
Abstract
Antibodies against viral pathogens represent promising therapeutic agents for the control of infection, and their antiviral efficacy has been shown to require the coordinated function of both the Fab and Fc domains1. The Fc domain engages a wide spectrum of receptors on discrete cells of the immune system to trigger the clearance of viruses and subsequent killing of infected cells1,2,3,4. Here we report that Fc engineering of anti-influenza IgG monoclonal antibodies for selective binding to the activating Fcγ receptor FcγRIIa results in enhanced ability to prevent or treat lethal viral respiratory infection in mice, with increased maturation of dendritic cells and the induction of protective CD8+ T cell responses. These findings highlight the capacity for IgG antibodies to induce protective adaptive immunity to viral infection when they selectively activate a dendritic cell and T cell pathway, with important implications for the development of therapeutic antibodies with improved antiviral efficacy against viral respiratory pathogens.
Similar content being viewed by others

Main
Several monoclonal antibodies to influenza virus epitopes from the globular head and the stalk domains of influenza hemagglutinin (HA) and neuraminidase (NA) (Fig. 1a) have been shown to confer broad and potent antiviral activity against diverse influenza strains5,6,7,8. These broadly protective monoclonal antibodies require Fc effector activity to provide full protection from lethal viral challenge, as mutant Fc domain variants that are unable to interact with Fc receptors (FcγRs) expressed on effector leukocytes are associated with reduced in vivo antiviral potency5,6. Although previous studies clearly demonstrated that broadly protective anti-influenza monoclonal antibodies depend on activating, but not inhibitory, FcγRs for activity5,6, the cell types and specific FcγRs that contribute to the antiviral activity of these monoclonal antibodies remains to be determined. The diversity of FcγR expression on immune cells, the structural complexity of the FcγR family, and the divergence of these receptors in different species (reviewed in ref. 9) pose particular challenges in resolving the mechanistic details of how FcγR dependence of anti-influenza antibodies result in enhanced protection in vivo.

a, Influenza virus antigens (HA and NA) and the names of the tested monoclonal antibodies. b, Fc variants with differential FcγR binding affinity were generated for anti-influenza monoclonal antibodies. WT, wild type. c, d, Fc variants of anti-HA stalk monoclonal antibodies FI6v3 (c) and FY1 (d) were administered intraperitoneally (4 mg kg−1 for FI6v3 and 2 mg kg−1 for FY1) to FcγR-humanized mice before challenge with influenza (H1N1 PR8) (n = 6 mice per group for PBS-treated; n = 10 for WT FI6v3, GA, ALIE, GRLR and WT FY1; n = 9 for FI6v3 GAALIE and FY1 V11; and n = 8 for FY1 GA, afucosylated (Afuc), and GAALIE in two independent experiments). Weight loss (left; mean ± s.e.m.) and survival curves (right) were compared to the corresponding wild-type human IgG1 antibody-treated group by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) (c: **P = 0.0072, #P = 0.0029, *P = 0.014, ***P = 0.0035) and log-rank (Mantel–Cox) test, respectively (c: *P = 0.019654; **P = 0.006759; ***P = 0.000303; d: *P = 0.0494, **P = 0.0045). NS, not significant.
To address this problem, we previously described a mouse model in which only human FcγRs are expressed in a pattern that recapitulates the expression pattern seen in human tissues as accurately as possible10. Despite the inherent limitations of the use of animal models to study human infection and disease, as well as differences in the absolute FcγR expression levels and pattern of expression between humans and FcγR-humanized mice (for example, the expression of FcγRIIb on monocytes and neutrophils that are commonly absent in humans), this strain represents a suitable platform to perform studies on human IgG antibody function with translational relevance to humans. This in vivo system is combined with anti-influenza antibodies (Fig. 1a) in which the human IgG1 Fc is expressed as a series of variants with selective binding affinity to specific human FcγRs (Fig. 1b, Extended Data Fig. 1a–j). These antibodies are administered to FcγR-humanized mice before lethal challenge with influenza virus (intranasally, with five mouse lethal doses at 50% (mLD50)) and weight loss and survival were monitored over 14 days. Mice treated with broadly protective monoclonal antibodies that target the stalk domain of HA (FI6v38 or FY17) showed enhanced protection when the Fc is modified to selectively engage the FcγRIIa receptor (GA variant11,12), alone or in combination with enhanced FcγRIIIa binding (GAALIE variant13) (Fig. 1c, d). Although FcγRIIa-enhanced variants (GA) of FY1 fully protect mice that express only human FcγRIIa under the conditions tested, they are unable to confer antiviral protection of FcγR-deficient mice, confirming the dependence on FcγRIIa engagement in the enhanced protection mediated by GA variants (Extended Data Fig. 5a–d). Similarly, the administration of a blocking monoclonal antibody with relative selectivity against FcγRIIa (clone IV.3) reduces the ability of FcγRIIa-enhanced variants (GA) of FI6v3 to protect FcγR-humanized mice against lethal influenza challenge (Extended Data Fig. 5e, f). Enhancing FcγRIIIa binding alone—using two complementary approaches: (i) protein engineering (ALIE variant12,14) or (ii) glycoengineering (afucosylated glycoforms15)—does not provide enhanced protection compared with the wild-type human IgG1 at the selected monoclonal antibody dose (determined based on titration studies that established the antibody dose at which wild-type IgG1 offers sub-optimal protection) (Extended Data Fig. 2). Consistent with previous studies that determined a key role for activating FcγR engagement in the anti-viral activity of the selected monoclonal antibodies5,6, no protection is observed when the Fc is modified to abrogate FcγR binding (GRLR variant) or engineered to engage the inhibitory FcγRIIb (V11 variant16) (Fig. 1c, d). None of these Fc modifications affect the in vitro neutralization activity and target antigen-binding specificity (Extended Data Fig. 3a–f) or cause protein aggregation and altered pharmacokinetics in vivo (Extended Data Figs. 1c, d, 4a). In addition, quantification of the serum levels of anti-influenza monoclonal antibodies on day 3 after infection revealed comparable levels among the different Fc domain variants, which indicates that the observed effects could not be attributed to differential monoclonal antibody half-lives or in vivo stability (Extended Data Fig. 4b, c).
