
Abstract
The lack of samples for generating standardized DNA datasets for setting up a sequencing pipeline or benchmarking the performance of different algorithms limits the implementation and uptake of cancer genomics. Here, we describe reference call sets obtained from paired tumor–normal genomic DNA (gDNA) samples derived from a breast cancer cell line—which is highly heterogeneous, with an aneuploid genome, and enriched in somatic alterations—and a matched lymphoblastoid cell line. We partially validated both somatic mutations and germline variants in these call sets via whole-exome sequencing (WES) with different sequencing platforms and targeted sequencing with >2,000-fold coverage, spanning 82% of genomic regions with high confidence. Although the gDNA reference samples are not representative of primary cancer cells from a clinical sample, when setting up a sequencing pipeline, they not only minimize potential biases from technologies, assays and informatics but also provide a unique resource for benchmarking ‘tumor-only’ or ‘matched tumor–normal’ analyses.
Similar content being viewed by others


Main
Accurate somatic mutation detection is essential for cancer genomics1,2,3,4 and precision cancer medicine5,6. Despite rapid advances in sequencing technologies, it remains challenging to detect somatic mutations accurately using next-generation sequencing (NGS). It is difficult to obtain consistent and concordant somatic mutation calls from individual platforms or pipelines7,8,9, hampering the development of personalized therapies. In addition, quality control of somatic mutation pipelines is often insufficient due to the lack of well-validated and publicly available reference samples and reference datasets. Therefore, paired tumor–normal reference samples with high-confidence somatic mutation calls are desirable and urgently needed10. As more sequencing technologies and bioinformatics pipelines are used to identify clinically actionable somatic mutations, the need grows even stronger5.
Although reference samples and call sets have been established for benchmarking germline variant calls by the Genome in a Bottle (GIAB) Consortium11,12, no such resource exists for benchmarking somatic variant calls. Thus, reference samples for germline variant calls are often used unwillingly to validate somatic mutation pipelines13,14,15. However, accurate detection of somatic mutations is much more challenging due to variable variant allele frequency (VAF), inter- and intratumor heterogeneity, prevalent copy number alterations (CNAs) and complex chromosomal rearrangements. Although gene-specific reference samples are available16,17 (https://www.horizondiscovery.com/reference-standards/type/oncospan), they are unsuitable to benchmark sequencing pipelines applied to the whole genome. Simulated sequence alignment data have been used to benchmark calling algorithms of single-nucleotide variants18 (SNVs) and structural variations19, but they are not feasible to benchmark sequencing technologies, sequencing protocols and alignment algorithms. The International Cancer Genome Consortium (ICGC) argued that real, not simulated, somatic mutations are preferred, and ICGC provided two whole-genome sequencing (WGS) benchmark datasets (from a chronic lymphocytic leukemia (CLL) case and a medulloblastoma (MB) case20). Similarly, a somatic WGS reference call set of a metastatic melanoma cell line has been provided21. Somatic mutations called in those two reference samples have not been successfully validated by orthogonal sequencing technologies. ICGC tried MiSeq and Ion Torrent for the CLL case, but the validation rate was low due to technical issues20. A recent landscape survey of current somatic reference samples by the Medical Devices Innovation Consortium did not identify any readily accessible tumor–normal reference samples for evaluating the somatic mutation calling accuracy on a whole-genome basis22.
To establish reference data and call sets for benchmarking somatic mutation calling, we extracted gDNA samples from a triple-negative breast cancer (TNBC) cell line (HCC1395) and a B lymphocyte-derived normal cell line (HCC1395BL) from the same donor from the American Type Culture Collection (ATCC). Unlike reference call sets derived from CLL and MB tumors with low mutation burden and very limited structural changes, the HCC1395 cell line is rich in genomic alterations (~40,000 SNVs, ~2,000 small insertions and deletions (indels), CNAs in ~56% of the genome and >256 complex genomic rearrangements23, an aneuploid genome and BRCAness24) and has been characterized previously using cytogenetic analysis25 and array-based comparative genomic hybridization26. Moreover, we preferred HCC1395 over a normal cell line with engineered mutations27,28,29 or synthetic DNA spike-in30 because it is difficult, if not impossible, to engineer a whole range of somatic mutations into host chromosomes to mimic the complexity and heterogeneity of a cancer genome.
Readers should note, however, that the genome of the HCC1395 cell line does not necessarily represent TNBC cancer genomes accurately. Unlike the 1000 Genomes Project31, which served as a detailed catalog of common human germline variants, our reference call sets are not designed to be a detailed catalog of somatic mutations of breast cancer. Rather, they include a wide range of mutations that occur naturally in vivo and later in cell cultures and, thus, are suitable to benchmark, develop and refine protocols and tools for somatic mutation detection. In addition, our reference call sets were developed with gDNA extracted from fresh cells and sequenced by Illumina short-read technology. Thus, they are not suitable for benchmarking different sequencing technologies or applications using different sample types, such as formalin-fixed paraffin-embedded (FFPE) samples or circulating tumor DNA (ctDNA).
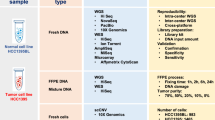
We sequenced the whole genomes of the paired tumor–normal cell lines using WGS (at 1,500× coverage) across seven sequencing centers. The sequencing reads were aligned, and somatic mutations were called by various bioinformatics pipelines. Thus, we minimized biases specific to the sequencing platforms and centers, or the bioinformatics algorithms, and created high-confidence mutation calls across the whole genome of the HCC1395 cell line, namely the ‘reference call set’. A subset of these mutation calls was validated by targeted sequencing (at 2,000× coverage), WES using HiSeq (at 2,500× coverage) and Ion Torrent (at 34× coverage) and long-read WGS by PacBio Sequel (at 40× coverage). In addition, we inferred subclones and heterogeneity of the HCC1395 cell line with bulk DNA sequencing. The results were confirmed by single-cell DNA sequencing analysis. Finally, we provided high-confidence germline and somatic call sets with clinically relevant annotations.
The established call sets have at least two advantages over the others: (1) the whole genomes of reference samples were sequenced with deeper coverage at a total of 1,500× and were validated by orthogonal sequencing platforms; consequently, high-confidence clonal and subclonal somatic mutations were called and validated; and (2) somatic mutations called from 378 datasets (21 sequencing replicates by three aligners and by six somatic mutation callers) were consolidated by two state-of-the-art machine learning-based somatic mutation classifiers (SomaticSeq32 and NeuSomatic33) to construct a high-confidence somatic reference call set, which alleviated calling errors specific to sequencing platform, sequencing site or bioinformatics algorithm.
Results
Study design with multiple sequencing platforms and various variant calling pipelines
gDNA was extracted from each of the matched cell lines (HCC1395 and HCC1395BL) in single batches for the study. To establish the high-confidence call sets for these batches of gDNA, we generated high-coverage short-read WGS data using multiple platforms at seven sequencing centers. We performed long-read WGS (with PacBio), WES (with HiSeq and Ion Torrent) and AmpliSeq (with MiSeq) to confirm our findings and used CytoScan microarray and 10x Genomics single-cell copy number variation (CNV) analysis to uncover the cytogenetic properties and heterogeneity of the two cell lines (Table 1).
The initial somatic mutation call sets were obtained from 21 replicates of HCC1395 and HCC1395BL cells with three aligners (BWA-MEM34, Bowtie2 (ref. 35) and NovoAlign (http://www.novocraft.com/products/novoalign/)) and six mutation callers (MuTect2 (ref. 13), SomaticSniper36, VarDict37, MuSE38, Strelka239 and TNscope40). We used SomaticSeq32 and NeuSomatic33 to score each mutation call based on evidence collected from the 378 call sets. In addition, two pooled datasets with 400× coverage from NovaSeq data of nine replicates and 350× coverage from the WGS-tumor content replicates were used to improve mutation calls with VAFs of <15%. Finally, we used NeuSomatic33 to confirm and complement the mutation calls with all WGS data with 1,500× coverage (Fig. 1).

Twenty-one sequencing replicates for the tumor (HCC1395) and normal (HCC1395BL) gDNA samples were performed at six sequencing centers. They were grouped into five groups shown as colored squares on the far left. The sequencing platforms used were HiSeq and NovaSeq, and the sequencing experiments were performed at Illumina (IL), Fudan University (FD), Novartis (NV), European Infrastructure for Translational Medicine (EA), National Cancer Institute (NC) and Loma Linda University (LL). Each of the 21 sequencing datasets was aligned with three aligners to create a total of 63 pairs of tumor–normal BAM files. For each of the 63 tumor–normal BAM files, we ran six somatic mutation callers (MuTect2, SomaticSniper, VarDict, MuSE, Strelka2 and TNscope) followed by SomaticSeq and NeuSomatic machine learning classifiers using models built specifically for these datasets. Initial tier was assigned to each variant call based on how consistently the variant was classified as a somatic mutation across different sequencing centers and aligners. Then, to rescue low-VAF variants into the reference call set, we ran the same variant-calling pipeline on two higher-depth datasets: an independent 350× replicate sequenced on a HiSeq at Genentech (GT) and a 400× replicate by combining nine NovaSeq replicates at Illumina (IL). High-confidence calls from those two datasets were used to promote less-reproducible low-VAF variants from the 21 sequencing replicates into the reference call set. Then, we combined all of our short-read sequencing data into a pair of 1,500× tumor–normal and used it to rescue additional low-VAF variants into the reference call set. Finally, we cross-referenced our Illumina short-read-based high-confidence calls with PacBio long-read sequencing data and removed a small number of calls that were inconsistent with each other. The high-confidence calls (labeled PASS) were considered ‘true’ somatic mutations, and genomic regions with low-confidence calls were removed from the high-confidence regions. Chr, chromosome; Pos, position.
