytotoxic T cell functions that are restricted by the binding and recognition of TCR complexes on T cells to antigen-presenting major histocompatibility complex (MHC) molecules on target cells, CAR-mediated cytotoxicity in CAR T cells is non-MHC restricted1. CAR T cells have resulted in durable remission in patients with relapse/refractory hematological malignancies. However, the severe toxicities, such as cytokine release syndrome (CRS) and neurotoxicity, and limited efficacy against solid tumors have prompted the engineering of alternative immune cells that have cytotoxic potential, such as NK cells1,2. Although all CAR T cell therapies approved by the US Food and Drug Administration (FDA) rely on autologous transfer of patient-derived and engineered CAR T cells to avoid graft-versus-host disease (GvHD), the FDA has also approved clinical trials of genetically modified allogeneic CAR T cells3. The development of hypoimmunogenic allogeneic CAR T cells is currently under investigation to enable large-scale engineering and transfusion of allogeneic CAR T cells from healthy donors.
In this Review, we compare the mechanisms of T cell and NK cell antitumor cytotoxicity, CAR design and engineering strategies, and discuss how tackling the challenges faced by CAR T cell and CAR NK cell therapies affect their future application, especially against solid tumors.
How T cells and NK cells fight cancer
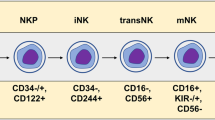
NK cells are innate immune cells that function as first-line defenders. Unlike T cells, NK cell antitumor function is independent of MHC molecules and does not require antigen priming4. NK cells express an array of activating receptors (aNKRs) and inhibitory receptors (iNKRs)5; depending on whether the coordination between aNKR and iNKR signaling favors NK cell activation or inhibition, it leads to either lysis of the target cell or immune self-tolerance, respectively. NK cell degranulation and target cell lysis occur as tumor cells downregulate self-MHC class-I molecules creating a state of ‘missing self’ or following recognition of tumor-associated stress ligands by aNKRs. NK cell cytotoxicity can also occur by conjugation of FasL to cognate receptors on tumor cells, via antibody-dependent cellular cytotoxicity (ADCC) that involves binding of CD16 on NK cells to antibody-bound target cells, or indirectly via release of immunogenic cytokines6,7,8.
Combat between the immune system and solid tumors involves an intricate network of innate and adaptive immune cells. The intratumoral abundance of these cells and orchestration of their signaling affects the fate of tumor progression or control. In a newly transformed solid tumor, NK cells can detect stress ligands on tumor cell subsets and eradicate these cells. Tumor-activated NK cells release XCL1, XCL2 and CCL5 chemokines that attract conventional type 1 dendritic cells (cDC1s), resulting in an inflammatory active microenvironment that is often referred to as a ‘hot tumor’9,10. Neoantigen-presenting or tumor-associated antigen (TAA)-presenting cDC1s prime CD8+ T cells in the tumor draining lymph nodes, enabling further T cell infiltration and MHC class-I-mediated tumor cell lysis. Polyclonal subsets of CD4+ and CD8+ tumor-infiltrating lymphocytes, including a subset of cytotoxic CD4+ T cells capable of MHC class-II-mediated tumor killing, with a diverse TCR repertoire and distinct memory differentiation and exhaustion phenotypes infiltrate solid tumors in response to this priming11,12,13.
Immune evasion can be facilitated by tumor cell secretion of prostaglandin E2, which not only blocks CD8+ cytotoxicity but also reduces NK cell survival, activity and secretion of the chemoattractants CCL5 and XCL1, which weakens NK cell-mediated recruitment of cDC1s to the tumor microenvironment (TME)9,14. Tumor cell-intrinsic sensitivity to interferon-γ (IFNγ) secretion by activated TAA-specific CD8+ T cells promotes the expression of PD-L1 and other immune checkpoint ligands on tumor cells, which become resistant to T cell cytotoxicity by engaging with cognate checkpoint receptors, such as PD-1 on T cells. In such cases, immune checkpoint blockade (ICB) can be an effective immunotherapy15; however, tumors can also evade T cell cytotoxicity and acquire resistance to ICB by reducing antigen presentation, thereby causing intratumoral T cells to exit into neighboring lymph nodes via CXCL12–CXCR4-mediated chemotaxis16. As iNKR conjugation to self-MHC class-I complexes favors NK cell inhibition, the loss of MHC class-I complexes in ICB-resistant tumor cells resensitizes intratumoral NK cells by limiting iNKR conjugation, thus enabling resumption of NK cell cytotoxicity10. However, not all intratumoral NK cells are cytotoxic. One study identified a pro-tumorigenic subset of Socs3hiCD11b−CD27− immature NK cells in patients with triple-negative breast cancer, which was associated with poor prognosis and could promote tumor progression and obstruct ICB efficacy in xenograft mouse models17.
Tumor-associated macrophages (TAMs) and myeloid-derived suppressor cells are recruited by tumor cells to create a hypoxic and immunosuppressive TME10,18. Anti-inflammatory M2-like macrophages inhibit NK cell degranulation and function by releasing suppressive transforming growth factor-β (TGFβ) and driving the upregulation of CD85j inhibitory receptors on NK cells18. Depending on the tumor type, malignancy stage and state of the TME, TAMs can enhance antitumor functions by cross-presentation of neoantigens to CD4+ T cells that acquire cytotoxic capacity19. TAMs can also aid in tumor evasion. Recent studies have shown that TAMs are the main consumers of glucose within the TME20; and by undergoing cell-intrinsic continuous glycolysis that leads to lactate accumulation within the TME, TAMs favor regulatory T (Treg) cell recruitment and CD8+ T cell expulsion19. Myeloid-derived suppressor cells express NK inhibitory ligands, secrete reactive oxygen species, contribute (along with tumor cells) to suppressive adenosine and TGFβ accumulation and activate Treg cells to release more TGFβ, a coordination of actions that has severe consequences for NK and T cell functions21. TME-abundant Treg cells and PD-1-expressing follicular regulatory T (TFR) differentiated cells, have potent immunosuppressive capacity to reduce tumor-infiltrating lymphocyte cytotoxicity and could further inhibit the efficacy of anti-PD-1 ICB22. Hence, enduring an immunosuppressive TME and overcoming tumor immunoevasive mechanisms is vital for successful CAR T or CAR NK cell therapy.
Advantages of CAR NK cells over CAR T cells
CAR NK cells have several advantages over CAR T cells that could be exploited to treat solid tumors. For example, CAR NK cells can leverage the innate antitumor capacity of NK cells while favoring more target-specific killing. This enables CAR NK cells to kill target-expressing cancer cells (CAR-mediated killing) and target-lacking cancer cells (CAR-independent/innate NK cytotoxicity-mediated), an advantage that is particularly important for treating solid tumors as these tumors are often characterized by antigen heterogeneity and escape to evade immune cell cytotoxicity23. As most NK cell cytotoxic capacity is diminished in patients with cancer during tumor progression and after curative regimens such as surgery, most CAR NK cells require an allogeneic source23,24. Clinical studies of allogeneic CAR NK cell-treated patients to date have recorded a lack of GvHD, owing to the lack of MHC class-I restriction25.
Although reversible, CRS is a life-threatening condition usually occurring in patients receiving CAR T cell therapy and is due to overactivation of effector immune cells and amplified levels of serum cytokines secreted either by CAR T cells or CAR T cell-activated macrophages23,26. These include IFNγ, tumor necrosis factor, granulocyte macrophage colony-stimulating factor, macrophage inflammatory protein-1 (MIP-1), monocyte chemoattractant protein-1 (MCP-1, also known as CCL2), and interleukin-6 (IL-6) and IL-10 (ref. 26). However, very low incidence of CRS has been documented in patients receiving CAR NK cells27. While the reason is not fully understood, the rarity in CRS cases after CAR NK cell therapy could be partially attributed to the divergent cytokine profile following NK cell activation, which includes IFNγ, tumor necrosis factor, granulocyte macrophage colony-stimulating factor and chemokines CCL1, MCP-1, MIP-1α (also known as CCL3), MIP-1β (CCL4), CCL5 and CXCL8 and/or the limited number of completed CAR NK cell clinical studies as compared to CAR T cell trials23,28,29.