To determine whether the dependence on FcγRIIa for the antiviral protection conferred by anti-HA stalk antibodies also extends to antibodies against other viral epitopes, we generated Fc domain variants for the 4G055 and 1A015 antibodies, which target the globular head of HA and exhibit differential neutralization and haemagglutination inhibition (HAI) activity, as well as for the broadly reactive anti-NA monoclonal antibody, 3C0517 (Extended Data Fig. 3g–o). Similar to anti-HA stalk antibodies, Fc variants with enhanced affinity for FcγRIIa (GA or GAALIE variants) demonstrate enhanced protective activity compared with their wild-type human IgG1 counterparts (Fig. 2, Extended Data Fig. 4d–f), which suggest that the FcγR mechanisms by which anti-influenza monoclonal antibodies confer protection against infection are conserved among antibodies with differential in vitro neutralization potency and epitope specificity.

a–c, Fc variants with differential FcγR affinity (Fig. 1b) were generated for the anti-HA head monoclonal antibodies 4G05 (a) and 1A01 (b), and the anti-NA monoclonal antibody 3C05 (c). FcγR-humanized mice were administered intravenously with Fc domain variants of 4G05 (0.5 mg kg−1) (n = 10 mice per group for antibody-treated groups; n = 5 for PBS group in two independent experiments) (a), 1A01 (2 mg kg−1) (n = 8 mice per group for WT, GA, ALIE and GAALIE groups; n = 6 for GRLR; n = 4 for PBS in two independent experiments) (b), and 3C05 (20 mg kg−1) (n = 12 mice per group for WT and GA groups; n = 10 for GRLR and ALIE; n = 9 for GAALIE; n = 6 for PBS in two independent experiments) (c) before lethal challenge with influenza (H1N1 Neth/09). Weight loss (left; mean ± s.e.m.) and survival curves (right) were compared to the corresponding wild-type unmodified IgG1-treated group by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) (a: *P = 0.0034, #P = 0.0052; ^P = 0.02; ¶P = 0.03; b: *P = 0.03, **P = 0.02, #P = 0.01, ^P = 0.001, ##P = 0.005, §P = 0.002, ^^P = 0.006, ¶P = 0.04, ^#P = 0.009; c: *P = 0.03, ^P = 0.02, #P = 0.04) and log-rank (Mantel–Cox) test, respectively (a: **P = 0.0042, b: **P = 0.0032, *P = 0.01285, c: *P = 0.0167, **P = 0.0055).
Our findings demonstrate that although FcγRIIa is the major receptor that drives the protective activity of anti-influenza monoclonal antibodies, FcγRIIIa has paradoxically limited contribution to the antibody-mediated protection, despite numerous studies that have previously determined that the cytotoxic clearance of malignant or virus-infected cells is predominantly mediated by FcγRIIIa2,12. In addition, despite the abundant expression of FcγRIIIa on alveolar macrophages at baseline as well as the influx of FcγRIIIa-expressing natural killer cells in response to infection (Extended Data Fig. 6a–c), selective engagement of this receptor does not enhance protection, suggesting that enhancing the clearance of viral particles by alveolar macrophages or killing of infected cells by natural killer cells does not improve the ability of these antibodies to protect against lethal influenza challenge.
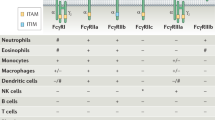
FcγRs can either activate (FcγRI, FcγRIIa and FcγRIIIa) or inhibit (FcγRIIb) cellular responses. Activating FcγRs trigger intracellular signalling subsequent to crosslinking of the extracellular ligand binding domains by IgG immune complexes by either intrinsic cytoplasmic ITAM motifs (FcγRIIa) or γ or ζ chain-associated ITAM motifs (FcγRIIIa), recruiting SYK family tyrosine kinases (reviewed in ref. 1). Because FcγRIIa and FcγRIIIa are redundantly expressed on a variety of innate immune cells, including monocytes and macrophages (Extended Data Fig. 6d–f), it is unlikely that the unique dependence on FcγRIIa engagement that results in enhanced antiviral protection is mediated by these cells. Although neutrophils co-express FcγRIIa and FcγRIIIb, the latter lacks intracellular signalling domains and has limited ability to mediate effector activities1, even when engaged by variants (ALIE and afucosylated) that exhibit increased affinity for this receptor. It is therefore possible that neutrophils, through specific activation of FcγRIIa, might contribute to the observed antiviral potency of FcγRIIa-enhanced variants. To test this hypothesis, we assessed whether or not neutrophil depletion influences the ability of FcγRIIa-enhanced variants (GA) of FY1 to protect FcγR-humanized mice from lethal influenza challenge. We found that depletion of neutrophils had no effect on the antiviral activity of FcγRIIa-enhanced FY1 (Extended Data Fig. 5h, i), which suggests that neutrophils are unlikely to contribute notably to the FcγRIIa-mediated antiviral protection we observe.
In contrast to alveolar macrophages and monocytes, conventional dendritic cells (such as cDC1 (CD11c+CD11b−CD103+) and cDC2 (CD11c+CD11b+CD103−) subsets) (Extended Data Fig. 7a–f) express FcγRIIa and the inhibitory receptor FcγRIIb, but not FcγRIIIa, and are found both at baseline and after infection in the lung (Extended Data Fig. 7b). To investigate the effect of FcγRIIa engagement by Fc-engineered monoclonal antibodies on the functional activity of the various dendritic cell subsets (cDC1, cDC2 and FcγRIII-expressing TNF/iNOS-producing dendritic cells (tipDCs) that emerge in response to influenza infection; defined as MHCII+CD11c+CD11b+CD103−Gr-1+) (Extended Data Fig. 7a), we analysed dendritic cells in the lungs of FcγR-humanized mice infected with influenza that have previously received Fc variants of the anti-HA stalk monoclonal antibody FI6v3. The treatment of mice with the FcγRIIa-enhancing variant (GAALIE) before influenza challenge resulted in dendritic cell maturation (Fig. 3a, b, Extended Data Fig. 7g–i, l–n), as seen by the increased frequency of CD80highCD86high cells on cDC1 and cDC2 subsets, and upregulation of CD40 expression in the cDC1 subset—the dendritic cell population specialized for cross-presentation and CD8 T cell stimulation18. By contrast, the same antibody (FI6v3) expressed either as wild-type or with an Fc modified to abrogate FcγR binding (GRLR) did not result in dendritic cell maturation. Unlike the cDC1 and cDC2 subsets, treatment with the GAALIE variant is not associated with increased maturation of tipDCs, which suggests a limited role for this population in the antibody-mediated protection against influenza infection conferred by FcγRIIa-enhanced variants (Extended Data Fig. 7i). Consistent with these in vivo data from FcγR-humanized mice, ex vivo stimulation of human monocyte-derived dendritic cells with IgG immune complexes revealed improved capacity for FcγRIIa-enhanced variants (GA and GAALIE) to induce cellular maturation (Extended Data Fig. 7j, k).