Definition of the somatic reference call set and high-confidence regions
We first combined call sets from the six callers for each of the 63 pairs of tumor–normal data (21 replicates by three aligners) and then used SomaticSeq to classify each variant call from each of the 63 pairs of tumor–normal data into PASS, REJECT or LowQual. All 63 SomaticSeq call sets were combined. Using cross-aligner and cross-sequencing center reproducibility, each variant was then assigned one of the four confidence levels: high confidence (HighConf), medium confidence (MedConf), low confidence (LowConf) and Unclassified. For low-VAF calls, three datasets (HiSeq dataset (350× coverage), NovaSeq dataset (400× coverage) and a combination of all WGS runs (1,500× coverage)) were used to salvage some LowConf and Unclassified calls as MedConf. A few HighConf calls that were inconsistent with the PacBio data were demoted to LowConf. The call set in its entirety was designated the ‘all-inclusive set’, which included LowConf and likely false-positive (Unclassified) calls. HighConf and MedConf calls in the all-inclusive set were grouped as the ‘somatic reference call set’ and treated as true positives (see Methods and Supplementary Information for details). A breakdown of the four confidence levels in the all-inclusive set is shown in Fig. 2a.

a, Breakdown of the somatic variant calls within the consensus callable regions based on the four labels HighConf, MedConf, LowConf and Unclassified. Variant calls labeled HighConf and MedConf are grouped into the reference call set; genomic positions with LowConf and Unclassified calls are removed from the high-confidence regions. b, Histogram of VAFs of the somatic variant calls. c, Average tumor purity fitting scores with 95% confidence intervals for the VAF of each SNV across the four different confidence levels versus the observed VAF in the tumor–normal titration series. The formula for fitting scores is described in equation 1 (see Methods for details). d, Scatter plot of VAFs observed in 21 WGS datasets versus an AmpliSeq targeted sequencing dataset. Solid shapes represent variants that were validated. Open shapes represent variants that were not validated. Sticks represent uninterpretable validation data. The diagonal dashed lines represent the 95% binomial confidence interval of the observed VAF given the actual VAF calculated based on 2,000× depth for AmpliSeq. The figure shows a very high correlation between VAFs estimated from the WGS data and AmpliSeq data for HighConf calls (Pearson’s R = 0.982). Many Unclassified data points lie at the bottom, implying that these calls were not real mutations, despite the large number of apparent variant-supporting reads in the all-inclusive set data; x axis, VAFs calculated from the all-inclusive set; y axis, VAFs calculated from the AmpliSeq data. e, Scatter plot of VAFs observed in WGS datasets versus Ion Torrent WES. The 95% binomial confidence intervals were calculated based on 34× depth for Ion Torrent. Pearson’s R = 0.930 for HighConf calls. f, Scatter plot of VAFs observed in WGS datasets versus 12 repeats of WES on the HiSeq platform; y axis, median VAFs calculated based on 12 HiSeq WES replicates. The 95% binomial confidence intervals were calculated based on 150× depth for HiSeq WES. Pearson’s R = 0.997 for HighConf calls. In d–f, the colors indicate the confidence level of the variant calls, whereas the shapes indicate their validation status.
Because NeuSomatic outperformed other callers in challenging situations, such as low-coverage, low-VAF and difficult genomic regions41, we applied NeuSomatic to each of the 63 call sets and created mutation classifiers as SomaticSeq did. The classification results from the decision tree-based adaptive-boosting approach used by SomaticSeq and the convolutional neural network-based deep learning algorithm used by NeuSomatic were highly correlated (Pearson’s R = 0.997) (Extended Data Fig. 1). We removed 526 SNVs and 60 indels that were discrepant between the two classifiers to improve precision and specificity of the somatic reference call set.
The 21 WGS runs used for the initial call set definition ranged from 50× to 100× coverage, and VAFs for most SNVs in the HighConf set were >5%. To improve sensitivity for low-VAF (<5%) variants, we pooled the short-read WGS sequencing data (1,500× coverage) to make NeuSomatic calls. We observed very high concordances for HighConf (99.6%) and MedConf (93.0%) SNVs between the initial call set and the call set from the 1,500× pooled data. Following manual inspection, most of the discordant calls (that is, classified as somatic mutations in the initial call set but not in the pooled dataset) were, indeed, likely true somatic mutations. They mostly consisted of dinucleotide/trinucleotide changes and low-VAF variants that were not confidently detected in the single 1,500× data. However, only 24.0% and 1.67% of LowConf and Unclassified SNVs in the initial call set, respectively, were called in the pooled dataset (Supplementary Table 1). In other words, LowConf and Unclassified SNVs were unlikely to be rediscovered in the pooled 1,500× dataset. Most of the Unclassified calls in the pooled call sets were complex variants (that is, more complex than simple dinucleotide/trinucleotide changes). In addition, HighConf and MedConf SNVs had high validation rates by AmpliSeq (Fig. 2b). Therefore, we used a conservative approach to allow low-VAF and low-signal variants into the somatic reference call set (that is, if they were supported by NeuSomatic in the 1,500× data in all three aligners (see Methods for details)). Consequently, 894 SNVs and 33 indels were ‘rescued’ into the somatic reference call set (Supplementary Data File 1).
It is well known that false positives are overwhelmingly enriched in genomic regions where alignment is challenging42. High GC content or low-complexity regions in the human genome cannot be covered adequately by current short-read technologies (Extended Data Fig. 2a). To obtain the callable regions in the reference genome, we ran GATK CallableLoci on each of the 63 BAM files to identify regions of low coverage (<10 reads), ultra-high coverage (more than eight times the mean coverage of the sample), mapping difficulty (mapping quality (MQ) < 20) and poor-quality reads (base quality (BQ) score < 20). From the consolidated CallableLoci results, we further removed the three chromosomal regions with normal loss of heterozygosity (LOH) and genomic regions with LowConf and Unclassified calls and created a list of high-confidence regions for the somatic reference call set, containing a total of 2.48 billion base pairs (bp) (Extended Data Fig. 2b). The derived regions were callable with short-read sequencing technologies (that is, Illumina platforms). Variant calls outside the high-confidence regions were labeled as ‘NonCallable’ in the all-inclusive set.
Validation of mutations in the somatic reference call set
To confirm HighConf and MedConf calls, we mixed tumor HCC1395 gDNA with normal HCC1395BL gDNA at different ratios to generate a range of admixtures that represented tumor purity levels of 100%, 75%, 50%, 20%, 10%, 5% and 0% and performed WGS with 350× coverage. We calculated the tumor purity fitting scores (described in the Methods, equation 1) for measuring the concordance between the expected VAF and the observed VAF across tumor purity levels (Fig. 2c). For real somatic mutations in copy number-neutral regions, the observed VAFs should scale linearly with the tumor fraction in the tumor–normal titration series and show a higher score but not for sequencing artifacts or germline variants. The fitting scores for HighConf and MedConf calls were much higher than LowConf and Unclassified calls across all VAF brackets, indicating that HighConf and MedConf calls contained notably more real somatic mutations than LowConf and Unclassified calls.
In addition, we randomly selected a number of mutation calls at HighConf, MedConf, LowConf and Unclassified in the all-inclusive set for validation by AmpliSeq with PCR enrichment in addition to Ion Torrent WES and HiSeq WES (SNVs, Fig. 2d–f; indels, Extended Data Fig. 3a–c). Simple rules were established to determine whether a call was validated, not validated or uninterpretable based on the existence of high-quality variant-supporting reads. We manually evaluated discordant calls and used additional evidence to resolve these discrepancies (see Methods for details). Combining the three validation experiments, the validation rates for SNVs and indels in the reference call set (HighConf and MedConf) were 99.93% (1,417/1,418) and 97.50% (78/80), respectively (Table 2).
PacBio long-read sequencing might cover the genomic regions where short reads cannot be mapped, especially in the high GC/AT or low-complexity genomic regions (Extended Data Fig. 2). Although its higher sequencing error rate than short-read sequencing makes it inappropriate for somatic mutation discovery (Supplementary Table 2), we used PacBio long-read sequencing to confirm 99.3% SNVs and 98.5% indels in HighConf. In addition, for high-VAF calls in low mapping score and low sequence complexity regions, we demoted their confidence level when they were not supported by PacBio data (P < 0.05 for one-sided two-proportion z test when PacBio had no supporting read), resulting in the removal of 33 SNVs and 11 indels from the reference call set due to discordance from PacBio data.
Discovery and validation of germline variants
For 21 WGS replicates with gDNA from HCC1395BL cells, we used three aligners (BWA-MEM, Bowtie2 and NovoAlign) and four germline variant callers (FreeBayes43, Real Time Genomics (RTG) (https://www.realtimegenomics.com/), DeepVariant44 and HaplotypeCaller45) to call SNVs and indels. We used a generalized linear mixed model (GLMM) to fit and consolidate various call sets (see the Methods for details). For SNVs and indels called at least four times, we estimated their SNV/indel calling probability (SCP) using the average over the four factors (sequencing center, sequencing replicate, aligner and caller). The frequency of SCPs showed a bimodal pattern (Fig. 3a). Most SNV calls (97%) had SCPs of <0.1 (57%) or >0.9 (40%). Only a few calls (3%) were between 0.1 and 0.9, indicating that when SNVs were called, 40% of them would be called as SNVs repeatedly, and the others would be called only infrequently.