Moreover, the feasible large-scale production of CAR NK cells from umbilical cord, stem cell differentiation, peripheral blood or NK cell lines is promising for future off-the-shelf production5,25. For example, in one study, approximately an 11-million-fold expansion was obtained for NK cells derived from cord blood, and more than 250-billion-fold expansion for NK cells derived from peripheral blood, by applying five stimulation rounds over 70 days30. In another study, up to 15,000-fold expansion of highly viable peripheral blood-derived NK cells could be achieved using a mix of irradiated autologous peripheral blood mononuclear cells, IL-2 and anti-CD3 antibody31.
CAR versus canonical T and NK cell immunological synapses
The type and features of the immunological synapse formed between CAR lymphocytes and target tumor cells is central to CAR lymphocyte antitumor efficacy (Fig. 1). The first encounter of CAR T cells with tumor cells involves identification and stable conjugation of CAR molecules on T cells with target antigen on tumor cells by forming an immunological synapse; however, the immunological synapse formed by CAR molecules is distinct from that of a canonical TCR in many aspects32. A classical mature immunological synapse is composed of three concentric zones of supramolecular activating clusters (SMACs), which are integral for TCR cytotoxic function33. In a lytic immunological synapse, central SMAC is composed of Lck kinase–TCR microclusters and is involved in initiating activation signaling cascades and release of lytic granules34. Peripheral SMAC governs LFA-1 integrin binding to ICAM-1 adhesion molecules on target cell surface and is thought to stabilize the synapse, and distal SMAC is mainly a ring of actin that contains glycocalyx protein repellers, such as CD43 and CD45, which protect T cells, and a corolla of CD2 whose interaction with CD58 domains on target cells enables additional costimulatory signaling35,36,37,38. However, in some cases, single-receptor engagement or membranous microcluster formations can trigger T cell activation sufficiently without the need for mature synapse formation39.

Helper T cell–APC interactions represent the prototype immunological synapse between a TCR and MHC. This immunological synapse that constitutes a central TCR–Lck complex (central SMAC), but lacks lytic granules, is surrounded by a peripheral LFA-1/ICAM-1 assembly (peripheral SMAC) and distal actin polymerization ring accompanied by CD45 and CD2 conjugations (distal SMAC). The CTL–tumor cell lytic synapse has similar SMAC clusters, but also involves lytic granule release within the central SMAC, following MTOC polarization. The NK lytic immunological synapse includes aNKR ligand binding as well as CD16-mediated ADCC with a low abundance/lack of inhibitory NK cell receptor–self-MHC class I conjugation and is surrounded by peripheral assembly of LFA-1/ICAM-1. NK cells contain lytic granules at resting stage and undergo ‘directed secretion’ only after a spatiotemporally organized signaling cascade that involves MTOC polarization. The immunological synapse in CAR T cells comprises convoluted CAR–Lck microclusters with less reliability on LFA-1 and lack of an adhesion ring. Likewise, the immunological synapse of CAR NK cells involves lytic granule release and multiple CAR–antigen clustering, but less actin organization at the synaptic cleft. A PDZ CAR NK cell–tumor cell lytic synapse has improved synapse organization as compared to CAR NK cells, characterized by accumulation of Scribble as well as a condensed synaptic area and enhanced clustering of activated ZAP70 with lytic granules. Created with BioRender.com.
On the other hand, the immunological synapse in CAR T cells does not involve mature synapse formation but rather comprises convoluted Lck microclusters, with less reliability on LFA-1 and lack of an adhesion ring. This less-organized synapse enables faster CAR-mediated engagement and subsequently faster release of signaling cascades as compared to the TCR and is associated with swifter cytotoxic granule secretion and successive CAR detachment from target cells32.
NK cell function depends on the balance in signaling between aNKRs and iNKRs. Inhibitory receptors such as killer immunoglobulin-like receptor (KIR) in humans and Ly49 in mice maintain self-MHC class-I tolerance against endogenous healthy cells, as well as having a pivotal role in promoting NK cell activation against low-MHC class-I-expressing cells in a process termed ‘education’40. During NK cell activation, the formation of the NK lytic immunological synapse starts with the SH2-domain-containing phosphotyrosine phosphatase-1 (SHP-1) forming small central clusters surrounded by LFA-1, and later by cytolytic granules clustering in central SMAC, while LFA-1 relocates into the periphery in a step that enables education of NK cell-activating synapses against low-MHC class-I-expressing targets41. Interestingly, distinct patterns of SHP-1 clustering in NK cell-activating synapses, including the degree of SHP-1 accumulation in the activating immune synapse and SHP-1 colocalization with F-actin and SLP-76 signaling adaptor, were seen in uneducated self-tolerant mouse Ly49A+ NK cells isolated from MHC class-I knockout mice versus MHC class-I molecule H2Dd-educated self-tolerant Ly49A+ NK cells from H2Dd transgenic mice40. The model suggested in the study proposes the accumulation of SHP-1 within the activating synapse leading to dephosphorylation of downstream signaling proteins to maintain ‘self-tolerance’. Once the activating synapse undergoes ‘education’ by iNKRs, SHP-1 is excluded from the activating synapse, thus releasing the inhibition, and enabling activation of downstream signaling and enhanced NK cell responsiveness40.
The general understanding of the multistep process of immunological synapse formation holds true for many immune cell interactions, including helper T cell–antigen-presenting cell (APC), cytotoxic T lymphocyte (CTL)–tumor cell, and NK cell–tumor cell or NK cell–healthy cell39. For lytic synapses, it starts with antigen recognition and receptor signaling and is followed by effector activation, microtubule organizing center (MTOC) polarization and degranulation, and is terminated by dissociation from lysed target cells, enabling immunological homeostasis and serial killing cascades32,39,42,43. However, studies have shown some kinetic and mechano-transduction differences between NK and CTL lytic synapses. Unlike T cells, in which lytic granules are induced by activation, NK cells contain lytic granules at resting stage, and thus follow precise spatiotemporal mechanisms of lytic granule secretion during activation and synapse formation in a process termed ‘directed secretion’39. NK cells can also form inhibitory synapses by clustering iNKRs, such as KIR, with self-determinants on healthy cells forming the supramolecular inhibitory cluster that blocks SMAC formation and protects healthy cells from lysis39,44. NK cell membrane protrusions contribute to inhibitory synapse formation and intercellular transfer of proteins45,46. In one study, intratumoral NK cells isolated from patients with liver cancer had fewer membrane protrusions compared to peritumor NK cells or peripheral NK cells from healthy donors. Tumor-induced alterations in serine metabolism lowered the levels of sphingomyelin in intratumoral NK cell membranes, resulting in dysregulated membrane protrusions that impaired lytic synapse formation and cytotoxicity. Blocking sphingomyelin catabolism increased the number of membrane protrusion-originated synapses and restored intratumoral NK cell lytic activity in vitro as well as in a humanized mouse model of liver cancer47.
Because the CAR lytic synapse requires a higher threshold for antigen recognition than a canonical immunological synapse to initiate killing and is more amenable for phosphatase-induced inactivation, scientists have hypothesized that by targeting cell polarization and provoking a more ordered assembly of the CAR immunological synapse, better CAR solid tumor cytotoxicity could be achieved34,48,49,50. Synapse-tuned CAR NK cells were generated by inserting the postsynaptic density-95, discs large and zona occludens-1 binding moieties (PDZbms) of cytotoxic and regulatory T cell molecule (CRTAM), a Scribble binding partner and a late-phase polarity protein that is involved in NK cell tumor immunosurveillance, into the C terminus of a CD28z CAR termed CAR PDZ NK48. Relative to regular CAR NK counterparts, these findings demonstrate enhanced avidity, antigen sensitivity, cytotoxicity, synapse strength and CAR PDZ NK cell polarization in vitro, as well as prolonged CAR PDZ NK cell survival and persistence in solid tumor mouse models. Also, CAR PDZ constructs, when tested in T cells, had enhanced effector functionality as compared to regular CAR T cells, both in vitro and in xenograft mouse models.