a–c, FcγR-humanized mice (n = 4 mice per group for all groups, except n = 3 for day 7 WT and GRLR) were treated with FI6v3 monoclonal antibody Fc variants (intraperitoneally 3 mg kg−1), infected with H1N1 (PR8), and then lung-resident dendritic cells and T cells were analysed on days 4 (n = 4 mice per group) or 7 (n = 3 mice per group for WT and GRLR, n = 4 for GAALIE in two independent experiments) after infection. One-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) was used to compare the frequency of mature conventional dendritic cell subsets (CD80highCD86high) on day 4 after infection (*P = 0.0384, **P = 0.0075, ***P = 0.0008, ****P = 0.0002) (a), CD40 expression in cDC1 subsets (*P = 0.0261, **P = 0.0139, ***P = 0.0061, ****P = 0.0007) (b), and the frequency of activated (CD44hiCD69+) CD8+ and CD4+ T cells (day 4 after infection) (^P = 0.0412, *P = 0.0232, **P = 0.0018, ***P = 0.0003) (c). MFI, mean fluorescence intensity. d–f, Fc variants of the FY1 monoclonal antibody (mAb) were administered intraperitoneally (2 mg kg−1) to FcγR-humanized mice before influenza challenge (H1N1 PR8). Isotype or anti-mouse CD8 monoclonal antibodies were administered on day 3 after infection (isotype-treated groups: n = 12 mice per group for WT, GAALIE and PBS groups, n = 8 mice for GA; anti-CD8-treated groups: n = 11 mice per group for WT, n = 8 for GA, n = 10 for PBS, and n = 12 for GAALIE in three independent experiments). e, f, Weight loss (mean ± s.e.m.) (e) and survival curves (f) were compared against the corresponding isotype-treated group by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) (*P = 0.03, ^P = 0.0220) and log-rank (Mantel–Cox) test, respectively (**P = 0.00199, *P = 0.0477).
Maturation of dendritic cells and the induction of the accessory molecules CD80, CD86 and CD40 in the virally challenged lung is a prerequisite to the activation of antigen-specific naive T cells. This would indicate that an anti-viral antibody modified to enhance dendritic cell maturation by FcγRIIa engagement can induce an adaptive response that results in the induction of protective T cell immunity. To explore this hypothesis, we characterized the T cell responses in the lungs of FcγR-humanized mice treated with anti-influenza monoclonal antibodies with selective FcγR-binding properties before viral challenge. We found that the GAALIE variant induced increased activation of both CD8+ and CD4+ T cells (Fig. 3c, Extended Data Fig. 8), whereas neither wild-type IgG1 nor the GRLR variant showed evidence of robust induction of T cell responses. To determine whether the observed induction of T cell activation also contributes to the enhanced protection we observed with the FcγRIIa-binding variants (Figs. 1, 2), we repeated the FY1 antibody pre-treatment and viral challenge protocol, modifying it to include a CD8+ or CD4+ cellular depletion step on day 3 after infection (Fig. 3d, Extended Data Fig. 10a–c). Depletion of CD8+, but not CD4+, T cells resulted in the loss of enhancement of the GA or GAALIE Fc variant, demonstrating that the improved protection observed for FcγRIIa-enhanced variants is mediated by the induction of protective CD8+ T cell responses (Fig. 3e, f, Extended Data Figs. 4g, 10a–e).
Because recent reports have suggested FcγRIIa expression by subsets of T cells19,20,21, we analysed FcγR expression in human peripheral T cells, as well as in T cells obtained from the blood, spleen or lung of naive or influenza-infected FcγR-humanized mice (Extended Data Fig. 9) to determine whether the improved protective activity of FcγRIIa-enhanced variants is the outcome of direct T cell activation by FcγR crosslinking. With the exception of a minor (10%) subset of CD8+ T cells in the lung of infected mice that is positive for FcγRIII (Extended Data Fig. 9l), T cells from human or FcγR-humanized mice were negative for all FcγRs, which suggests that the antibody-mediated anti-viral protection is not due to direct effects on T cells.
Because FcγRIIa-enhanced variants induce the activation of both CD8+ and CD4+ T cells, we tested whether these effects also drive improved antiviral IgG responses. IgG responses were characterized in mice treated with wild-type or GAALIE variants of the non-neutralizing anti-HA monoclonal antibody 1A01 at a dose that both variants offer comparable protection. Treatment with either wild-type or FcγRIIa-enhanced variants resulted in robust and comparable IgG responses against HA and NP from homologous or heterologous virus strains, as well as serum HAI activity (Extended Data Fig. 10f–m).