a, Histogram of SCP for germline variants identified by four callers from 63 BAM files. b, VAF scatter plot of germline SNVs by the call set and AmpliSeq data. Pearson’s R = 0.986 for SCP = 1 call. c, VAF scatter plot of germline SNVs by the call set and Ion Torrent WES. Pearson’s R = 0.758 for SCP = 1 call. In b and c, colors indicate the calling probability of the germlines variants, whereas shapes indicate their validation status.
We validated a subset of germline variants using AmpliSeq and Ion Torrent. For the highest confidence calls (SCP = 1; that is, they were called in each combination of replicate, aligner and caller), the validation rates for SNVs and indels were 99% and 98% for AmpliSeq and 98% and 97% for Ion Torrent (Supplementary Table 3). Of the 11 SNVs with SCP values <0.5, none were confirmed by AmpliSeq.
Most validated germline variants had VAFs around 50% or 100% (Fig. 3b,c). By contrast, a considerable number of lower-confidence germline SNVs were clustered around a VAF of 20% and could not be validated (Fig. 3b). VAF scatter plots for indels were qualitatively similar to those of the SNVs (Extended Data Fig. 4).
Heterogeneity of the HCC1395 cancer cell line
Previous studies of the TNBC cell line revealed large-scale genomic instability and ploidy changes24, which were confirmed by our karyotype and cytogenetic analysis of the HCC1395 cell line (Extended Data Figs. 5 and 6). The WGS CNA analysis by ascatNgs46 consistently revealed numerous chromosome gains and losses as well as LOH events (Extended Data Fig. 7). In addition, we observed peaks of VAF at 0.15 and 0.08, suggesting that these SNVs were from subclones (Extended Data Fig. 8).
We performed clonality analysis47 leveraging WGS and WES data derived from bulk DNA as well as using single-cell data from 10x Genomics Chromium CNV analysis. The phylogenetic tree (Fig. 4a) generated by PhyloWGS48 suggests a branching evolution that is consistent with previous reports49,50,51,52; the clonal expansion was further confirmed by superFreq47 (Extended Data Fig. 9a). Specifically, HCC1395 cells showed some typical characteristics of breast cancer tumor development, including a clonal driver mutation in TP53, clonal copy neutral loss of heterozygosity (NLOH) at chromosome 17q and an early driver mutation in PIK3CA. We detected most of the point mutations in driver genes and almost all of the somatic CNAs involving driver genes present in the most recent common ancestor clone S1, suggesting that most of the driver events are clonal53,54. In addition, the branching phylogenetic tree suggests that the tumor continues clonal evolution, resulting in two major branches (beginning with nodes at S2 and S8) either in vivo or in cell culture.

a, The inferred tumor phylogenetic tree. The subclone S1 represents the most recent common ancestor (MRCA) of all tumor cells, and S2 to S10 represent the subclones with various cancer cell fractions (for example, S2: 60%). The edges represent the evolutionary relationships between subclones. Subclones S3 and S6 are not shown given that their cancer cell fractions were less than 10%. Most point mutations are in driver genes (labeled beside the edges) present in the MRCA. Using the 10x Genomics Single Cell CNV Solution, integer-scaled CNA profiles were obtained across the genomes of 638 HCC1395BL cells (b) and 1,270 HCC1395 cells (c). Noisy cells and cells in the S phase of the cell cycle were removed. The complete linkage method was used for hierarchical clustering. Each row represents a cell being sequenced; similar cells were clustered together based on CNVs. Chromosome-scale gains are in orange and losses are in blue in the heat maps.
We further inferred both clonal and subclonal CNAs using subHMM55 (Extended Data Fig. 9b). The estimated purity based on CNAs was 99%, as expected for a typical breast cancer cell line. The existence of subclonal CNAs was also observed in single-cell gDNA sequencing of 1,270 cells for HCC1395 and 638 cells for HCC1395BL (Fig. 4b,c). Integer-scaled CNA profiles across the genomes showed substantial subclonal CNAs in the TNBC cell line (Fig. 4c). Our CNA analyses revealed that HCC1395 is a heterogeneous cell line.
Discussion
Our reference samples and their call sets can have broad applications in benchmarking sequencing pipelines. For example, we used the reference call sets to benchmark WGS and WES in companion studies56,57, which compared various experimental conditions to determine factors encompassing both analytical technology platforms and bioinformatic methods that affect the reproducibility and accuracy of somatic mutation detection. In particular, the variants in high-confidence regions can help to assess detection sensitivity and specificity. Our reference samples have also been used to benchmark single-cell RNA-sequencing technologies58, and the associated data are available to the community59. The characteristics of genetic alterations in HCC1395 cells are in line with what have been described in a large cohort study60 (Extended Data Fig. 10). The changes in ploidy observed in HCC1395 cells are common for breast cancers61. Therefore, the large number of somatic mutations and CNAs in HCC1395 cells provides the genomic representativeness desired in a reference gDNA sample. In addition, many variants and mutations in HCC1395 cells have clinical implications. In the coding regions, 207 somatic mutations in the reference call set are documented in the COSMIC database, and 8 germline variants are annotated as pathogenic in the ClinVar database. Interestingly, we found nonsense germline variants and somatic mutations in BRCA1 and BRCA2 genes, respectively, and hotspot somatic mutations in the TP53 and FGFR1 genes (Supplementary Data File 2). Hence, our reference call sets might also be valuable to benchmark bioinformatics pipelines for the detection of mutations with targeted panels.
Genetic materials derived from the paired tumor–normal cell lines are sustainable for community usage. We chose cell lines over real tissues to extract gDNA as somatic reference samples because real tissues are not renewable. To address the same consideration, the GIAB Consortium, which has been leading the efforts in establishing standard reference materials for germline variation detection, chose to use cell lines rather than real tissues from healthy individuals11,62. In addition, the spatial heterogeneity of tumor tissue63,64, for which different slices of the same tissue might have different mutation profiles, hampers the assessment of sequencing assay reproducibility. To mitigate genetic drift during the expansion of cell lines65 and related concerns, GIAB generated a large quantity of gDNA from normal cell lines as reference material so that DNA aliquots could be distributed to the community. However, new material will eventually need to be qualified when the current stock of DNA is exhausted.
An alternative approach adopted by this study to establish cell substrate as a source of reference materials is through a cell banking tier system, which has been fully documented66. In this study, we made a large batch of gDNA from each of the two cell lines and distributed aliquots to the sequencing centers to develop reference call sets. The remaining gDNA can be used for validating or benchmarking any emerging sequencing technologies or protocols. Should the need for additional gDNA arise, cell aliquots derived from the same batch of the reserved master cells can be used for the production of gDNA, and genetic drift can be characterized with a bridging study using our gDNA materials and the reference call sets. We propose the call sets reported here as a reference and expect that the reference call sets and high-confidence regions might be further refined as sequencing technologies and data analysis tools improve.
There are several limitations with the somatic reference call sets reported here. First, reference somatic mutations were called in regions of the genome limited to those that could be analyzed with the Illumina sequencing platforms. Although we used Ion Torrent as an orthogonal method to validate some mutations, other mutations were validated on the Illumina sequencing platform. PacBio long-read sequencing with modest sequencing depths was also applied for validation; however, its sequencing error rates were higher than some low-VAF variants in the reference call sets (Supplementary Table 2). Second, the reference genome alignment-based approach might also leave undetected somatic mutations within regions that are susceptible to variations in a personal genome. Indeed, our ongoing study using a personal genome (that is, de novo assembly of the genome for the normal cell line) indicated that extra new mutations could be identified using a personal genome as the reference genome67. Third, reproducibility was used as a proxy for defining a reference call set that can minimize biases/artifacts from library preparation, sequencing machine and bioinformatics pipeline. Our reference call set was defined with a tumor–normal match approach for gDNA derived from fresh cell cultures. To benchmark the performance of other sample types, such as FFPE or ctDNA, consideration needs to be taken to watch out for artifacts due to DNA damage (for FFPE)56 or clonal hematopoiesis (for ctDNA)68. Fourth, our somatic reference call set is not meant to be a comprehensive catalog of genomic alterations in breast cancer but, instead, is meant to serve as an analytical benchmark reference with many types of genomic alterations.
In summary, we established well-characterized somatic mutation and germline reference call sets for a pair of tumor–normal cell lines and shared them with the scientific community so that they can be used for evaluating NGS sequencing analysis pipelines. The diverse sequencing data generated from multiple platforms at multiple sequencing centers can help tool developers to build more accurate and robust artificial intelligence models for somatic mutation detection. The preserved gDNA and master cells can be used to develop standard reference materials in the future for assay development, qualification, validation and proficiency testing. Furthermore, the methodology used in this study can be extended to establish reference materials and reference datasets for additional sample types.
Methods
Cell lines and DNA extraction
HCC1395 human breast carcinoma cells (expanded from ATCC CRL-2324) were cultured in ATCC-formulated RPMI-1640 medium (ATCC, 30-2001) supplemented with fetal bovine serum (FBS; ATCC, 30-2020) to a final concentration of 10%. Cells were maintained at 37 °C with 5% CO2 and were subcultured every 2–3 d, per ATCC recommended procedures, using 0.25% (wt/vol) trypsin–0.53 mM EDTA solution (ATCC, 30-2101) until appropriate densities were reached. HCC1395BL human B lymphoblast Epstein–Barr virus-transformed cells (expanded from ATCC CRL-2325) were cultured in ATCC-formulated Iscove’s Modified Dulbecco’s medium (ATCC, 30-2005) supplemented with FBS to a final concentration of 20%. Cells were maintained at 37 °C with 5% CO2 and were subcultured every 2–3 d, per ATCC recommended procedures, using centrifugation with subsequent resuspension in fresh medium until appropriate densities were reached. Final cell suspensions were centrifuged and resuspended in PBS for nucleic acid extraction.