Together, future engineering strategies need to balance lytic synapse stability, threshold of antigen recognition and kinetics of intercellular conjugation to fine-tune next-generation CAR designs to treat solid tumors and optimize on-tumor off-target toxicities.
CAR T versus CAR NK cell engineering
Similarities, discrepancies and advances in CAR design
Compared to TCRs, CARs are synthetic receptors that bestow CAR T cells with target-specific cytotoxicity in a non-MHC restricted mechanism. The first generation of CAR constituted an extracellular single-chain variable fragment (scFv) linked by a hinge domain and transmembrane domain to a C-terminal CD3ζ activation domain that relays non-TCR outside-in activating signals into the T cell1,51. However, clinical efficacy of first generation CAR T cells was limited, possibly due to limited engraftment and cytotoxicity52. Later modifications of CAR molecules, aiming at improving CAR T cell cytotoxicity, cytokine production and proliferation capacity in response to target stimulation, involved insertion of one (second generation) or two (third generation) intracellular costimulatory (ICS) domains upstream of CD3ζ53,54 (Fig. 2a). Notably, genetic alterations in any of the CAR modular domains or linker structures can affect the affinity, potency and persistence of CAR T cells51. The six currently FDA-approved CAR T cell therapies targeting CD19 or BCMA have resulted in durable remission in many patients with hematological malignancies55. However, additional engineering strategies have been implemented to create next-generation CAR T cells for solid tumors and to enhance CAR T cell cytotoxicity, persistence, infiltration and safety.

a, Canonical TCR signaling involves sequential activation of Lck, CD3ζ and ZAP70 and phosphorylation of SLP-76 and LAT, which form a signaling complex with GADS and PLCγ leading to T cell activation. First generation CAR bridges a recombinant extracellular (Exc) domain via a hinge and TM domains to an intracellular CD3ζ as the main activation signal to promote CAR cell activation. In second and third generation CAR, signaling is supported by one or two ICS domains. LINK CAR T cells enable gated activation of two scFv CARs, the signals for which are dependent on SLP-76 complexing with LAT as ICS domains, only when both cognate antigens are presented on tumor cells and bind to scFv CARs. b, CAR cell dysfunction could be blocked by engineering DN-TGFβ-RII alone or complexed to an IL-15 receptor agonist (HCW9218); DN-PD-1 as an ICB; or by genetic deletion of negative regulators in T cells or NK cells. Methods that enhance CAR T cell cytotoxicity include engineering TRUCK CARs that enable CAR-induced cytokine secretion, inserting a cytokine receptor as an additional ICS, engineering constitutively active IL-7 receptor (C7R), or by overexpression of orthoIL-2/orthoIL-2Rβ. Methods that enhance CAR NK cytotoxicity include engineering strategies to increase cytokine payload or ex vivo expansion of CAR NK cells with K562/4-1BBL/membrane-bound IL-21 (mbIL-21) or K562/membrane-bound IL-15 (mb15)/4-1BBL feeder cells, or medium supplemented with IL-2, IL-15 and/or IL-21. Memory-like CAR NK cells have improved persistence and metabolic fitness and can be generated by stimulation with IL-12, IL-15 and IL-18. Examples of multiple-targeting CAR designs to overcome antigen heterogeneity and loss include IL-13/anti-EphA2 Tan-CAR T cells or a multi-targeting CAR NK cell that coexpresses a tumor-cleavable anti-CD73 scFv linked to anti-GD2 CAR along with NKG2D CAR. Binding of anti-CD73 scFv to CD73 blocks the generation of immunosuppressive adenosine from adenosine monophosphate (AMP). CAR T cell infiltration and persistence could be enhanced by overexpressing chemokine receptors or heparanase, which degrades proteoglycan and other extracellular matrix (ECM) proteins, along with a CAR. αFAP-scFv CAR T cells target FAP on cancer-associated fibroblasts to mitigate FAP-induced ECM remodeling. Replacing scFv with FAP on CAR NK cells improves ECM degradation and increases NK cell extravasation and infiltration. CAF, cancer-associated fibroblast; FAP, fibroblast activation protein; MDSC, myeloid-derived suppressor cell. Created with BioRender.com.
CAR NK cells have the CAR backbone utilized for CAR T cell engineering, with more variations in transmembrane and ICS domains7 (Fig. 2a). Replacement of scFv with the extracellular domain of aNKR, as in NKG2D CAR T cells and CAR NK cells, can increase potency against conventional NK cell targets56. Nanobodies are single-domain camelid antibody fragments that outperform conventional monoclonal antibodies, owing to higher stability, improved TME infiltration and easier genetic engineering. Both for CAR T cells and CAR NK cells, replacing the CAR scFv domain with a nanobody has been shown to be preclinically effective against hematological and solid tumors57. A major attribute of CAR NK cells that is lacking in CAR T cells is MHC-independent cytotoxicity, which enables allogeneic NK cells from healthy donors to be used as a source for CAR NK cell expansion and possible generation and storage of cryopreserved ‘off-the-shelf’ cellular products7. However, rejection of adoptively transferred allogeneic CAR NK cells could occur as a result of host immune T and NK cell responses5. To date, most CAR T cells have been generated from autologous apheresis products before adoptive transfer to patients. Allogeneic adoptive transfers can have fatal consequences, such as GvHD, and are prone to host-versus-graft rejections due to MHC incompatibility between donor and recipient. However, genetic deletions of TCR as well as MHC class-I-related and MHC class-II-related genes from healthy donor allogeneic CAR T cells along with overexpression of HLA-E in induced pluripotent stem (iPS) cell-derived CAR T cells permits the generation of universal CAR T cells, which can withstand the recipient host immune system without causing GvHD58,59. Although genetic deletion of MHC class I in donor cells provides protection from recipient allogeneic T cells, it creates a ‘missing-self’ signal exposing donor cells to recipient NK cell attack. Thus, strategies to evade recipient NK cell cytotoxicity involve additional overexpression of HLA-E, HLA-G or CD47 to generate hypoimmunogenic CAR T or CAR NK cells5.
An additional major challenge for successful implementation of CAR T and CAR NK cell therapy is the risk of severe off-tumor toxicities due to shared antigen recognition of normal tissues2,60. To mitigate this problem, several logic-gated and switch-receptor CAR T cells have been developed1. The most recent is a reversible Boolean-logic AND-gated intracellular network (LINK) CAR in which CD3ζ was replaced with the intracellular proximal downstream signaling molecules LAT and SLP-76 to generate CD19-28TM-LAT and HER2-8TM-SLP-76 CAR T cells (Fig. 2a). CD19+HER2+ target cell-induced stimulation of LINK CARs outperformed previously developed logic-gated platforms in terms of improved in vitro and in vivo effector functions and restricted off-tumor on-target toxicities61. Likewise for CAR NK cells, SENTI-202 is a novel ‘OR- and NOT-logic-gated’ CAR NK cell candidate that has been engineered to target acute myeloid leukemia (AML)-associated targets, FLT3 or CD33, by an OR-gated activating CAR, while sparing healthy hematopoietic stem cells from off-tumor toxicity by a NOT-gated EMCN-targeting inhibitory CAR. This would eventually enhance AML leukemic stem cell/blast tumor clearance, while mitigating the need for bone marrow transplantation62.