Through interactions with effector leukocytes, antibodies against viral antigens can enhance disease and contribute to specific histopathologic manifestations. Although this phenomenon, termed antibody-dependent enhancement, has been shown specifically for flaviviruses, such as dengue22, clinical experience from severe cases of viral respiratory infections, such as influenza and SARS-CoV-2, also raise the possibility that antibodies might have a pathogenic role through uncontrolled or inappropriate amplification of local inflammatory responses. For example, studies during the 2009 influenza pandemic have shown that severe disease was associated with evidence of IgG-mediated inflammation in the lung parenchyma through IgG immune complex and complement deposition to the lung tissue23. Similarly, severe cases of COVID-19 disease are often characterized by excessive pro-inflammatory cytokine expression and T cell responses, as well as by clinical manifestations that resemble secondary haemophagocytic lymphohistiocytosis24. Given that Fc-engineered variants with increased affinity for FcγRIIa can enhance adaptive T cell responses by activation of FcγRIIa-expressing dendritic cells, it is important to determine whether such variants could also modulate disease pathogenesis through inappropriate amplification of host inflammatory responses that are elicited in response to virus infection. To determine whether FcγRIIa-enhanced variants could lead to severe disease, we assessed the in vivo activity of these variants in FcγR-humanized mice with established influenza infection (Fig. 4a). Mice were infected with influenza and 3 days after infection, FY1 monoclonal antibody (either wild-type or GAALIE) was administered at different doses (5–15 mg kg−1). Although wild-type IgG1 FY1 was unable to rescue mice from lethal influenza infection, GAALIE variants exhibited a dose-dependent therapeutic benefit. These findings suggest that the unique FcγR-binding profile of the GAALIE variant (increased for activating FcγRIIa and FcγRIIIa receptors, but reduced for the inhibitory FcγRIIb receptor), and its ability to stimulate CD8 T cell responses has no pathogenic consequences, instead it provides meaningful and robust protection from established infection (Fig. 4b, c).

a–c, FcγR-humanized mice (n = 6 mice per group for antibody-treated groups; n = 3 for PBS-treated in two independent experiments) were infected with influenza (H1N1 PR8), and FY1 Fc variants were administered intraperitoneally 3 days after infection at the indicated dose. b, c, Weight loss (mean ± s.e.m.) (b) and survival curves (c) of GAALIE-treated mice were compared with the PBS-treated group at the corresponding antibody dose by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) (*P = 0.0456, ^P = 0.041, **P = 0.0014, ^^P = 0.0003, #P = 0.0005, ***P < 0.0001) and log-rank (Mantel–Cox) test, respectively (**P = 0.000911). d–h, The protective activity of LS and GAALIE–LS variants of FY1 was evaluated in a model of antibody-mediated prophylaxis. FcγR/FcRn-humanized mice (n = 8 mice per group for GAALIE–LS at 1.6 and 0.4 mg kg−1; n = 10 for LS at 0.1 mg kg−1, n = 3 for PBS, n = 9 for the remaining treatment groups in two independent experiments) were administered intravenously with the indicated dose of FY1 2 days before influenza challenge (H1N1 PR8). e, f, Weight loss (mean ± s.e.m.) (e) and survival curves (f) of GAALIE–LS-treated mice were compared against the LS group at the corresponding antibody dose by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) (*P < 0.001, ^P = 0.01, #P = 0.02, §P = 0.006, ¶P = 0.002) and log-rank (Mantel–Cox) test, respectively (*P = 0.0283, **P = 0.0266, ***P = 0.00377, ****P = 0.000143). h, The enhanced potency conferred by the GAALIE–LS variant was quantified by plotting the maximum weight change after infection against the serum antibody concentration at the time of challenge. Data were fitted by nonlinear regression analysis (four-parameter, variable slope).
In addition to their therapeutic potential, monoclonal antibodies engineered for increased FcγRIIa affinity could provide long-term prophylaxis from influenza infection, especially when combined with Fc domain mutations (for example, the LS (M428L/N434S) variant25) that increase affinity for human FcRn and extend IgG half-life in vivo25. Using a mouse model of antibody-mediated prophylaxis of influenza infection (Fig. 4d), we compared the ability of LS (enhanced for FcRn) and GAALIE–LS (enhanced for FcRn, FcγRIIa and FcγRIIIa) variants of FY1 to protect FcγR- or FcRn-humanized mice from influenza infection. At all doses tested (0.1–1.6 mg kg−1), GAALIE–LS variants demonstrated superior protective activity over their LS counterparts (Fig. 4e–g, Extended Data Fig. 4h). In addition, quantification of the protective activity of the two FY1 Fc variants over a wide range of doses revealed that the GAALIE–LS variant exhibited at least 5.5-fold improvement in in vivo antiviral potency (Fig. 4h), which suggests that Fc engineering for increased affinity to specific FcγRs represents a promising approach that could substantially improve the clinical efficacy of antiviral monoclonal antibodies.
IgG antibodies can mediate pleiotropic effects, resulting from the diversity of Fc binding molecules that engage the Fc domain. The Fc domain is structurally diverse, the consequence of subclasses and Fc glycosylation, and results in differential Fc receptor binding activities for various Fc structural variants (reviewed in ref. 1). This natural heterogeneity contributes to the efficacy of polyclonal IgG responses to viral infections, and provides a mechanism for the recognition of diverse viral epitopes and triggering of several effector pathways. The development of monoclonal antibodies for the selective binding to specific neutralizing viral epitopes can now be coupled to Fc modifications to facilitate the engagement of specific FcγRs to optimize the potency of these therapeutic agents. As demonstrated for anti-tumour therapeutic antibodies, enhancing their affinity for FcγRIIIa results in improved phagocytosis of tumour cells by macrophages (antibody-dependent cell phagocytosis, ADCP), as well as enhanced killing by natural killer cells (antibody-dependent cell cytotoxicity, ADCC), which translates to improved clinical efficacy26. On the basis of these findings, it has been presumed that enhancing the ability of the Fc domain to engage and activate FcγRIIIa would also lead to increased therapeutic efficacy of protective anti-viral antibodies. However, this does not seem to be the case. Antibody treatment of HIV infection has been shown to induce a CD8+ response both in chronically infected macaques and humans, which contributes to the control of viraemia27,28. Our present results show that selective engagement of the activating FcγR on dendritic cells, FcγRIIa, by a variety of anti-influenza monoclonal antibodies results in the induction of a protective CD8+ response, mechanistically similar to the ‘vaccinal’ response we have observed for anti-tumour antibody treatment12. The ability of an antibody to not only couple to innate effector responses through its Fc domain, but also induce an adaptive response by engaging and activating dendritic cells, provides a potent new approach to the design of therapeutic antibodies for the prevention and treatment of viral diseases. This approach to Fc engineering is particularly relevant to pandemic viruses, such as influenza and SARS-CoV-2. Neutralizing antibodies to these viruses, engineered to enhance dendritic cell activation and CD8+ T cell responses, as shown here for the GAALIE variant, are predicted to provide considerable enhancement of protection by stimulating a variety of synergistic immunological pathways.