All cellular genomic material was extracted using a modified phenol–chloroform–isoamyl alcohol extraction approach. Cell pellets were resuspended in TE, lysed in a 2% Triton X-100/0.1% SDS/0.1 M NaCl/10 mM Tris/1 mM EDTA solution and extracted with a mixture of glass beads and phenol–chloroform–isoamyl alcohol. After multiple rounds of extraction, the aqueous layer was treated further with chloroform–isoamyl alcohol and underwent RNAse treatment and DNA precipitation using sodium acetate (3 M, pH 5.2) and ice-cold ethanol. The final DNA preparation was resuspended in TE, and aliquots were distributed to sequencing centers for NGS and microarray analysis.
Cell line karyotyping
Karyotyping was performed by Cell Line Genetics as described previously69. Cells were treated with Colcemid (Gibco) for 40 min, exposed to 0.075 M KCl for 23 min at 37 °C and then fixed with 3:1 methanol:glacial acetic acid. Slides were stained with Leishman’s stain before observation. During observation, roughly 20 metaphase cells were counted on the microscope, and numerical and structural chromosome aberrations were recorded. An analysis of 5–10 band cells was performed at the microscope using a ×100 objective with an effort to karyotype at least two cells from each clone.
WGS and WES on Illumina platforms
See methods section in ref. 56.
PacBio library preparation and sequencing
Fifteen micrograms of material was sheared to 40 kilobases (kb) with Megarupter (Diagenode). Per the Megarupter protocol, the samples were diluted to below 50 ng µl−1. A 1× AMPure XP bead cleanup was performed. Samples were prepared as outlined in the PacBio protocol titled ‘Preparing >30 kbp SMRTbell Libraries Using Megarupter Shearing and Blue Pippin Size-Selection for PacBio RS II and Sequel Systems’. After library preparation, the library was run overnight for size selection using the Blue Pippin (Sage). The Blue Pippin was set to select a size range of 15–50 kb. After collection of the desired fraction, a 1× AMPure XP bead cleanup was performed. The samples were loaded on the PacBio Sequel (Pacific Biosciences) following the protocol titled ‘Protocol for loading the Sequel’. The recipe for loading the instrument was generated by the PacBio SMRTlink software v5.0.0. Libraries were prepared using Sequel chemistry kits v2.1, a SMRTbell template kit 1.0 SPv3, a magbead v2 kit for magbead loading, sequencing primer v3 and SMRTbell cleanup columns v2. Libraries were loaded between 16:00 and 20:00.
AmpliSeq deep sequencing
Criteria for picking variants
The variant call format (VCF) files were filtered to generate a list of variant coordinates that could be uploaded into Illumina Design Studio. The list of somatic variants was selected based on the following criteria: all variant call confidence tiers were represented equally, low and high VAFs were covered and all chromosomes were represented. The logic behind picking the variants was to have an equal number of variants per tier while equally representing all four confidence levels. The selection process also attempted to ensure representation of all chromosomes across all tiers but did not require that all chromosomes be represented in each tier. The TNBC cell line HCC1395 has significant structural rearrangements and ploidy changes, resulting in severe overrepresentation of variants from chromosomes 6, 16 and X. Therefore, variants from these three chromosomes were downweighted in random sampling to ensure that variants from other chromosomes had a statistical chance of being selected. Germline variants were also included to determine the accuracy of the AmpliSeq panel with reference to previously characterized variants in cell line HCC1395. At the end of this process, a total of 2,477 variants (somatic and germline SNVs and indels) were selected to enter into the AmpliSeq design software.
The 2,477 variants’ chromosomal positions were expanded to 300-bp regions to upload into Illumina Design Studio, selecting for a 275-bp amplicon size. The design algorithm failed to generate complete coverage for 1,109 of the 300-bp regions, leaving 1,368 regions with the following makeup: 641 germline variants, 77 germline indels, 554 somatic variants and 96 somatic indels. The 1,368 300-bp amplicon regions were submitted to Illumina for panel manufacturing.
Library construction and sequencing
The HCC1395 and HCC1395BL libraries were prepared in triplicate as specified in the Illumina protocol (document number 1000000036408 v04) following the two oligo pools workflow with 10 ng of input gDNA per pool. The numbers of amplicons per pool were 1,517 and 1,506, respectively. The libraries were quality checked using an Agilent TapeStation 4200 with the DNA HS 1000 kit and quantitated using a Qubit 3.0 and DNA high-sensitivity assay kit. The libraries were applied to a MiSeq v2.0 flowcell. They were then amplified and sequenced with a MiSeq 300 cycle reagent cartridge with a read length of 2 × 150 bp. The MiSeq run produced 7.3 gigabases (gb) (94.5%) at ≥Q30. The number of reads passing filtering was 48,886,352 of 52,601,972 total (96.4%).
Sequence alignment and variant validation analysis for AmpliSeq
We aligned the FASTQ files with BWA-MEM and counted the number of variant-supporting reads and total reads for each variant position with MQ ≥ 40 and BQ ≥ 30 cutoffs. The total sequencing depths for the tumor and normal samples were approximately 2,000×. The following rule was applied to the AmpliSeq data to determine whether a call was deemed confirmed, not confirmed or uninterpretable:
-
If tumor and normal depth were both ≥600× and if variant depth in the tumor was ≥100 and normal variant depth was <10 → validated.
-
Else, if normal depth was ≥600× and if variant reads in normal consisted of ≥10% of the total depth → not validated (germline false positive).
-
Else, if tumor total depth was ≥1,000 and tumor variant depth was <5, or if tumor total depth was ≥1,000 and tumor variant depth consisted of <0.1% of the total depth → not validated (no variant reads in tumor when there should be).
-
Else, if tumor or normal total depth was ≤50 → uninterpretable.
-
Else → manual inspection on Integrative Genome Viewer (IGV) to make a determination.
In addition, we used IGV to study calls that were
-
1.
Originally annotated as ‘confirmed’ but had a large difference in VAF calculated from the truth set data and AmpliSeq data and were reannotated if needed.
-
2.
HighConf calls that were ‘not confirmed’, although no annotation was actually changed.
-
3.
Unclassified calls that were ‘confirmed’ and reannotated if needed.
When there was manual reannotation after IGV inspection, a comment was left in the final column in the validation file.
Ion Torrent WES
Library construction and sequencing
A SureSelect Target Enrichment Reagent kit, PTN (G9605A), SureSelect Human All Exon v6 + UTRs (5190-8881), Herculase II Fusion DNA Polymerase (600677) from Agilent Technologies and an Ion Xpress Plus Fragment kit (4471269, Thermo Fisher Scientific) were combined to prepare libraries according to the manufacturer’s guidelines (user guide: SureSelect Target Enrichment System for Sequencing on Ion Proton, version C0, December 2016, Agilent Technologies). Before, during and after library preparation, the quality and quantity of gDNA and/or libraries were evaluated using a Qubit fluorometer 2.0 with a dsDNA HS Assay kit (Thermo Fisher Scientific) and an Agilent Bioanalyzer 2100 with a high sensitivity DNA kit (Agilent Technologies). For sequencing the WES libraries, the Ion S5 XL Sequencing platform with Ion 540-Chef kit (A30011, Thermo Fisher Scientific) and the Ion 540 Chip kit (A27766, Thermo Fisher Scientific) were used. One sample per 540 Chip kit was sequenced, generating up to 60 million reads with an average length of 200 bp.
Sequence alignment and analysis of Ion Torrent data
Raw reads were first filtered for low-quality reads and trimmed to remove adapter sequences and low-quality bases. This step was performed using the BaseCaller module of the Torrent Suite software package v5.8.0 (Thermo Fisher Scientific). Low-quality reads were retained from further analysis in the raw signal processing stage. Low-quality bases were trimmed from the 5′ end if the average quality score of the 16-base window fell below 16 (Phred scale), cleaving 8 bases at once. Processed reads were mapped to the GRCh38 reference genome by the TMAP module of the Torrent Suite software package using the default map4 algorithm with recommended settings.
Variant validation analysis
The following rule was applied to interpret WES validation by both Ion Torrent and HiSeq with the same MQ ≥ 40 and BQ ≥ 30 thresholds as used for AmpliSeq data.
-
If tumor variant depth was >2 and tumor VAF was >10 times the normal VAF → confirmed.
-
If normal total depth was >20 and normal VAF was >10% → not confirmed (germline).
-
If the expected value for variants in reads in the tumor is >5 based on truth set VAF and tumor total depth but there was no such read → not confirmed (no signal when signal was expected).
-
Else → uninterpretable.
Bioinformatics pipelines
In this section, we describe the bioinformatics analysis methods to generate the high-confidence somatic mutation call set. The overall schematic is shown in Fig. 1. The commands and source codes are documented at our project site at http://sites.google.com/view/seqc2.