Clinical trials investigating CAR T and CAR NK cells against hematological tumors have been discussed elsewhere5,7,63. Here we focus on CAR T or CAR NK cell-based clinical trials that target a wide array of solid tumors, extracted from Clinicaltrials.gov as of 1 July 2023, with glioblastoma, breast cancer and ovarian cancer being at the top of the list of targeted tumors. For CAR T cells, the total number of trials against solid tumors is 340 (23 completed, 23 active/not recruiting, 165 actively recruiting or enrolling by invitation, 67 with unknown status, 33 suspended/terminated/withdrawn and 29 not yet recruiting). Meanwhile, 19 solid tumor-targeting CAR NK cell-based trials have been reported (9 actively recruiting, 6 with unknown status, and 4 not yet recruiting). Table 1 lists all CAR NK cell trials and completed CAR T cell trials targeting solid tumors. While clinical data from CAR T cell therapies fall short of reproducing the durable response rates for blood tumors, data for CAR NK cells are pending. For both CAR T and CAR NK cells, intratumoral homing and persistence within the solid tumor milieu are major obstacles yet to be overcome. Clinical data on CAR NK cell homing within solid tumors is lacking; however, input inferred from the first CAR NK cell trial utilizing CD19 CAR NK cells armed with human IL-15 demonstrates persistence of infused CAR NK cells for at least 1 year, albeit at low levels, in peripheral blood of patients25. Overexpressing DNAM-1 and/or NKG2D in NK cells are among the proposed strategies to enhance intratumoral infiltration of NK cells and have shown effectiveness against patient-derived sarcoma specimens in vitro64.
Enhancing antitumor cytotoxicity of CAR T and CAR NK cells, especially against solid tumors, is a growing area of research. Tactics used by researchers include the engineering of costimulatory domains that enhance the activating intracellular signaling of CAR-modified cells. Although the TCR is the only primary receptor mediating T cell cytotoxicity, many transmembrane costimulatory molecules, such as CD28, 4-1BB, ICOS and OX-40, can amplify signaling capacity downstream of the TCR for superior cytotoxic efficacy. Each of these molecules favors distinct signaling pathways that affect CAR T cell cytotoxicity, proliferation and persistence, once included within the ICS domain of the CAR constructs53,65. Meanwhile, NK cells have a variety of activating and inhibitory receptors that recruit distinct downstream signaling mediators, and whose fine-tuning of signaling cues controls the activating versus inhibitory action of NK cells. Studies have shown that tailoring CAR engineering to NK cells using NK-specific signaling domains, such as 2B4 and DAP10 or DAP12, as ICS domains could increase CAR NK cell antitumor efficacy and IFNγ secretion, compared to T cell-tailored CAR constructs65,66 (Fig. 2a).
Overcoming solid tumor evasion is another actively expanding area in CAR-based cellular engineering. Methods to overcome CAR T and CAR NK cell dysfunction include overexpression of dominant negative receptors (DNRs) of TGFβ (DN-TGFβ-RII) or PD-1 (DN-PD-1), or genetic deletions of negative CAR cell regulators, including PD-1 and diacylglycerol kinases15,53,67,68,69,70,71. Orthogonal and/or membrane-bound cytokine/cytokine receptor engineering strategies have also been used to enhance CAR T or CAR NK cell antitumor functions5,25,53,54,72,73,74,75,76,77,78,79,80,81. Multi-targeting and tandem-targeting of TAAs by either CAR T or CAR NK cells can enhance efficacy against solid tumors and associated antigen heterogeneity or loss, as compared to single CARs53,82,83,84,85. Tactics to improve CAR T or CAR NK cell trafficking and infiltration through the stromal barrier include overexpression of chemokine receptors, generation of FAP-scFv CAR T or FAP-overexpressing CAR NK cells, or overexpression of heparanase7,23,86,87,88,89. Figure 2b summarizes prominent engineering approaches that have been investigated.
Combination immunotherapies that enhance antitumor efficacy
Cancer immunotherapies are a broad collection of treatment modalities that aim to engage and boost immune cell effector functions for tumor eradication. Figure 3a depicts prominent models of CAR T and CAR NK combination immunotherapies.

a, Anti-CD3 and anti-CD3/anti-CD28 activating monoclonal antibody-conjugated beads enhance CAR T cell expansion. Targets for CAR NK cell-activating monoclonal antibodies (mAbs) and nanobodies (Nbs) include costimulatory molecules and aNKR, alone or in combination with overexpression of high-affinity CD16 (ha-CD16). Blocking monoclonal antibodies and nanobodies either target immune checkpoint receptors or ligands (ICB), iNKR or IL-6 receptor (IL-6R). Examples of FDA-approved blocking monoclonal antibodies include tocilizumab, atezolizumab, nivolumab and ipilimumab. Lenalidomide enhances cytotoxicity and proliferation of CAR T cells and, by increasing aNKR ligand expression on multiple myeloma (MM) cells, augments CAR NK cell cytotoxicity in combination with KIR blocking monoclonal antibody (IPH2101). OV19t oncolytic viruses induce truncated-CD19 (CD19t) expression by solid tumor cells that become amenable to anti-CD19 CAR T cell cytotoxicity, whereas OV-15C induces expression and secretion of human IL-15/IL-15 receptor alpha sushi domain fusion protein (IL-15–suIL-15Rα) by tumor cells, which improves antitumor efficacy of anti-EGFR CAR NK cells. Examples of immune engagers include BiHC (anti-HER2-Nb/anti-CD3-scFv BiTE) or blinatumomab (anti-CD19-scFv/anti-CD3-scFv BiTE) for CAR T cells, and anti-EGFR-Nb/anti-CD16-Nb BiKE or anti-CD16-Nb/recombinant IL-15/anti-CLEC12A-Nb TriKE that combines ADCC with TAA-specific killing by CAR NK cells. b, Viral CAR transduction leads to CAR integration within T cells or NK cells. c, Examples of nonviral CAR engineering strategies include mRNA delivery by electroporation or lipid nanoparticles, DNA nanovector platform and transposons as well as CRISPR–Cas9 and base editing as gene editing tools. Advanced strategies under development include the use of biomaterials for in situ CAR delivery and expansion as well as targeted delivery and expansion of CAR T cell-based live microrobots (M-CAR T cells), created by conjugation of anti-CD3/anti-CD28 beads with CAR T cells via sequential magnetic acoustic actuation in preclinical models. Created with BioRender.com.
Recombinant monoclonal antibodies, or nanobodies as alternatives, could be used a number of ways to enhance CAR T and CAR NK cell functions57. Activating monoclonal antibodies function as specific agonists for activating receptors or costimulatory molecules, such as CD28, 4-1BB and SLAMF7, important for T cell or NK cell expansion and persistence5,57,90. Tumor-specific monoclonal antibodies are used alone or in conjunction with overexpression of high-affinity CD16 to redirect CAR-expressing cells toward tumor cells and/or enhance ADCC functions, respectively, via opsonization of the Fc portion of antibody-bound tumor cells91,92. Blocking monoclonal antibodies (such as those used in ICB) target an immunosuppressive TME by blocking immune checkpoint receptors or ligands, such as TIM3, CTLA4 and PD(L)-1 (refs. 53,93). Although FDA-approved ICB has provided durable remission in some patients with solid tumors as a monotherapy, its failure in many others is contingent on tumor-specific primary and secondary resistance mechanisms94. NK inhibitory receptor blocking monoclonal antibodies promote NK functions by mimicking the signal of ‘missing self’ that usually occurs upon downregulation or loss of inhibitory ligands5. Other blocking monoclonal antibodies target inflammatory cytokine receptors. For example, tocilizumab is an FDA-approved blocking monoclonal antibody that targets IL-6 receptor (IL-6R) and is used for management of CAR T cell-induced CRS95. Immune engagers, such as BiTE and TriTE (for T cells), and BiKE, TriKE and TetraKE (for NK cells), are bi-specific or multi-specific T cell or NK cell engagers that combine scFv, nanobodies and/or other molecules such as cytokines to facilitate targeted tumor eradication by CAR-expressing cells57,96,97,98,99.
Lenalidomide is an immunomodulator of the E3 ubiquitin-ligase pathway that has versatile immune effects. Besides the induction of cell cycle arrest in tumor cells, lenalidomide can modulate the TME by blocking angiogenesis and suppressing Treg cells, as well as mediate the restoration of lymphoma-associated defects in the immunological synapse100,101. In combination with T cells, lenalidomide is a costimulatory agent that enhances cytotoxicity and cytokine secretion of anti-CD133 or anti-HER2 CAR T cells against solid tumors101. In a phase I clinical trial, lenalidomide promoted NK cell expansion, and in combination with anti-inhibitory KIR therapy enhanced steroid-sparing cytotoxicity against multiple myeloma102.