Methods
Viruses, cell lines and mouse strains
The A/Puerto Rico/8/34 (PR8) and A/Netherlands/602/09 (Neth09) H1N1 viruses were grown as previously described6. MDCK cells (ATCC) were maintained at 37 °C, 5% CO2 in DMEM supplemented with 10% FBS, 50 U ml−1 penicillin and 50 μg ml−1 streptomycin (ThermoFisher). Expi293F cells (ThermoFisher) were maintained at 37 °C, 8% CO2 in Expi293 expression medium (ThermoFisher) supplemented with 10 U ml−1 penicillin and 10 μg ml−1 streptomycin. FcγRIIa- and FcγRIIIa-expressing NFAT reporter cells were purchased from Promega and have been authenticated by the vendor by STR analysis. All cell lines tested negative for mycoplasma contamination. All in vivo experiments were performed in compliance with federal laws and institutional guidelines and have been approved by the Rockefeller University Institutional Animal Care and Use Committee. Mice were bred and maintained at the Comparative Bioscience Center at the Rockefeller University at a controlled ambient temperature environment with 12-h dark/light cycle. C57BL/6 mice were purchased from The Jackson Laboratory. FcγR-humanized mice (mFcγRα−/−, Fcgr1−/−, hFcγRI+, hFcγRIIaR131+, hFcγRIIb+, hFcγRIIIaF158+, and hFcγRIIIb+) and FcγR knockout mice (mFcγRα−/−; Fcgr1−/−) were generated in the C57BL/6 background and extensively characterized in previous studies10. Human FcγRIIa-only mice (hFcγRIIa+) were generated in a mouse FcγR-deficient background (C57BL/6; FcγRα−/−; Fcgr1−/−)10. FcRn-humanized mice (B6.Cg-Fcgrttm1Dcr Tg(FCGRT)32Dcr/DcrJ) were purchased from The Jackson Laboratory and are deficient in mouse FcRn and express human FcRn as a transgene29,30. FcγR/FcRn-humanized mice were generated by crossing the FcγR-humanized strain to the FcRn-humanized mice31.
Cloning, expression and purification of recombinant IgG antibodies
For the generation of Fc domain variants of human IgG1 Fc domain variants, site-directed mutagenesis using specific primers was performed as previously described4. Recombinant antibodies were generated by transient transfection of Expi293 or Expi-CHO cells with heavy and light chain expression plasmids using previously described protocols13. Before transfection, plasmid sequences were validated by direct sequencing (Genewiz). Recombinant IgG antibodies were purified from cell-free supernatants by affinity purification using protein G or protein A sepharose beads (GE Healthcare). Purified proteins were dialysed in PBS, filter-sterilized (0.22 μm), and purity was assessed by SDS–PAGE followed by SafeStain blue staining (ThermoFisher). All antibody preparations were more than 90% pure and endotoxin levels were less than 0.05 EU mg−1, as measured by the limulus amebocyte lysate assay. For the generation of afucosylated Fc domain variants, CHO cells were transfected with heavy chain and light chain expression plasmids in the presence of 100 μM 2-fluorofucose peracetate32. To confirm the absence of fucose, glycans were released with PNGase F, labelled with Waters RapiFluor-MS, cleaned up with a HILIC microElution plate, injected onto a Waters Glycan BEH Amide column, using a Thermo Vanquish UHPLC with FLD detection. Chromatograms were integrated and the relative contribution of each glycan calculated as a percentage. Peaks were identified by mass spec using a Thermo Q Exactive Plus mass spectrometer and through comparison to the NIST monoclonal antibody standard. Size exclusion chromatography (SEC) was performed using a Superdex 200 Increase 10/300GL column (GE Healthcare) on an Äkta Pure 25 HPLC system and analysed using Unicorn v.6.3 software.
ELISA-based FcγR binding assay
Recombinant human FcγR ectodomains (Sinobiological) (5 μg ml−1) were immobilized into high-binding 96-well microtitre plates (Nunc), and after overnight incubation at 4 °C, plates were blocked with PBS plus 2% (w/v) BSA and 0.05% (v/v) Tween20 for 2 h. IgG immune complexes were prepared by incubating for 1 h at 4 °C Fc variants of the anti-NP (4-hydroxy-3-nitrophenylacetyl) monoclonal antibody Ab 3B62 with NP-BSA (27 conjugation ratio, Biosearch Technologies) at a molar ratio of 1:10 (antigen:antibody). IgG immune complexes or monomeric IgG (for FcγRI) were serially diluted (1:3 consecutive dilutions in PBS starting at 10 μg ml−1 (IgG concentration) for immune complexes or 1 μg ml−1 for monomeric IgG) and applied to FcγR-coated plates. After 1 h incubation at room temperature, bound IgG was detected using horseradish peroxidase (HRP)-conjugated goat F(ab’)2 anti-human IgG (1 h; 1:5,000; Jackson Immunoresearch). Plates were developed using the TMB (3,3′,5,5′-tetramethylbenzidine) two-component peroxidase substrate kit (KPL) and reactions were stopped with the addition of 1 M phosphoric acid. Absorbance at 450 nm was immediately recorded using a SpectraMax Plus spectrophotometer (Molecular Devices) and background absorbance from negative control samples was subtracted. Data were collected and analysed using SoftMax Pro v.7.0.2 software (Molecular Devices).