Read alignment
For each of the paired-end read files (that is, FASTQ 1 and 2 files) generated by Illumina sequencers (HiSeq, NovaSeq and MiSeq platforms), we first trimmed low-quality bases and adapter sequences using Trimmomatic70 (performed at Frederick National Laboratory for Cancer Research). The resulting sequences were then mapped with three different aligners (that is, BWA-MEM, Bowtie2 and NovoAlign) to create three BAM files. Picard Tools (http://broadinstitute.github.io/picard/) was then used to mark PCR and optical duplicates in the BAM files (except for AmpliSeq data from MiSeq).
The Ion Torrent reads were first trimmed for low-quality bases and adapter sequences using the BaseCaller module of the Torrent Suite software package v5.8.0. Low-quality bases were trimmed from the 5′ end if the average quality score of the 16-base window fell below a BQ of 16, cleaving 8 bases at once. The processed reads were then mapped with TMAP using the default map4 algorithm with otherwise default settings. Picard Tools was then used to mark PCR and optical duplicates on the BAM files.
Building the center- and aligner-specific SomaticSeq classifiers
BAM files produced from the same sequencing centers and platform and aligned with the same aligner were grouped into one data group. For instance, the three pairs of HiSeq BAM files at IL aligned with BWA-MEM were grouped into one data group, and the same reads aligned with NovoAlign were grouped into another data group under NovoAlign (Fig. 1). BAM files that came from LL, NC and EA were grouped into ‘Others’. Hence, there were five data groups for each aligner: (1) HiSeq at IL, (2) NovaSeq at IL, (3) HiSeq at FD, (4) HiSeq at NV and (5) HiSeq at Others. SNV and indel SomaticSeq classifiers were built for each of the first four data groups because we took advantage of multiple sequencing replicates of the normal genome to build machine learning classifiers that remove data group-specific artifacts as false positives. Because three aligners were used, 12 SNVs and 12 indel center- and aligner-specific classifiers were created. For the Others groups, data from the first four groups were combined to simulate aligner-specific (but not center-specific) artifacts for it.
For each data group that has intracenter sequencing replicates, we designated normal replicate 1 as the normal and normal replicate 2 as the pseudotumor. BAMSurgeon was used to spike approximately 100,000 in silico SNVs and 20,000 in silico indels into the pseudotumor to create an in silico tumor18. SomaticSeq’s tumor–normal workflow was then used for the in silico tumor–normal pairs (Supplementary Fig. 1a). Briefly, six somatic mutation callers were incorporated into the workflow: MuTect2 (ref. 13), SomaticSniper36, VarDict37, MuSE38, Strelka2 (ref. 39) and TNscope40. About 100 genomic and sequencing features were extracted for each variant call using SomaticSeq32. Every mutation call that was not spiked in by BAMSurgeon was labeled a false positive, and only the in silico variants were labeled true positives.
Two training sets were created for each data group. Training set A designated normal replicate 2 as the in silico tumor versus normal replicate 1. Training set B designated normal replicate 3 as the in silico tumor versus normal replicate 2. Different mutations were spiked into the two different training sets. We have done cross-validation between these two sets, that is, classifiers built on training set A were used to classify in silico tumor–normal pairs in training set B and vice versa. Cross-validations were to ensure that SomaticSeq classifiers could filter out false positives in different datasets from the same data group reliably and not just the same dataset it was trained on.
Supplementary Fig. 1b displays the classification accuracy of cross-validation from the 12 × 2 = 24 SNV cross-validations. PASS calls had SomaticSeq SCORE values ≥0.7, and, conversely, REJECT calls had SCORE values ≤0.1. Calls with 0.1 < SCORE < 0.7 were labeled LowQual. In the 24 cross-validations, the classification sensitivity (that is, true positives classified as PASS in the combined call set from the six callers), specificity, positive predictive value and negative predictive value were 98.39%, 99.23%, 99.52% and 99.86% for SNVs and 96.96%, 98.03%, 98.08% and 99.67% for indels, respectively. A few in silico mutations were not detected by any of the six callers. They are discussed in the Supplementary Materials. For each data group, training sets A and B were combined to make one SNV and one indel SomaticSeq classifier for later use. For the Others groups, training sets from the other four data groups were combined to make an SNV and an indel classifier.
Building aligner-specific NeuSomatic classifiers
For NeuSomatic, instead of creating center-specific and variant type-specific (that is, SNV and indel separately) classifiers, one NeuSomatic-Ensemble and one NeuSomatic-Standalone classifier were created for each aligner by combining all the simulated data from all the data groups.
Make somatic mutation calls in each HCC1395 versus HCC1395BL pair
The same six somatic mutation callers were run on each pair of tumor–normal BAM files performed on the Cancer Genomics Cloud (CGC) by Seven Bridges Genomics. The corresponding SomaticSeq and NeuSomatic classifiers (described previously) were used to classify mutations for each of the 63 call sets, creating somatic SNV and indel VCF files for each tumor–normal pair (Supplementary Fig. 3a). The histograms of the variant scores (Supplementary Fig. 3b,c) for the real datasets are qualitatively similar to the cross-validation of the training datasets shown in Supplementary Fig. 1b,c.
Determine confidence level for initial somatic mutation call with SomaticSeq
The schematic of this process is shown in Fig. 1. The four confidence levels (that is, HighConf, MedConf, LowConf and Unclassified) of the somatic mutation calls were annotated primarily based on the three aligner-centric classifications of a somatic mutation call. The aligner-centric classification of a call for each aligner was in turn determined by the five data group scores within this aligner. Each data group score was in turn determined by a call’s reproducibility across the sequencing replicates within the data group.
The algorithm is described in detail in the Supplemental Material. For example, if a call was classified as a PASS in two replicates and LowQual in the other replicate for HiSeq at IL with BWA-MEM (a data group), it would have had a data group score of +3 (Supplementary Fig. 7). If the five data group scores added up to at least 6, it would have been considered ‘Strong Evidence’ for BWA-centric classification (Supplementary Fig. 7). If this call was deemed ‘Strong Evidence’ for BWA-, Bowtie2- and NovoAlign-centric classifications, it would have been considered a HighConf call (Supplementary Fig. 7). For low-VAF (≤15%) calls initially annotated as LowConf and Unclassified, additional 350× and 400× (combined nine NovaSeq at IL replicates) datasets were used to potentially rescue those calls into MedConf if they were deemed PASS in those high-depth datasets. The 15% VAF threshold for confidence-level recalibration was determined after examining AmpliSeq deep sequencing data when it was determined that for non-HighConf calls with VAF ≤ 15%, they were far more likely to be true positives than false positives. HighConf and MedConf calls were grouped into the truth set. Calls in the truth set had many samples classified as PASS across different aligners and sequencing centers with very few (if any) REJECTS.
Determine high-confidence genomic regions and somatic mutation reference call set
We have labeled four different confidence levels in our entire call set (the super set): HighConf, MedConf, LowConf and Unclassified. HighConf and MedConf calls were considered as the reference call set used to benchmark pipelines along with the high-confidence genomic regions. Twenty-base pair regions spanning LowConf and Unclassified calls were not included in the high-confidence region to ensure that there were no false negatives in the high-confidence regions. The technical details of the workflow are described in detail in the Supplemental Material. The commands and future updates will be documented on the Working Group’s project website at https://sites.google.com/view/seqc2. In this section, we will describe general methods to categorize the somatic mutation in the following seven steps.
-
1.
Use corresponding SomaticSeq classifiers to score somatic mutation calls in each of the 63 tumor–normal data sets. Combine the call sets and keep all mutation calls that have been scored ≥0.7 in at least 1 of the 63 datasets.
-
2.
Assign initial confidence levels based on how consistent a mutation call has been classified by SomaticSeq as PASS across the three aligners and six sequencing centers as well as false-positive markers, such as MQ, read-to-reference mismatches, BQ and inconsistent VAF movement with the 350× tumor–normal titration series.
-
3.
Promote the initial confidence levels and rescue additional low-VAF calls using 350× and 400× SomaticSeq datasets.
-
4.
Modify confidence levels of calls when SomaticSeq and NeuSomatic classifications showed significant discrepancies or when the SomaticSeq classifications themselves were not consistent across the 63 datasets. Rescue additional calls that were captured by NeuSomatic but not SomaticSeq.
-
5.
Use the 1,500× NeuSomatic tumor–normal call sets (combining all short-read WGS reads) to promote low-VAF calls and rescue low-VAF calls that were not captured previously.
-
6.
For HighConf and MedConf calls that are not supported by PacBio data (P < 0.05, two-proportion z test), demote them to LowConf if the variant in question is in a low mapping or low sequence complexity region.
-
7.
Finally, label HighConf and MedConf calls in the consensus-callable regions as PASS. Create high-confidence regions by removing 20-bp regions spanning LowConf and Unclassified positions and the germline LOH of the three chromosome arms (chromosome 6p, chromosome 16q and chromosome X) from the consensus-callable regions.
Validate somatic mutations with tumor titration series fitting scores
We divided SNVs into k = 13 groups based on the VAF, Mk = 0.00, 0.05, 0.10,…, 0.55 and ≥0.60 at 100% purity. For the kth group, there are nk SNVs, each of which has seven VAF \({Y_{ij}^{{\textrm{observed}}}}\), i = 1, 2,…, 7 diluted purities 100%, 75%, 50%, 20%, 10%, 5% and 0%. For SNVs in the kth group, we fitted a linear mixed model because each SNV has its VAF calculated at diluted purities and obtained the best unbiased predictor \(Y_{ij}^{{\textrm{fitted}}}\). We calculated the relative squared error
The mean relative squared error (MRSE) is simply the average of relative squared error for SNVs in the kth group,
Tumor purity fitting score is the inverse of MRSE. MRSE was applied to every somatic SNV call in the super set. The ‘error bars’ were calculated by removing data points whose \(Y_{ij}^{{\textrm{observed}}} - Y_{ij}^{{\textrm{fitted}}}\) values were 1 s.d. below the mean (upper bar) or 1 s.d. above the mean (lower bar).