Oncolytic viruses are a group of engineered viruses that can enhance tumor immunogenicity and dendritic cell-induced NK cell activity, as well as enable tumor-targeted delivery of transgenes7,103. Combined intratumoral treatment of an oncolytic virus expressing truncated-CD19 (OV19t) and anti-CD19 CAR T cells resulted in solid tumor regression103. Likewise, a combination of an oncolytic virus expressing human IL-15/IL-15 receptor alpha sushi domain fusion protein (IL-15–suIL-15Rα) complex (OV-IL15C) and anti-EGFR CAR NK cells provoked synergistic antitumor cytotoxicity and persistence of CAR NK cells and improved survival in a mouse model of glioblastoma75.
For all those combination therapies, preclinical and clinical evaluations are ongoing for blood and solid tumors. While ICB clinical evaluation in combination with CAR T cells seems promising, its role in CAR NK cytotoxicity is debatable7. Nanobody-based therapies might soon predominate in the immunotherapy era against solid tumors due to an ability to penetrate the dense stroma; however, the fast clearance of nanobodies from circulation necessitates additional engineering57. Cytokine armoring for enhancing cytotoxicity and persistence of both CAR-based therapies has proven effective, with more flexible opportunities yet to be explored7,51. Eventually, the multimodal mechanisms mediated by different cancer immunotherapies in enhancing CAR-based therapies, factoring in possible off-tumor toxicities, is critical for future selection of the best combination immunotherapies alongside CAR T and CAR NK cells.
Challenges that affect cytotoxicity and persistence in solid tumors
Sources of T cells for ex vivo expansion include peripheral blood and iPS cell-derived populations, with very few documented reports utilizing umbilical cord expanded T cells as these cells tend to be enriched in Treg cells after expansion59,104,105. Despite the prominent success of CAR T cell therapy against hematological malignancies, it falls short against solid tumors1. Aside from the induced exhaustion of CAR T cell products following long-term ex vivo expansions that affect memory phenotype, longevity and proliferative capacity, the first barrier to impede CAR T cell success in treating solid tumors is the dense stroma formed by cancer-associated fibroblasts that inhibits CAR T cell infiltration to the tumor site106,107. Intratumoral CAR T cells that bypass this barrier could become exhausted after continuous stimulation with target antigen and/or immunosuppressive tumor and associated TME108. Tumor escape can happen as the tumor tries to evade effective intratumoral CAR T cell cytotoxicity by undergoing mutations leading to either antigen loss or antigen heterogeneity3,109. Intratumoral CAR T cells can also have reduced proliferation capacity and loss of persistence as they are expelled to lymph nodes due to compounded intrinsic and extrinsic factors108,109.
Unlike T cells, which can live for months, NK cells have a short lifespan, with a half-life of less than 10 d in young adults, a limiting factor that highlights the need for repeated infusion of NK cell products to maintain efficacy and persistence in vivo110. Moreover, the sensitivity of expanded NK cells for freezing and thawing affects both cytotoxicity and survival after adoptive transfer3,110. A recorded sixfold decrease in the fraction of migratory NK cells in a three-dimensional-tumor model after thawing, with severe effects on cytotoxic efficacy, could partially explain the impaired persistence of cryopreserved CAR NK cells in vivo111. Studies are currently investigating the optimization of cryopreservation protocols, including utilizing alternatives to dimethyl sulfoxide, a cryoprotectant traditionally used in freezing cells, to enable the preservation of integrity, viability, function and potency of CAR T and CAR NK cells after thawing and facilitate future off-the-shelf production and cryopreservation112.
Ex vivo expansion of NK cells requires priming with cytokines to prevent NK cell exhaustion and senescence6. NK cells can be expanded from multiple sources for use in clinical testing, such as peripheral blood, umbilical cord, iPS cells and immortalized NK cell lines such as NK-92 cells; each source having specific limitations and advantages5. The generation of CAR NK cells mirrors the ex vivo expansion protocols of unedited NK cells, albeit with additional viral or nonviral engineering of the latter to express CAR constructs4. Those expansion protocols rely on continuous cytokine stimulation with or without coactivation by feeder cells5,6,7,76,77,81. Although expanded CAR NK cells provide prominent cytotoxicity against target cells in vitro, studies have shown that their dependence on cytokine and activating signals might be the reason for the state of dysfunction and reduced longevity after adoptive transfer to patients, especially as those signaling cues become sparse within the TME113.
NK cells are exposed to many tumor-induced and TME-mediated cytokines and metabolites that can reduce NK cell persistence and function. Analysis of blood and solid tumor samples from patients has shown that tumor-mediated and TME-mediated NK cell dysfunction, as well as impaired cytotoxicity, persistence and longevity, can be summarized by five main mechanisms: exhaustion, suppression, anergy, plasticity and defective proliferation6,112. Tumor-associated factors that lead to NK cell exhaustion include persistent activation of NK cells by tumor cells leading to reduced effector functions. Tumor-associated and TME-associated release of TG-β and other suppressive metabolites induces downregulation of aNKR; decreased lytic granule release; defective cytokine production; and overexpression of iNKR. Anergy could be due to excessive inhibitory signaling or to continuous stimulation of aNKR during lack of licensing from iNKRs6,113. Since NK cells belong to group 1 innate lymphoid cells (ILC1s), tumor-associated factor-induced plasticity between ILC1 and ILC3 subclasses may permit the conversion of conventional cytotoxic NK cells to the less cytotoxic or ILC1-like phenotype, bearing pro-tumorigenic potential6,114. NK cell dysfunction is also accompanied by defective proliferative capacity and decreased number of NK infiltrating cells6,110,115.
Current engineering strategies for efficient CAR T and CAR NK cell development include armoring these cells through additional genetic manipulations or combination therapies that address T and NK cell dysfunction and persistence.
Delivery technologies to enhance CAR T and CAR NK cells
Traditional generation of CAR T cells involves viral-vector delivery of CAR constructs to enable high transduction efficiency and stable expression of a CAR. The most widely used vectors are lentivirus, retrovirus or adeno-associated virus vectors4. Compared with T cells, NK cells are more difficult to engineer, due to inherent antiviral capacity, higher sensitivity to apoptosis and limited expansion potential4,116. As the lentivirus envelope directly affects transduction efficiency in primary NK cells, lentiviruses could be pseudotyped with vesicular stomatitis virus G (VSV-G), gibbon ape leukemia virus envelope (GALV), feline endogenous retrovirus envelope protein (RD114-TR) or baboon envelope (BaEV), with the latter demonstrating superior transduction efficiency comparable to that of retroviruses4,117,118 (Fig. 3b). However, increased immunogenicity, possible insertional mutagenesis, limited insert size, heightened cost for good manufacturing practice-grade viral production and associated regulatory complications are major drawbacks that have motivated exploration of nonviral delivery routes as cost-effective alternatives4,119. The nonviral transposon platforms Sleeping Beauty and PiggyBac enable safer integration of CAR constructs but with lower efficiency than viral systems4. Nonviral delivery of mRNA-based CAR constructs by either electroporation or lipid nanoparticles generates high but transient CAR expression efficiency116,120. Scientists have also developed an easy, versatile, affordable, nonviral and nonintegrating DNA nanovector platform that enables both in vitro and in vivo nonchromosomal CAR expression in T cells with lack of immunogenicity or genotoxicity121. Additional nonviral gene editing platforms include CRISPR–Cas9 that enable site-directed integration of CAR and other favorable genes or deletions of immune cell negative regulators, TCR or MHC-related genes, respectively122. In one study, fratricide-resistant dual CAR T cells were generated by combining base editing of T cell-related genes with CAR-lentiviral transduction123. Finally, biomaterial delivery vehicles permit site-directed transfer of edited or unedited immune cells, with options for viral or nonviral editing in situ, without prior ex vivo expansion, whereas CAR T cell-based live microrobots enable targeted delivery and activation of CAR T cells by magnetic acoustic actuation (Fig. 3c)1,124.