Surface plasmon resonance
All experiments were performed with a Biacore T200 SPR system (GE Healthcare) at 25 °C in HBS-EP+ buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3.4 mM EDTA, 0.005% (v/v) surfactant P20). IgG antibodies (clone 3B62) were immobilized on Series S Protein G sensor chip (GE Healthcare) at a density of 2,000 response units (RU). Serial dilutions of recombinant soluble human FcγRI ectodomain (Sinobiological) were injected to the flow cells at 20 μl min−1, with the concentration ranging from 250 to 0.488 nM (1:2 successive dilutions). Association time was 60 s followed by a 900-s dissociation step. At the end of each cycle, sensor surface was regenerated with a glycine HCl buffer (10 mM, pH 2.0; 50 μl min−1, 30 s). Background binding to blank immobilized flow cells was subtracted, and affinity constants were calculated using BIAcore T200 evaluation software v.2.0 (GE Healthcare) using the 1:1 Langmuir binding model.
Determination of antibody-dependent activation of human FcγRIIIa
Monoclonal antibodies were serially diluted in ADCC assay buffer (Promega). Target cells (A549-H1 HA, A/California/04/2009) were added in a white flat-bottom 96-well plate at 1.25 × 104 cells per well in 25 μl, then serially diluted antibodies were added to each well (25 μl per well), and the antibody and cell mixture was incubated for 10 min at room temperature. Effector cells (Jurkat-FcγRIIIa) for the ADCC Bioassay are thawed and added at a cell density of 7.5 × 104 per well in 25 μl (effector to target ratio of 6:1). Plates were incubated for 20 h at 37 °C with 5% CO2. Activation of human FcγRIIIa (V158 or F158 variants) in this bioassay results in the NFAT-mediated expression of the luciferase reporter gene. Luminescence is therefore measured with a luminometer (Synergy H1, Biotek) using the BioGlo Luciferase Assay Reagent according to the manufacturer’s instructions. The data (that is, specific FcγRIIIa activation) are expressed as the average of relative luminescence units (RLU) over the background by applying the following formula: (RLU at concentration x of monoclonal antibodies − RLU of background).
Determination of antibody-dependent activation of human FcγRIIa
Monoclonal antibodies were serially diluted in ADCP assay buffer (Promega). Target cells (A549-H1HA, A/California/04/2009) were added in a white flat-bottom 96-well plate at 104 cells per well in 25 μl, then serially diluted antibodies were added to each well (25 μl per well), and the antibody and cell mixture was incubated for 10 min at room temperature. Effector cells (Jurkat-FcγRIIa) for the ADCP Bioassay are thawed and added at a cell density of 5 × 104 per well in 25 μl (effector to target ratio of 5:1). Plates were incubated for 20 h at 37 °C with 5% CO2. Activation of human FcγRIIa (H131 variant) in this bioassay results in the NFAT-mediated expression of the luciferase reporter gene. Luminescence is therefore measured with a luminometer (Synergy H1, Biotek) using the BioGlo Luciferase Assay Reagent according to the manufacturer’s instructions. The data (that is, specific FcγRIIa activation) are expressed as the average of relative luminescence units (RLU) over the background by applying the following formula: (RLU at concentration x of antibodies − RLU of background).
ADCC assay
Natural killer cells were freshly isolated from whole EDTA blood using the MACSxpress NK isolation kit following the manufacturer instruction. Monoclonal antibodies were serially diluted tenfold in AIM-V medium from 1 μg ml−1 to 0.001 μg ml−1. Target cells (A549-H1HA, A/California/04/2009) were added in a round-bottom 384-well plate at 7.5 × 103 cells per well in 23 μl, then serially diluted antibodies were added to each well (23 μl per well), and the antibody–cell mixture was incubated for 10 min at room temperature. After incubation, human natural killer cells were added at a cell density of 4.5 × 104 per well in 23 μl (effector to target ratio of 6:1). Control wells were also included that were used to measure maximal lysis (containing target cells with 23 μl of 3% Triton X-100) and spontaneous lysis (containing target cells and effector cells without antibody). Plates were incubated for 4 h at 37 °C with 5% CO2. Cell death was determined by measuring lactate dehydrogenase (LDH) release using an LDH detection kit (Roche) according to the manufacturer’s instructions. Using a kinetic protocol, the absorbance at 490 nm and 650 nm was measured once every 2 min for 8 min. The percentage of specific lysis was determined by applying the following formula: (specific release − spontaneous release)/(maximum release − spontaneous release) × 100.
Anti-HA, NA and NP ELISA
Recombinant HA (Influenza A H1N1 (A/California/04/2009 or A/Puerto Rico/8/34) or H3N2 (A/x31)), or NA (A/California/04/2009) or NP (H1N1 (A/California/04/2009) or H3N2 (A/x31)) (Sinobiological) (3 μg ml−1) were immobilized into high-binding 96-well microtitre plates (Nunc) and after overnight incubation at 4 °C, plates were blocked with PBS plus 2% (w/v) BSA and 0.05% (v/v) Tween20 for 2 h. After blocking, plates were incubated for 1 h with serially diluted IgG antibodies or serum samples (1:3 consecutive dilutions in PBS starting at 1 μg ml−1 for monoclonal antibodies or 1:10 for serum samples), followed by HRP-conjugated goat anti-human IgG (minimal cross-reactivity to mouse IgG) for human monoclonal antibodies or goat anti-mouse IgG (minimal cross-reactivity to human IgG) for serum samples (1 h; 1:5,000; Jackson Immunoresearch). Plates were developed using the TMB two-component peroxidase substrate kit (KPL) and reactions were stopped with the addition of 1 M phosphoric acid. Absorbance at 450 nm was immediately recorded using a SpectraMax Plus spectrophotometer (Molecular Devices) and background absorbance from negative control samples was subtracted. Data were collected and analysed using SoftMax Pro v.7.0.2 software (Molecular Devices).