Determine callable regions
The consensus high-confidence region was determined in the following manner:
-
1.
GATK CallableLoci was run on each tumor and normal (63 × 2 = 126) BAM file.
-
2.
Within each data group:
-
a.
Determine callable regions in the majority of the tumor replicates.
-
b.
Determine callable regions in the majority of the normal replicates.
-
c.
Consider the callable regions of the data group to be the intersection of the regions in (a) and (b).
-
a.
-
3.
Within each of the three aligners, consider regions deemed callable by the majority (that is, three of five) of the data groups to be the callable regions of the aligner.
-
4.
Consider regions deemed callable by the majority (that is, two of three) of the aligners to be the consensus-callable regions. Label somatic mutation calls outside the consensus regions ‘NonCallable’ as a value in FLAGS in the INFO field.
Determine germline variants
We used four germline variant callers (freebayes43, RTG, DeepVariant44 and HaplotypeCaller45), which were readily available on the National Institutes of Health (NIH) Biowulf cluster. We ran each of them using the default parameters or parameters recommended by the user’s manual. Specifically, for HayplotypeCaller, we included a flag for ‘–stand_call_conf 30’ and used dbsnp build 146 on the GRCh38 reference genome. For DeepVariant, we used model.cpkt for WGS, which was provided by DeepVariant (gs://deepvariant/modelsDeepVariant/0.5.1/DeepVariant-inception_v3-0.5.1+data-wgs_standard/).
To consolidate all the call sets, we fit a GLMM for each SNV that was sequenced at different sites on various replicates, aligned by three aligners and called by four callers. The SNVs considered were called at least four times by various combinations of factors (including sites, replicates, aligners and callers). We defined the SNV call probability Pmnjk as the probability of an SNV being called by a given aligner (α1m) and a caller (α2n) at a site (α3j) and on a replicate within a site (α4k). We modeled Pmnjk as
Here, β is a fixed effect, and α1m, α2n, α3j and α4k are normally distributed random effects with zero means and variances \(\alpha _1^2\), \(\alpha _2^2\), \(\alpha _3^2\) and \(\alpha _4^2\), respectively. The SNV call probability averaged across factors is defined as \(P = \frac{{e^\beta }}{{1 + e^\beta }}\), and variance explained by each factor is defined as \(\frac{\alpha_i^2}{\alpha_1^2+\alpha_2^2+\alpha_3^2+\alpha_4^2}\), where i = 1, 2, 3 and 4. All these parameters were estimated by the R package lme4.
SNV annotation and TMB benchmarking
SnpSift was used to link dbSNP build 146 and COSMIC v85 identifiers, and information to the somatic variant calls and ClinVar (version 20190305) to the germline variant calls8. SnpEff was used to annotate genes and their functional effects (that is, missense, synonymous and so on) of the variants71.
When counting non-synonymous variants, only missense_variant (319), stop_gained (16), stop_lost (1) and structural_interaction_variant (6) were counted. We used University of California Santa Cruz (UCSC) genome track GRCh38-based GENCODE v29 coding exons to obtain the coding regions (http://genome.ucsc.edu/cgi-bin/hgTables?command=start).
When hg19 coordinates were given by oncology panel vendors for target regions, we used https://www.ncbi.nlm.nih.gov/genome/tools/remap to remap them into hg38. The region size of the panels (as well as UCSC coding regions) had the three LOH arms (chromosome 6, chromosome 16 and chromosome X arm losses in normal) subtracted, and the mutation rates were calculated based on those region sizes.
CNV analysis with WGS
We used the CNA detection tool ascatNgs (version 4.2.1), which contains the Cancer Genome Project’s Singularity workflow implementation of the ascatNgs46. The tool was available on the NIH high-performance computing resource (Biowulf). The BAM files were generated using BWA-MEM, and CNA was performed using the default parameters of ascatNgs recommended by the user’s manual with a flag ‘-protocol WGS’ specific for WGS data. The run command is ‘ascat.pl -tumour tumor.bam -norm normal.bam -reference hg38.fa -snp_gc Snp_panel -protocol WGS’; the ASCAT SNP_panel was generated using a provided ascatSnpPanelGenerator Perl script with human genome reference hg38.
Cell line clonality analysis
To determine the clonality of HCC1395 and HCC1395BL cells, we performed somatic SNV and CNA analysis using superFreq47 on capture WES datasets. Mapped and markDuplicate BAM files of a pair of HCC1395 and HCC1395BL datasets were used as input, and BAM files of the remaining replicates of the HCC1395BL library were used to filter background. An analysis was run using the superFreq default parameters. The clonality of each somatic SNV was calculated based on the VAF, accounting for local copy number. The SNVs and CNAs undergo hierarchical clustering based on the clonality and uncertainty across replicates for the tumor sample.
We inferred both clonal and subclonal CNA profiles using subHMM. First, we calculated log odds ratio (log OR) and log ratio (log R) values of variant allele counts for tumor versus normal. Second, log OR and log R were adjusted by library size and GC normalization. Third, subHMM embedded adjusted log OR and log R values into a hidden Markov model (HMM) and simultaneously conducted the segmentation and genotype mixture modeling to identify both clone/subclone region and region-specific clone/subclone genotype and clonal proportion in a heterogeneous tumor sample. Note that subHMM allows the subclonal genotype and proportion to vary across different subclonal regions. Hence, multiple subclonal regions with distinct clonal proportions are considered as distinct subclones.
Single-cell CNV and heterogeneity analysis
HCC1395 and HCC1395BL cells were cultured as described above. Cells (500,000) of each culture were suspended in 1 ml of suspension medium (10% DMSO in cell culture medium). Cells were collected the next day for single-cell CNV analysis via the 10x Genomics Chromium Single Cell CNV Solution (CG000153), which produces single-cell DNA libraries ready for Illumina sequencing, according to manufacturer’s recommendations (10x Genomics). Libraries were sequenced on an Illumina HiSeq 4000 using standard manufacturer’s protocols (Illumina) for 2 × 150-bp paired-end reads. Demultiplexing BCL files from the sequencing run and CNV analysis was performed using 10x Genomics Cell Ranger DNA version 1.1 software. CNV and heterogeneity visualization analysis was performed via 10x Genomics Loupe scDNA browser.
Affymetrix Cytoscan HD microarray
We obtained DNA of two reference cell lines from ATCC (HCC1395, SCCRL2324_ D; HCC1395BL, SCCRL2325_D). DNA concentration was measured spectrophotometrically using a Nanodrop (Life technology), and integrity was evaluated with a TapeStation 4200 (Agilent). Two hundred and fifty nanograms of gDNA was used to proceed with the Affymetrix CytoScan Assay kit (Affymetrix). The workflow consisted of restriction enzyme digestion with NspI, ligation, PCR, purification, fragmentation and end labeling. DNA was then hybridized for 16 h at 50 °C on a CytoScan array (Affymetrix), washed and stained in the Affymetrix Fluidics Station 450 (Affymetrix) and then scanned with the Affymetrix GeneChip Scanner 3000 G7 (Affymetrix). Data were processed with ChAS software (version 3.3). Array-specific annotation (NetAffx annotation release 36, built with human hg38 annotation) was used in the analysis workflow module of ChAS. Karyoview plot and segments data were generated with default parameters.
Disclaimer
This is a research study, not intended to guide clinical applications. The views presented in this article do not necessarily reflect current or future opinion or policy of the US Food and Drug Administration. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services. Any mention of commercial products is for clarification and not intended as endorsement.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All raw data (FASTQ files) are available on NCBI’s SRA database (SRP162370). The call set for somatic mutations in HCC1395 cells, VCF files derived from individual WES and WGS runs, BAM files for BWA-MEM alignments and source codes are available on NCBI’s ftp site (ftp://ftp-trace.ncbi.nlm.nih.gov/ReferenceSamples/seqc/Somatic_Mutation_WG). Some alignment files (BAM) are also available on Seven Bridges’ CGC platform under SEQC-II project. gDNA tested in the current study was prepared by ATCC using cell expansions from master banks of cells for the HCC1395 (ATCC, CRL-2324) and HCC1395BL (ATCC, CRL-2325) cell lines. gDNA aliquots from these preparations were distributed to the sequencing centers to perform WGS and WES as described. For remaining gDNA aliquots, contact the corresponding authors. Contact ATCC for additional materials related to the HCC1395 and HCC1395BL cell lines.
Software and code availability
The code to create somatic reference call set v1.2 is deposited on GitHub under a BSD 2-Clause open-source license tagged at https://github.com/bioinform/somaticseq/tree/seqc2_v1.2. A snapshot can also be downloaded at https://github.com/bioinform/somaticseq/archive/seqc2_v1.2.tar.gz.
References
Gall, J. G. Human genome sequencing. Science 233, 1367–1368 (1986).
Garraway, L. A. & Lander, E. S. Lessons from the cancer genome. Cell 153, 17–37 (2013).
Bailey, M. H. et al. Comprehensive characterization of cancer driver genes and mutations. Cell 173, 371–385 (2018).
ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020).
Hyman, D. M., Taylor, B. S. & Baselga, J. Implementing genome-driven oncology. Cell 168, 584–599 (2017).
Berger, M. F. & Mardis, E. R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 15, 353–365 (2018).