Conclusion
The past decade has unveiled a revolutionary triumph in the engineering of CAR-based immunotherapies, with six FDA-approved CAR T cell therapies that have enabled decade-long remissions from hematological malignancies in some pediatric and adult patients. However, the prevalence of relapse after CAR T cell infusion in patients with hematological malignancies is variable, and achieving significant numbers of complete remissions against solid tumors has not yet been achieved. Factors such as CAR T and CAR NK cell exhaustion, hypo-responsiveness and rejection by the host immune system are thought to be major contributors of tumor resistance and relapse in many patients7,106. Thus, achieving optimal CAR-based immune cell functionality requires tackling the multiple challenges that these cell therapies face alongside optimization of clinical administration settings and leveraging of combinatorial immunotherapeutic modalities.
Although some CAR NK and CAR T cell-based preclinical studies demonstrated effective cytotoxicity against solid tumors, clinical data are sparse for CAR NK cells, while CAR T cells are yet to succeed clinically3,5. Future therapeutic regimens should manipulate engineering strategies and combination therapies to tackle the challenges hindering CAR T and CAR NK cell efficacy against solid tumors. These include surmounting tumor-induced antigen heterogeneity or loss; fostering long-term intratumoral antigenic encounter for effective on-tumor cytotoxicity; sustaining better persistence and retention of CAR-expressing cells intratumorally; enduring hypoxic and immunosuppressive TME; and maintenance of memory-like CAR cell repertoires for longer lasting and more tumor-specific cytotoxicity. Additional challenges linked to lytic synapse formation and stability, such as immune cell-target recognition, binding kinetics and potency should be addressed. Methods to rid recipients of hyperactive or toxic CAR cells, including logic-gated and switch CAR designs besides other novel approaches, should continue to be examined until a best-in-class platform is achieved. Eventually, successful development of efficient CAR therapies against solid tumors will probably require a holistic combinatorial approach, rather than single targeting of individual impediments.
Future ‘off-the-shelf’ production of allogeneic universal CAR T cell products from MHC-mismatched healthy donors is needed to facilitate global delivery of already FDA-approved products to patients at critical stages of illness, within underprivileged communities, or at medical institutions that lack CAR T cell development facilities. Building on the MHC-unrestricted cytotoxicity, reduced off-tumor on-target toxicities, and ‘off-the-shelf’ production amenability of allogeneic CAR NK cells, we envision the next decade will bring further research findings that help in the optimization of NK cell expansion workflows; mitigate NK cell exhaustion, improve post-thawing recovery and enhance post-adoptive transfer cytotoxicity and longevity. However, as allogeneic CAR NK cells can promote immunogenicity and undergo later rejection by host immune cells5, additional genetic manipulations reminiscent of those applied to universal CAR T cells are required. Novel strategies that create optimal feeder-free expansions should also be adopted to enable mass production of CAR NK cells and circumvent both regulatory and consistency complications.
Most clinically available data regarding CAR T or CAR NK cell antitumor efficacy have been extrapolated from analysis of heavily pretreated patients at advanced stages of cancer, while lacking evidence of effectiveness as first-line therapies at earlier stages of tumor progression. Hence, further investigation of the optimal time, dosage, route and frequency of administration of CAR-based therapies for each tumor type would better inform clinical decisions and expand the scope of treatment-eligible patients. Another major drawback in CAR-based cellular therapies is the considerably elevated cost of treatment, including symptomatic management of accompanying side effects. Future steps that should help alleviate heightened costs include optimizing the ‘off-the-shelf’ production and cryopreservation of ‘universal’ engineered CAR products, mitigating off-tumor on-target toxicity, shortening ex vivo expansion workflows and optimization of nonviral CAR delivery120 as well as fine-tuning in vivo kinetics and the potency of individual CAR designs.
References
Irvine, D. J., Maus, M. V., Mooney, D. J. & Wong, W. W. The future of engineered immune cell therapies. Science 378, 853–858 (2022).
Lee, D. W. et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 25, 625–638 (2019).
Lu, H., Zhao, X., Li, Z., Hu, Y. & Wang, H. From CAR-T cells to CAR-NK cells: a developing immunotherapy method for hematological malignancies. Front. Oncol. 11, 720501 (2021).
Schmidt, P., Raftery, M. J. & Pecher, G. Engineering NK Cells for CAR therapy-recent advances in gene transfer methodology. Front. Immunol. 11, 611163 (2020).
Berrien-Elliott, M. M., Jacobs, M. T. & Fehniger, T. A. Allogeneic natural killer cell therapy. Blood 141, 856–868 (2023).
Coyle, K. M., Hawke, L. G. & Ormiston, M. L. Addressing natural killer cell dysfunction and plasticity in cell-based cancer therapeutics. Cancers 15, 1743 (2023).
Laskowski, T. J., Biederstädt, A. & Rezvani, K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer 22, 557–575 (2022).
Ramírez-Labrada, A. et al. All About (NK cell-mediated) death in two acts and an unexpected encore: initiation, execution and activation of adaptive immunity. Front. Immunol. 13, 896228 (2022).
Böttcher, J. P. et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 172, 1022–1037 (2018).
Huntington, N. D., Cursons, J. & Rautela, J. The cancer-natural killer cell immunity cycle. Nat. Rev. Cancer 20, 437–454 (2020).
Pai, J. A. et al. Lineage tracing reveals clonal progenitors and long-term persistence of tumor-specific T cells during immune checkpoint blockade. Cancer Cell 41, 776–790 (2023).
Zheng, L. et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021).
Oh, D. Y. et al. Intratumoral CD4+ T cells mediate anti-tumor cytotoxicity in human bladder cancer. Cell 181, 1612–1625(2020).
Zelenay, S. et al. Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162, 1257–1270 (2015).
Cherkassky, L. et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 126, 3130–3144 (2016).
Steele, M. M. et al. T cell egress via lymphatic vessels is tuned by antigen encounter and limits tumor control. Nat. Immunol. 24, 664–675 (2023). This study unveils the CXCR4–CXCL12 axis as an important target for enhancing intratumoral T cell retention in vivo.
Thacker, G. et al. Immature natural killer cells promote progression of triple-negative breast cancer. Sci. Transl. Med. 15, eabl4414 (2023). This study uncovers a ‘regulatory-like’ immature NK cell population that could be targeted to improve efficacy of immunotherapies against triple-negative breast cancer.
Nuñez, S. Y. et al. Human M2 macrophages limit NK cell effector functions through secretion of TGF-β and engagement of CD85j. J. Immunol. 200, 1008–1015 (2018).
Kloosterman, D. J. & Akkari, L. Macrophages at the interface of the co-evolving cancer ecosystem. Cell 186, 1627–1651 (2023).
Reinfeld, B. I. et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288 (2021).
Zalfa, C. & Paust, S. Natural killer cell interactions with myeloid derived suppressor cells in the tumor microenvironment and implications for cancer immunotherapy. Front. Immunol. 12, 633205 (2021).
Eschweiler, S. et al. Intratumoral follicular regulatory T cells curtail anti-PD-1 treatment efficacy. Nat. Immunol. 22, 1052–1063 (2021).
Maalej, K. M. et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol. Cancer 22, 20 (2023).
Tai, L. H., Zhang, J. & Auer, R. C. Preventing surgery-induced NK cell dysfunction and cancer metastases with influenza vaccination. Oncoimmunology 2, e26618 (2013).
Liu, E. et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 382, 545–553 (2020).
Xiao, X. et al. Mechanisms of cytokine release syndrome and neurotoxicity of CAR T-cell therapy and associated prevention and management strategies. J. Exp. Clin. Cancer Res. 40, 367 (2021).
Zhang, X. et al. Cytokine release syndrome after modified CAR-NK therapy in an advanced non-small cell lung cancer patient: a case report. Cell Transplant. 31, 9636897221094244 (2022).
Klingemann, H. Are natural killer cells superior CAR drivers? Oncoimmunology 3, e28147 (2014).