Microneutralization assay
The neutralizing activity of anti-HA and NA monoclonal antibody Fc variants was evaluated in microneutralization assays, using previously described protocols33. Virus input was titrated to maximize the signal-to-noise ratio. Fc domain variants (starting concentration at 100 μg ml−1 followed by 1:3 serial dilutions) and viruses (1.8 × 103 pfu ml−1 for A/Puerto Rico/8/34 and 3.2 × 104 pfu ml−1 for A/Netherlands/602/09) were prepared in DMEM supplemented with 50 U ml−1 penicillin, 50 μg ml−1 streptomycin, 25 mM HEPES and 1 μg ml−1 TPCK-treated trypsin (Sigma). The virus–antibody mixture was pre-incubated for 1 h at 37 °C and added to a monolayer of MDCK cells (70–80% confluent in 96-well plates). After incubation at 37 °C for 1 h to allow for virus adsorption, the cell monolayer was washed three times with PBS and re-incubated for 18–20 h at 37 °C with medium (DMEM supplemented with 50 U ml−1 penicillin, 50 μg ml−1 streptomycin, 25 mM HEPES and 1 μg ml−1 TPCK-treated trypsin) containing monoclonal antibodies (at equivalent concentrations as during the virus co-incubation). Cells were fixed with 80% (v/v) acetone, blocked with 5% (w/v) non-fat milk diluted in PBS for 30 min at room temperature, and quenched with 3% (v/v) hydrogen peroxide (in PBS) by incubating for a further 20 min at room temperature. Cells were stained with biotinylated anti-NP antibody (EMD Millipore; 1:2,000), followed by HRP-conjugated streptavidin (Jackson Immunoresearch; 1:5,000). Plates were developed using the TMB two-component peroxidase substrate kit (KPL) and reactions stopped with the addition of 1 M phosphoric acid. Absorbance at 450 nm was immediately recorded using a SpectraMax Plus spectrophotometer (Molecular Devices) and background absorbance from negative control samples was subtracted. Data were collected and analysed using SoftMax Pro v.7.0.2 software (Molecular Devices).
Haemagglutination inhibition assay
HAI activity was evaluated using previously described protocols34. In brief, serum samples (diluted 1:3 in cholera filtrate) or Fc domain variants of antibodies (starting concentration at 100 μg ml−1 followed by 1:3 serial dilutions) and viruses (A/Puerto Rico/8/34 or A/Netherlands/602/09; 107 pfu ml−1) were incubated in V-bottom 96 microtitre plates for 30 min at room temperature. Turkey red blood cells (0.75% (v/v); Rockland) were added to the antibody–virus mixture, mixed gently, and incubated for 30 min at room temperature. Plates were scored for the number of wells exhibiting HAI activity.
Quantification of serum IgG levels
Blood from mice was collected into gel microvette tubes, serum was fractionated by centrifugation (10,000g, 5 min) and stored at −20 °C. IgG levels in serum samples were determined by ELISA following previously published protocols13,31.
Mouse influenza infection models
No statistical method was used to predetermine sample size. On the basis of preliminary studies that determined experimental variation in survival after infection and antibody treatment, we performed power calculations and determined that at least n = 6 mice per group is sufficient to detect differences among experimental groups (powered at 80% for 5% significance level; survival assessed by log-rank (Mantel–Cox) test). Mice (females; 6–12 weeks old) were anaesthetized with a ketamine (75 mg kg−1) and xylazine (15 mg kg−1) mixture (administered inraperitoneally) and viruses (diluted in PBS) were administered intranasally (five mLD50) in 30 μl. After infection, mice were monitored daily, and their weights were recorded for 14 days. Death was determined by a 20% body weight loss threshold that was authorized by the Rockefeller University Institutional Animal Care and Use Committee. Mice were randomized based on age and weight. Before treatment, we ensured that the mean weight and age were comparable among the various treatment groups. Whenever possible (not always, due to limited access of laboratory staff in BSL-2 animal facilities), the treatment groups were blinded to the person involved in monitoring mouse survival and weight upon challenge (applicable to data presented in Figs. 1d, 4). For antibody-mediated prophylaxis, antibodies were administered intraperitoneally or intravenously 4 h before virus challenge (except for experiments with FcRn/FcγR-humanized mice, in which antibodies were administered 2 days before infection), whereas for antibody-mediated therapy, antibodies were administered on day 3 after infection. Antibody dose was calculated as mg kg−1. For FcγRIIa-blocking experiments, recombinant anti-FcγRIIa (clone IV.3) expressed as human IgG1 GRLR variant to abrogate FcγR binding or isotype control (anti-hapten (NP) monoclonal antibody; clone 3C13) was administered (80 μg) intranasally to mice 1 day after virus challenge.
Ex vivo stimulation of human monocyte-derived dendritic cells
Leukocyte packs (buffy coats) were purchased from the New York Blood Center, mononuclear cells were isolated by Ficoll gradient centrifugation, and CD14+ monocytes were purified using CD14 microbeads (Miltenyi Biotech). Cells were cultured at 37 °C, 5% CO2 for 6 days in RPMI supplemented with 50 U ml−1 penicillin, 50 μg ml−1 streptomycin, 10% heat-inactivated FBS, 100 ng ml−1 GM-CSF, and 100 ng ml−1 IL-4. Cells were then seeded in 96-well plates at 8 × 104 cells per well in RPMI supplemented with 50 U ml−1 penicillin, 50 μg ml−1 streptomycin, 5% ultra-low IgG, heat-inactivated FBS and stimulated overnight with IgG immune complexes (100 μg ml−1). The following immune complexes were used: (i) heat-aggregated IgG complexes generated by incubation of IgG (1A01 monoclonal antibody) at 63 °C for 60 min; or (ii) anti-NP:NP-BSA immune complexes generated as described for the ELISA-based FcγR binding assay. After overnight stimulation, dendritic cell maturation was assessed by flow cytometry. Cells were stained with the following fluorochrome-conjugated antibodies (diluted at 1:100 unless otherwise stated): anti-DC-SIGN-BrilliantViolet 421, anti-CD40-BrilliantViolet 510, anti-human FcγRI-BrilliantViolet 605 (clone 10.1), anti-CD11b-BrilliantViolet 650, anti-CD86-BrilliantViolet 711, anti-human FcγRIIa-FITC (clone IV.3, used at 1:50 dilution), anti-CD11c-PerCP-Cy5.5, anti-human FcγRIIIa/b-PE (clone 3G8), anti-CD80-PE/Cy7, anti-human FcγRIIb-Dylight 650 (clone 2B6) (used at 10 μg ml−1), anti-HLA-DR-AlexaFluor700, and anti-CD83-APC/Cy7. Samples were collected on an Attune NxT flow cytometer (ThermoFisher) using Attune NxT software v.3.1.2 and analysed using FlowJo (v.10.6) software.