Hofmann, A. L. et al. Detailed simulation of cancer exome sequencing data reveals differences and common limitations of variant callers. BMC Bioinformatics 18, 8 (2017).
Krøigård, A. B., Thomassen, M., Lænkholm, A.-V., Kruse, T. A. & Larsen, M. J. Evaluation of nine somatic variant callers for detection of somatic mutations in exome and targeted deep sequencing data. PLOS ONE 11, e0151664 (2016).
Shi, W. et al. Reliability of whole-exome sequencing for assessing intratumor genetic heterogeneity. Cell Rep. 25, 1446–1457 (2018).
Kim, S. Y. & Speed, T. P. Comparing somatic mutation-callers: beyond Venn diagrams. BMC Bioinformatics 14, 189 (2013).
Zook, J. M. et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol. 32, 246–251 (2014).
Zook, J. M. et al. An open resource for accurately benchmarking small variant and reference calls. Nat. Biotechnol. 37, 561–566 (2019).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Xu, H., DiCarlo, J., Satya, R. V., Peng, Q. & Wang, Y. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics 15, 244 (2014).
Chen, Z. et al. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Sci. Rep. 10, 3501 (2020).
WHO Reference Panel 1st International Reference Panel for Genomic KRAS Codons 12 and 13 Mutations NIBSC code: 16/250 (National Institute for Biological Standards and Control, 2020).
Huo, Z., Tu, J., Lee, D.-F. & Zhao, R. Engineering mutation clones in mammalian cells with CRISPR/Cas9. Methods Mol. Biol. 2108, 355–369 (2020).
Ewing, A. D. et al. Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat. Methods 12, 623–630 (2015).
Lee, A. Y. et al. Combining accurate tumor genome simulation with crowdsourcing to benchmark somatic structural variant detection. Genome Biol. 19, 188 (2018).
Alioto, T. S. et al. A comprehensive assessment of somatic mutation detection in cancer using whole-genome sequencing. Nat. Commun. 6, 10001 (2015).
Craig, D. W. et al. A somatic reference standard for cancer genome sequencing. Sci. Rep. 6, 24607 (2016).
MDIC SRS Report: Somatic Variant Reference Samples for NGS. (Medical Device Innovation Consortium, 2019).
Stephens, P. J. et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 462, 1005–1010 (2009).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Gazdar, A. F. et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int. J. Cancer 78, 766–774 (1998).
Staaf, J. et al. Segmentation-based detection of allelic imbalance and loss-of-heterozygosity in cancer cells using whole genome SNP arrays. Genome Biol. 9, R136 (2008).
Suzuki, T., Tsukumo, Y., Furihata, C., Naito, M. & Kohara, A. Preparation of the standard cell lines for reference mutations in cancer gene-panels by genome editing in HEK 293T/17 cells. Genes Environ. 42, 8 (2020).
Jia, S. et al. A novel cell line generated using the CRISPR/Cas9 technology as universal quality control material for KRAS G12V mutation testing. J. Clin. Lab. Anal. 32, e22391 (2018).
Tian, X. et al. CRISPR/Cas9—an evolving biological tool kit for cancer biology and oncology. NPJ Precis. Oncol. 3, 8 (2019).
Blackburn, J. et al. Use of synthetic DNA spike-in controls (sequins) for human genome sequencing. Nat. Protoc. 14, 2119–2151 (2019).
Auton, A. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Fang, L. T. et al. An ensemble approach to accurately detect somatic mutations using SomaticSeq. Genome Biol. 16, 197 (2015).
Sahraeian, S. M. E. et al. Deep convolutional neural networks for accurate somatic mutation detection. Nat. Commun. 10, 1041 (2019).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Larson, D. E. et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317 (2012).
Lai, Z. et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 44, e108 (2016).
Fan, Y. et al. MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 17, 178 (2016).
Kim, S. et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594 (2018).
Freed, D., Pan, R. & Aldana, R. TNscope: accurate detection of somatic mutations with haplotype-based variant candidate detection and machine learning filtering. Preprint at bioRxiv https://doi.org/10.1101/250647 (2018).
Sahraeian, S. M. E., Fang, L. T., Mohiyuddin, M., Hong, H. & Xiao, W. Robust cancer mutation detection with deep learning models derived from tumor–normal sequencing data. Preprint at bioRxiv https://doi.org/10.1101/667261 (2019).
Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30, 2843–2851 (2014).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at https://arxiv.org/abs/1207.3907 (2012).
Poplin, R. et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 36, 983–987 (2018).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at bioRxiv https://doi.org/10.1101/201178 (2018).
Raine, K. M. et al. ascatNgs: identifying somatically acquired copy-number alterations from whole-genome sequencing data. Curr. Protoc. Bioinformatics 56, 15.9.1–15.9.17 (2016).
Flensburg, C., Sargeant, T., Oshlack, A. & Majewski, I. SuperFreq: integrated mutation detection and clonal tracking in cancer. PLoS Comput. Biol. 16, e1007603 (2020).
Deshwar, A. G. et al. PhyloWGS: reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol. 16, 35 (2015).
Nik-Zainal, S. et al. The life history of 21 breast cancers. Cell 149, 994–1007 (2012).
Wang, Y. et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 512, 155–160 (2014).
Yates, L. R. et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 21, 751–759 (2015).
Gerstung, M. et al. The evolutionary history of 2,658 cancers. Nature 578, 122–128 (2020).
Greaves, M. & Maley, C. C. Clonal evolution in cancer. Nature 481, 306–313 (2012).
McGranahan, N. & Swanton, C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell 168, 613–628 (2017).
Choo-Wosoba, H., Albert, P. S. & Zhu, B. A hidden Markov modeling approach for identifying tumor subclones in next-generation sequencing studies. Biostatistics https://doi.org/10.1093/biostatistics/kxaa013 (2020).
Xiao, W. & The Somatic Mutation Working Group of the SEQC-II Consortium. Towards best practice in cancer mutation detection with whole-genome and whole-exome sequencing. Nat. Biotechnol. https://doi.org/10.1038/s41587-021-00994-5 (2021).
Zhao, Y. et al. Whole genome and exome sequencing reference datasets from a multi-center and cross-platform benchmark study. Preprint at bioRxiv https://doi.org/10.1101/2021.02.27.433136 (2021).
Chen, W. et al. A multicenter study benchmarking single-cell RNA sequencing technologies using reference samples. Nat. Biotechnol. https://doi.org/10.1038/s41587-020-00748-9 (2020).
Chen, X. et al. A multi-center cross-platform single-cell RNA sequencing reference dataset. Sci. Data 8, 39 (2021).
Nik-Zainal, S. et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54 (2016).
Storchova, Z. & Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 121, 3859–3866 (2008).
Zook, J. M. et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 3, 160025 (2016).
Morrissy, A. S. et al. Spatial heterogeneity in medulloblastoma. Nat. Genet. 49, 780–788 (2017).
Araf, S. et al. Genomic profiling reveals spatial intra-tumor heterogeneity in follicular lymphoma. Leukemia 32, 1261–1265 (2018).
Ben-David, U. et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330 (2018).
Abraham, J. in Handbook of Transnational Economic Governance Regimes (eds. Tietje, C. & Brouder, A.) 1041–1053 (Brill Nijhoff, 2010).
Xiao, C. et. al. Personalized genome assembly for accurate cancer somatic mutation discovery using cancer-normal paired reference samples. Preprint at bioRxiv https://doi.org/10.1101/2021.04.09.438252 (2021).
Ptashkin, R. N. et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol. 4, 1589–1593 (2018).
Meisner, L. F. & Johnson, J. A. Protocols for cytogenetic studies of human embryonic stem cells. Methods 45, 133–141 (2008).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2012).