Paul, S. & Lal, G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front. Immunol. 8, 1124 (2017).
Whang, M. et al. Large-scale expansion and engineering of human NK cells sourced from peripheral blood versus umbilical cord blood. J. Immunother. Cancer 10, A401 (2022).
Min, B. et al. Optimization of large-scale expansion and cryopreservation of human natural killer cells for anti-tumor therapy. Immune Netw. 18, e31 (2018).
Davenport, A. J. et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc. Natl Acad. Sci. USA 115, E2068–E2076 (2018). This study identifies an immature immunological synapse formed by CAR molecules in CAR-T cells as compared to canonical TCR.
Monks, C. R., Freiberg, B. A., Kupfer, H., Sciaky, N. & Kupfer, A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature 395, 82–86 (1998).
Watanabe, K., Kuramitsu, S., Posey, A. D. Jr. & June, C. H. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front. Immunol. 9, 2486 (2018).
Al-Aghbar, M. A., Jainarayanan, A. K., Dustin, M. L. & Roffler, S. R. The interplay between membrane topology and mechanical forces in regulating T cell receptor activity. Commun. Biol. 5, 40 (2022).
Somersalo, K. et al. Cytotoxic T lymphocytes form an antigen-independent ring junction. J. Clin. Invest. 113, 49–57 (2004).
Potter, T. A., Grebe, K., Freiberg, B. & Kupfer, A. Formation of supramolecular activation clusters on fresh ex vivo CD8+ T cells after engagement of the T cell antigen receptor and CD8 by antigen-presenting cells. Proc. Natl Acad. Sci. USA 98, 12624–12629 (2001).
Stinchcombe, J. C., Bossi, G., Booth, S. & Griffiths, G. M. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 15, 751–761 (2001).
Orange, J. S. Formation and function of the lytic NK-cell immunological synapse. Nat. Rev. Immunol. 8, 713–725 (2008).
Schmied, L. et al. SHP-1 localization to the activating immune synapse promotes NK cell tolerance in MHC class I deficiency. Sci. Signal. 16, eabq0752 (2023).
Davis, D. M. & Dustin, M. L. What is the importance of the immunological synapse? Trends Immunol. 25, 323–327 (2004).
Mukherjee, M., Mace, E. M., Carisey, A. F., Ahmed, N. & Orange, J. S. Quantitative imaging approaches to study the CAR immunological synapse. Mol. Ther. 25, 1757–1768 (2017).
Jenkins, M. R. et al. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J. Exp. Med. 212, 307–317 (2015).
Masilamani, M., Nguyen, C., Kabat, J., Borrego, F. & Coligan, J. E. CD94/NKG2A inhibits NK cell activation by disrupting the actin network at the immunological synapse. J. Immunol. 177, 3590–3596 (2006).
Williams, G. S. et al. Membranous structures transfer cell surface proteins across NK cell immune synapses. Traffic 8, 1190–1204 (2007).
McCann, F. E. et al. The size of the synaptic cleft and distinct distributions of filamentous actin, ezrin, CD43, and CD45 at activating and inhibitory human NK cell immune synapses. J. Immunol. 170, 2862–2870 (2003).
Zheng, X. et al. Tumors evade immune cytotoxicity by altering the surface topology of NK cells. Nat. Immunol. 24, 802–813 (2023). This study introduces a mechanism of tumor evasion in which tumor cells alter sphingomyelin content on intratumoral NK cells, leading to perturbed membrane protrusions and decreased cytotoxicity.
Chockley, P. J., Ibanez-Vega, J., Krenciute, G., Talbot, L. J. & Gottschalk, S. Synapse-tuned CARs enhance immune cell anti-tumor activity. Nat. Biotechnol. https://doi.org/10.1038/s41587-41022-01650-41582 (2023). This study introduces an approach by which tuning the synapse of CAR T or CAR NK cells could enhance their efficacy and intratumoral survival against solid tumors.
Lin, J. & Weiss, A. The tyrosine phosphatase CD148 is excluded from the immunologic synapse and down-regulates prolonged T cell signaling. J. Cell Biol. 162, 673–682 (2003).
Anikeeva, N. et al. Efficient killing of tumor cells by CAR-T cells requires greater number of engaged CARs than TCRs. J. Biol. Chem. 297, 101033 (2021).
Labanieh, L. & Mackall, C. L. CAR immune cells: design principles, resistance and the next generation. Nature 614, 635–648 (2023).
Kershaw, M. H. et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin. Cancer Res. 12, 6106–6115 (2006).
Tian, Y., Li, Y., Shao, Y. & Zhang, Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J. Hematol. Oncol. 13, 54 (2020).
Kagoya, Y. et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat. Med. 24, 352–359 (2018).
Dagher, O. K., Schwab, R. D., Brookens, S. K. & Posey, A. D. Jr. Advances in cancer immunotherapies. Cell 186, 1814–1814 (2023).
Curio, S., Jonsson, G. & Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 1, ltab018 (2021).
Maali, A. et al. Nanobodies in cell-mediated immunotherapy: on the road to fight cancer. Front. Immunol. 14, 1012841 (2023).
Lin, H., Cheng, J., Mu, W., Zhou, J. & Zhu, L. Advances in universal CAR-T cell therapy. Front. Immunol. 12, 744823 (2021).
Wang, B. et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat. Biomed. Eng. 5, 429–440 (2021).
Doan, A. & Pulsipher, M.A. Hypogammaglobulinemia due to CAR T cell therapy. Pediatr. Blood Cancer 65, e26914 (2018).
Tousley, A. M. et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature 615, 507–516 (2023). This study introduces a gated-CAR platform in whichreplacing CD3ζ with downstream intracellular molecules such as SLP-76 and LAT enhanced the antitumor activity and mitigated on-target off-tumor toxicities.
Garrison, B. S. et al. FLT3 OR CD33 NOT EMCN logic gated CAR-NK cell therapy (SENTI-202) for precise targeting of AML. Blood 138, 2799 (2021).
Zhang, X., Zhu, L., Zhang, H., Chen, S. & Xiao, Y. CAR-T cell therapy in hematological malignancies: current opportunities and challenges. Front. Immunol. 13, 927153 (2022).
Sayitoglu, E. C. et al. Boosting natural killer cell-mediated targeting of sarcoma through DNAM-1 and NKG2D. Front. Immunol. 11, 40 (2020).
Ruppel, K. E., Fricke, S., Köhl, U. & Schmiedel, D. Taking Lessons from CAR-T cells and going beyond: tailoring design and signaling for CAR-NK cells in cancer therapy. Front. Immunol. 13, 822298 (2022).
Wrona, E., Borowiec, M. & Potemski, P. CAR-NK cells in the treatment of solid tumors. Int. J. Mol. Sci. 22, 5899 (2021).
Kloss, C. C. et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 26, 1855–1866 (2018).
Chaudhry, K. et al. Co-transducing B7H3 CAR-NK cells with the DNR preserves their cytolytic function against GBM in the presence of exogenous TGF-β. Mol. Ther. Methods Clin. Dev. 27, 415–430 (2022).
Liu, X. et al. A novel dominant-negative PD-1 armored anti-CD19 CAR T cell is safe and effective against refractory/relapsed B cell lymphoma. Transl. Oncol. 14, 101085 (2021).
Liu, B. et al. Bifunctional TGF-β trap/IL-15 protein complex elicits potent NK cell and CD8+ T cell immunity against solid tumors. Mol. Ther. 29, 2949–2962 (2021).
Jung, I. Y. et al. CRISPR/Cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. 78, 4692–4703 (2018).
Aspuria, P. J. et al. An orthogonal IL-2 and IL-2Rβ system drives persistence and activation of CAR T cells and clearance of bulky lymphoma. Sci. Transl. Med. 13, eabg7565 (2021).
Zhang, Q. et al. A human orthogonal IL-2 and IL-2Rβ system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci. Transl. Med. 13, eabg6986 (2021).