Acknowledgements
We thank J. Zook of the National Institute of Standards and Technology for advice in establishing reference samples and truth sets, S. Gowrisankar of Novartis and S. Chacko of the Center for Information Technology, NIH, for their assistance with data transfer and J. Ye of Sentieon for providing the Sentieon software package. We also thank D. Goldstein of the Office of Technology and Science at the National Cancer Institute (NCI), NIH, and L. Amundadottir of the Division of Cancer Epidemiology and Genetics, NCI, NIH, for the sponsorship and the use of the NIH Biowulf cluster, R. Phillip, Y. Hu, S. Liang and Y. Li of the Center for Devices and Radiological Health, US FDA, for their advice on study design and manuscript writing, J. Collins and E. Stahlberg of Biomedical Informatics and Data Science Directorate at Frederick National Laboratory for Cancer Research for reviewing the manuscript and providing suggestions and Seven Bridges Genomics for providing storage and computational support on the CGC. B.Z. was supported by the Intramural Research Program of the NIH, NCI, Division of Cancer Epidemiology and Genetics. Y. Zhao, K.T., T.S., B.T., J.S. and Y.K. were supported by the Frederick National Laboratory for Cancer Research and through the NIH fund (contract number 75N910D00024). L. Shi and Y. Zheng were supported by the National Natural Science Foundation of China (31720103909), the National Key R&D Project of China (2018YFE0201600) and Shanghai Municipal Science and Technology Major Project (2017SHZDZX01). E.R. was supported by the European Union through the European Regional Development Fund (2014-2020.4.01.15-0012). The CGC has been funded in whole, or in part, by Federal funds from the NCI, NIH (HHSN261201400008C), and ID/IQ Agreement number 17×146 under contract number HHSN261201500003I. C.X. and S. Sherry were supported by the Intramural Research Program of the National Library of Medicine, NIH. This work also used the computational resources of the NIH Biowulf cluster (http://hpc.nih.gov). Original data were also backed up on the servers provided by Center for Biomedical Informatics and Information Technology (CBIIT), NCI. The genomic work performed at the Loma Linda University (LLU) Center for Genomics was funded in part by the NIH grant S10OD019960, the American Heart Association grant 18IPA34170301, the Ardmore Institute of Health grant 2150141 and C.A. Sims’ gift to LLU Center for Genomics. We acknowledge TopEdit for linguistic editing and proofreading during the preparation of this manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
The study was conceived and designed by W.X., C.W., L. Shi, H.H., E.D., Z.T., B.N., W.T. and R.J. Biosample preparation was performed by L.K., K.L., M. Mars and T.H. NGS library preparation and sequencing was performed by W.C., Z.C., S. Stanbouly, K.I., H.J., E.J., G.P.S., Y. Zheng, B.T., Y.Y., J.S., Y.K., M. Mehat, V.P., M.S., T.H., E.P., R.K., J.D., P.V., R.M., D.G., S.K., E.R., A.S., J.N., U.L., J.W., J.L., P.D.H., C.C., S.M., J.S., J.F., D.B. and C.E.M. Data analysis was performed by L.T.F., W.X., B.Z., Y. Zhao, Z.Y., L.R., C.L., O.D.A., L. Song, J.L., T.S., K.T., D.M., C.N., M.C., S.M.E.S., M. Mohiyuddin, Y.G., L.Y., H.L., M.P., Z.L., W.S.L., J.K., J.A., E.T., V.Z., T.M. and J.T. Data management was performed by W.X., C.X. and S.T.S. The manuscript was written by L.T.F., B.Z., W.X., R.K., M. Moos, C.X., S.T.S. and Y. Zhao. W.X. managed the project.
Corresponding authors
Ethics declarations
Competing interests
L.T.F., S.M.E.S., M. Mohiyuddin, Y.G., L.Y. and H.L. are employees of Roche Sequencing Solutions Inc. L.K., K.L. and M. Mars are employees of ATCC, which provides cell lines and derivative materials. E.J., G.P.S. and O.D.A. are employees of Illumina Inc. V.P. and M.S. are employees of Novartis Institutes for Biomedical Research. T.H., E.P. and R. Kalamegham are employees of Genentech (a member of the Roche group). Z.L. is an employee of Sentieon Inc. R. Kusko is an employee of Immuneering Corp. C.C., S.M. and J.S. are employees of 10x Genomics. All other authors declare no competing interests.
Additional information
Peer review information Nature Biotechnology thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 3D scatter plot shows the consistency of SomaticSeq and NeuSomatic classification of somatic variant calls.
3D scatter plot for number of PASS classifications by SomaticSeq, NeuSomatic-E, and VAF for (a) SNV (R = 0.997) and (b) indel calls (R = 0.925). (c) The subset of SNV calls that were re-sequenced by AmpliSeq. Solid markers are deemed ‘validated.’ Open markers are deemed ‘not validated.’ Stars/crosses are deemed uninterpretable. HighConf calls generally have many PASS calls and a full range of VAF. MedConf have fewer PASS calls and tend to have lower VAF. Unclassified calls have a full range of VAF, which means their somatic signals were poor-quality.
Extended Data Fig. 2 Genome coverage and high-confidence regions.
(a) Genome coverage by reads from three technologies. Inner track: PacBio. Middle track: 10X Genomics. Outer track: Illumina HiSeq. (b) Genome regions coverage by short reads in comparison to NA12878. Outer black Track: Gene density plot. Middle orange track: NA12878. Inner blue track: the callable regions in HCC1395.
Extended Data Fig. 3 Validation of somatic indels.
(a) Validation of indels by AmpliSeq. R = 0.989 for HighConf calls. (b) Validation of indels by WES with Ion Torrent. R = 0.767 for HighConf calls. (c) Validation of indels by WES with HiSeq. R = 0.990 for HighConf calls. (d) Histogram of indel sizes. The dashed lines on the diagonal for (a), (b), and (c) are the 95% binomial confidence-interval of observed VAF given the actual VAF, calculated based on depths of 2000X for AmpliSeq, 34X for Ion Torrent, and 100X for WES, respectively. (d) shows the indel lengths of the somatic indels in the reference call set.
Extended Data Fig. 4 Validation of germline indels.
Germline indel scatter plots comparing VAF super set to confirmed VAF. (a) VAF scatter plot of germline indels by WGS super set and AmpliSeq. (b) VAF scatter plot of germline indels by truth set and Ion Torrent WES.
Extended Data Fig. 5 Karyotyping of HCC1395 and HCC1395BL.
(a) Karyotype of HCC1395. Cytogenetic analysis was performed on ten G-Banded metaphase cells from HCC1395. Analysis pointed to a hypertetraploid line with chromosome counts ranging from 64-79 and gain of 38-63 unidentifiable marker chromosomes. (b) Karyotype of HCC1395BL. Cytogenetic analysis was performed on ten G-banded metaphase cells from HCC1395BL. All ten cells showed loss of a chrX and an unbalanced whole arm translocation between the long-arm of chr6 at band q10 and the short-arm of chr16 at band p10. This resulted in a net loss of one copy of the short-arm of chr6 and loss of one copy of the long-arm of chr16. The abnormal chromosome could be placed in either a chr6 or chr16 locus as we were unable to determine if the centromere belongs to chr6 or chr16 (inset figure).
Extended Data Fig. 6 Cytogenetic analysis with Affymetrix Cytoscan HD microarray.
Cytogenetic analysis with Affymetrix Cytoscan HD microarray. (a) Cytogenetic view of HCC1395. (b) Cytogenetic view of HCC1395BL. The losses of chr6p, chr16q, and chrX were confirmed.
Extended Data Fig. 7 Variant allele frequencies across the genome.
(a) VAF of truth set germline SNVs in HCC1395BL. The copy numbers of HCC1395BL were predicted by Affymetrix Cytoscan HD microarray. (b) VAF of the truth set germline SNV positions (discovered in HCC1395BL) in HCC1395. (c) VAF of the truth set somatic SNVs in HCC1395. The copy numbers of HCC1395 were predicted by ascatNgs.
Extended Data Fig. 8 Variant allele frequencies of somatic mutations.
(a) VAFs of somatic SNVs and indels in the reference call sets. (b) VAFs of reference SNVs in different copy number states as predicted by ascatNgs.
Extended Data Fig. 9 Tumor sample HCC1395 CNV and Clonality Analysis.
Tumor sample HCC1395 CNV and Clonality Analysis. (a) Clonality analysis from WES data using SuperFreq for tumor cell line HCC1395. The clonality of each somatic SNV was calculated based on the VAF, accounting for local copy number. The SNVs and CNAs were evaluated with hierarchical clustering based on the clonality and uncertainty across replicates for HCC1395. The river plot shows the relative distribution of multiple subclones in HCC1395. The main cancer clone (blue) and the two subclones (red and green) appeared in early time of clonal evolution, while subclone (orange) and its descendant (peak) appeared in the late event of the clonal evolution. (b) The main- and sub-clonal somatic copy number profiles using subHMM38 from the Illumina WGS data set. Main-clonal genotype: upper panel; sub-clonal genotype: middle panel; sub-clonal proportion: bottom bar plot. Each colored block represents the genotype of somatic copy number alterations (SCNAs) in the corresponding position of the chromosome. The chromosomes are separated by vertical dash lines. Genotype of SCNAs: deletion (DEL), homozygous deletion (HOMD), hemizygous deletion loss of heterozygosity (DLOH), copy neutral loss of heterozygosity (NLOH), diploid heterozygous (HET), gain of one allele (GAIN), amplified loss of heterozygosity (ALOH), allele-specific copy number amplification (ASCNA), balanced copy number amplification (BCNA), and unbalanced copy number amplification (UBCNA).
Extended Data Fig. 10 Number of somatic mutations detected in HCC1395 and 560 triple negative and non-triple negative breast cancers from previous literature.
Number of somatic mutations detected in HCC1395 and 560 triple negative and non-triple negative breast cancers from previous literature59.
Supplementary information
Supplementary Information
Supplementary Figs. 1–7, Tables 1–10 and sections on (1) somatic mutations, (2) germline variants, (3) detailed method to produce the reference call set, (4) software used and (5) statistical methods.
Supplementary Data 1
Tables of manually curated somatic variant calls: a record of manually inspected variant sites.
Supplementary Data 2
Tables of somatic coding variants and germline variants in ClinVar.
Supplementary Data 3
Tables of somatic variant calls where the validation platform showed discrepancy with the reference data sets
Rights and permissions
About this article

Cite this article
Fang, L.T., Zhu, B., Zhao, Y. et al. Establishing community reference samples, data and call sets for benchmarking cancer mutation detection using whole-genome sequencing. Nat Biotechnol 39, 1151–1160 (2021). https://doi.org/10.1038/s41587-021-00993-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-021-00993-6
This article is cited by
-
COSAP: Comparative Sequencing Analysis Platform
BMC Bioinformatics (2024)
-
Improving somatic exome sequencing performance by biological replicates
BMC Bioinformatics (2024)
-
Reference Materials for Improving Reliability of Multiomics Profiling
Phenomics (2024)
-
The Quartet Data Portal: integration of community-wide resources for multiomics quality control
Genome Biology (2023)
-
Haplotype-resolved assemblies and variant benchmark of a Chinese Quartet
Genome Biology (2023)