Thomas, S. & Abken, H. CAR T cell therapy becomes CHIC: ‘cytokine help intensified CAR’ T cells. Front. Immunol. 13, 1090959 (2022).
Ma, R. et al. An oncolytic virus expressing IL15/IL15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 81, 3635–3648 (2021).
Imai, C., Iwamoto, S. & Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 106, 376–383 (2005).
Denman, C. J. et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE 7, e30264 (2012).
Shum, T. et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 7, 1238–1247 (2017).
Adachi, K. et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 36, 346–351 (2018).
Chen, Y. et al. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin. Cancer Res. 25, 2915–2924 (2019).
Romee, R. et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 8, 357ra123 (2016).
Wang, J. et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc. Natl Acad. Sci. USA 118, e2107507118 (2021).
Hegde, M. et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 126, 3036–3052 (2016).
Bielamowicz, K. et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro. Oncol. 20, 506–518 (2018).
Schmidts, A. et al. Tandem chimeric antigen receptor (CAR) T cells targeting EGFRvIII and IL-13Rα2 are effective against heterogeneous glioblastoma. Neurooncol. Adv. 5, vdac185 (2023).
Caruana, I. et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 21, 524–529 (2015).
Jin, L. et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 10, 4016 (2019).
Bughda, R., Dimou, P., D’Souza, R. R. & Klampatsa, A. Fibroblast activation protein (FAP)-targeted CAR-T cells: launching an attack on tumor stroma. Immunotargets Ther. 10, 313–323 (2021).
Weiner, L. M. inventor. Fibroblast activation protein modulation to alter immune cell migration and tumor infiltration. US patent application no. 22/165,019 (2022).
Storti, P. et al. Novel approaches to improve myeloma cell killing by monoclonal antibodies. J. Clin. Med. 9, 2864 (2020).
Rataj, F. et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br. J. Cancer 120, 79–87 (2019).
Capuano, C. et al. Harnessing CD16-mediated NK cell functions to enhance therapeutic efficacy of tumor-targeting mAbs. Cancers 13, 2500 (2021).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Invest. 128, 4654–4668 (2018).
Sharma, P. et al. Immune checkpoint therapy-current perspectives and future directions. Cell 186, 1652–1669 (2023).
Le, R. Q. et al. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist 23, 943–947 (2018).
Xing, J. et al. BiHC, a T-cell-engaging bispecific recombinant antibody, has potent cytotoxic activity against Her2 tumor cells. Transl. Oncol. 10, 780–785 (2017).
Goebeler, M. E. & Bargou, R. Blinatumomab: a CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk. Lymphoma 57, 1021–1032 (2016).
van Faassen, H. et al. Incorporation of a novel CD16-specific single-domain antibody into multispecific natural killer cell engagers with potent ADCC. Mol. Pharm. 18, 2375–2384 (2021).
Arvindam, U. S. et al. A trispecific killer engager molecule against CLEC12A effectively induces NK-cell mediated killing of AML cells. Leukemia 35, 1586–1596 (2021).
Gandhi, A. K. et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4(CRBN.). Br. J. Haematol. 164, 811–821 (2014).
Wang, Z. et al. Lenalidomide enhances CAR-T cell activity against solid tumor cells. Cell Transplant. 29, 963689720920825 (2020).
Benson, D. M. Jr. et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin. Cancer Res. 21, 4055–4061 (2015).
Park, A. K. et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci. Transl. Med. 12, eaaz1863 (2020).
Liu, D. D. et al. Umbilical cord blood: a promising source for allogeneic CAR-T cells. Front. Oncol. 12, 944248 (2022).
Motwani, K. et al. Human regulatory T cells from umbilical cord blood display increased repertoire diversity and lineage stability relative to adult peripheral blood. Front. Immunol. 11, 611 (2020).
Zhu, X., Li, Q. & Zhu, X. Mechanisms of CAR T cell exhaustion and current counteraction strategies. Front. Cell Dev. Biol. 10, 1034257 (2022).
Arpinati, L. & Scherz-Shouval, R. From gatekeepers to providers: regulation of immune functions by cancer-associated fibroblasts. Trends Cancer 9, 421–443 (2023).
Finck, A. V., Blanchard, T., Roselle, C. P., Golinelli, G. & June, C. H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 28, 678–689 (2022).
Brookens, S. K. & Posey, A. D. Jr. Chimeric antigen receptor T-cell therapy: current perspective on T cell-intrinsic, T cell-extrinsic, and therapeutic limitations. Cancer J. 29, 28–33 (2023).
Nayar, S., Dasgupta, P. & Galustian, C. Extending the lifespan and efficacies of immune cells used in adoptive transfer for cancer immunotherapies—a review. Oncoimmunology 4, e1002720 (2015).
Mark, C. et al. Cryopreservation impairs 3-D migration and cytotoxicity of natural killer cells. Nat. Commun. 11, 5224 (2020).
Yao, X. & Matosevic, S. Cryopreservation of NK and T cells without DMSO for adoptive cell-based immunotherapy. BioDrugs 35, 529–545 (2021).
Judge, S. J., Murphy, W. J. & Canter, R. J. Characterizing the dysfunctional NK cell: assessing the clinical relevance of exhaustion, anergy, and senescence. Front. Cell. Infect. Microbiol. 10, 49 (2020).
Heinrich, B. et al. The tumour microenvironment shapes innate lymphoid cells in patients with hepatocellular carcinoma. Gut 71, 1161–1175 (2022).
Zhang, W., Zhao, Z. & Li, F. Natural killer cell dysfunction in cancer and new strategies to utilize NK cell potential for cancer immunotherapy. Mol. Immunol. 144, 58–70 (2022).
El-Mayta, R., Zhang, Z., Hamilton, A. G. & Mitchell, M. J. Delivery technologies to engineer natural killer cells for cancer immunotherapy. Cancer Gene Ther. 28, 947–959 (2021).
Colamartino, A. B. L. et al. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front. Immunol. 10, 2873 (2019).
Ojeda, P. Biological engineering of natural killer cells for cellular therapy against cancer. Master’s thesis, Harvard Medical School (2020).
Balke-Want, H. et al. Non-viral chimeric antigen receptor (CAR) T cells going viral. Immunooncol. Technol. 18, 100375 (2023).
Moretti, A. et al. The past, present, and future of non-viral CAR T cells. Front. Immunol. 13, 867013 (2022).
Bozza, M. et al. A nonviral, nonintegrating DNA nanovector platform for the safe, rapid, and persistent manufacture of recombinant T cells. Sci. Adv. 7, eabf1333 (2021).
Stadtmauer, E. A. et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367, eaba7365 (2020).
Georgiadis, C. et al. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia 35, 3466–3481 (2021).
Tang, X. et al. Magnetic-acoustic sequentially actuated CAR T cell microrobots for precision navigation and in situ antitumor immunoactivation. Adv. Mater. 35, e2211509 (2023). This study uses biomedical engineering strategies that enable the use of CAR T cells as live microrobots that could be targeted and activated in situ.
Acknowledgements
A.D.P. is supported by funding from the V Foundation, Lustgarten Foundation and Veterans Affairs (I01 BX006247).
Author information
Authors and Affiliations
Contributions
O.K.D. and A.D.P. contributed to the writing and editing of the manuscript. O.K.D. designed the figures.
Corresponding authors
Ethics declarations
Competing interests
A.D.P. is an inventor on patents related to CAR T cell therapies. The other authors declare no competing interests.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Nick Bernard, in collaboration with the Nature Immunology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Dagher, O.K., Posey, A.D. Forks in the road for CAR T and CAR NK cell cancer therapies. Nat Immunol 24, 1994–2007 (2023). https://doi.org/10.1038/s41590-023-01659-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-023-01659-y
This article is cited by
-
In situ genetic engineering of host T-cells based on acellular scaffold strategy: a big but also small step for solid tumor immunotherapy
Military Medical Research (2024)
-
Combinational delivery of TLR4 and TLR7/8 agonist enhanced the therapeutic efficacy of immune checkpoint inhibitors to colon tumor
Molecular and Cellular Biochemistry (2024)