In vivo neutrophil, CD8+ or CD4+ T cell depletion
Neutrophils, CD8+ or CD4+ cells were depleted in mice by administration of anti-Gr-1, anti-CD8 or anti-CD4 monoclonal antibodies, respectively. To establish the efficiency of antibody-mediated cell depletion, FcγR-humanized mice were injected intravenously with 150 μg anti-mouse Gr-1 monoclonal antibody (clone RB6-8C5; rat IgG2b; Bioxcell), anti-mouse CD8α monoclonal antibody (clone 2.43; rat IgG2b; Bioxcell), anti-mouse CD4 monoclonal antibody (clone GK1.5; rat IgG2b; Bioxcell), or isotype control (clone LTF-2; rat IgG2b; Bioxcell). The abundance of neutrophils, CD8+ and CD4+ T cells in peripheral blood was determined at various time points after antibody administration by flow cytometry. Baseline CD8+ and CD4+ T cell frequencies were determined in blood samples obtained before antibody administration. For the flow cytometry analysis, fluorescently conjugated monoclonal antibodies targeting Ly6G (clone 1A8; Biolegend), the β subunit of mouse CD8 (clone YTS156.7.7; Biolegend) or epitopes non-overlapping with the GK1.5 epitope (clone RM4-4; Thermofisher) were used to avoid competition with the depleting antibodies. Neutrophil, CD8+ or CD4+ T cell depletion of influenza-infected mice was performed using the aforementioned conditions and depleting antibodies or isotype were administered intravenously (150 μg) on day 1 (for neutrophils) or 3 (for CD8+ or CD4+ T cells) after infection.
Processing of mouse tissues and flow cytometry analysis
Mice were euthanized and lungs were perfused by injection of PBS (containing 10 U ml−1 heparin) into the right cardiac ventricle. Lungs were excised and homogenized using the gentleMACS dissociator (mouse lung dissociation kit, Miltenyi), according to the manufacturer’s recommendations. Spleens were homogenized by mechanical shearing. After lysis of red blood cells (RBC lysis buffer; Biolegend), single-cell suspensions were labelled with the LIVE/DEAD Fixable Near-IR (ThermoFisher) and resuspended in PBS containing 0.5% (w/v) BSA and 5 mM EDTA. Cells were labelled with mixtures of fluorescently labelled antibodies including (all used at 1:250 dilution unless otherwise stated): (i) for the characterization of the FcγRIIa expression of the FcγRIIa+ mouse strain: anti-CD11c-eFluor506, anti-CD11b-BrilliantViolet605, anti-SiglecF-SuperBright645, anti-CD8β-BrilliantViolet711, anti-MHCII- BrilliantViolet785, anti-human FcγRIIa (clone IV.3)-FITC (used at 1:50 dilution), anti-Gr-1-PerCP/Cy5.5, anti-CD3-PE, anti-CD103-PE/eFluor610, anti-NK1.1-PE/Cy7, anti-CD4-AlexaFluor647, and anti-CD19-AlexaFluor700; (ii) for the evaluation of FcγR expression on innate effector leukocytes: anti-CD11c-eFluor506, anti-human FcγRI (clone 10.1)-BrilliantViolet605 (used at 1:100 dilution), anti-SiglecF-SuperBright645, anti-Ly6G-BrilliantViolet711, anti-CD11b-BrilliantViolet785, anti-human FcγRIIa (clone IV.3)-FITC (used at 1:50 dilution), anti-Ly6C-PerCP/Cy5.5, anti-human FcγRIIIa/b (clone 3G8)-PE (used at 1:100 dilution), anti-CD103-PE/eFluor610, anti-NK1.1-PE/Cy7, and anti-human FcγRIIb (clone 2B6)-Dylight650 (used at 10 μg/ml); (iii) for the evaluation of FcγR expression and activation status of DCs: anti-CD11c-eFluor506, anti-human FcγRI (clone 10.1)-BrilliantViolet605 (used at 1:100 dilution), anti-SiglecF-SuperBright645, anti-CD80-BrilliantViolet711, anti-CD11b-BrilliantViolet785, anti-human FcγRIIa (clone IV.3)-FITC (used at 1:50 dilution), anti-Gr-1-PerCP/Cy5.5, anti-human FcγRIIIa/b (clone 3G8)-PE (used at 1:100 dilution), anti-CD103-PE/eFluor610, anti-CD86-PE/Cy7, anti-human FcγRIIb (clone 2B6)-Dylight650 (used at 10 μg ml−1), and anti-MHCII-AlexaFluor700; (iv) for the evaluation of CD8 or CD4 depletion: anti-CD3e-eFluor506, anti-CD19-BrilliantViolet605, anti-CD8β-BrilliantViolet711, anti-CD11b-PE, anti-NK1.1-PE/Cy7, anti-CD4-FITC, anti-Gr-1-PerCP/Cy5.5, anti-NKp46-eFluor660, and anti-B220-APC/eFluor780; (v) for the evaluation of neutrophil depletion: anti-CD11c-eFluor506, anti-CD19-BrilliantViolet605, anti-SiglecF- SuperBright645, anti-Ly6G-BrilliantViolet711, anti-CD11b-BrilliantViolet785, anti-CD8a-FITC, anti-Ly6C-PerCP/Cy5.5, anti-CD3-PE, anti-NK1.1-PE/Cy7, anti-CD4-AlexaFluor647, anti-CD45-AlexaFluor700; (vi) for the assessment of FcγR expression on T cells: anti-B220-BrilliantViolet421, anti-CD3-BrilliantViolet510, anti-human FcγRI (clone 10.1)-BrilliantViolet605 (used at 1:100 dilution), anti-CD8-BrilliantViolet711, anti-CD4-BrilliantViolet785, anti-human FcγRIIa (clone IV.3)-FITC (used at 1:50 dilution), anti-NK1.1-PerCP/Cy5.5, anti-human FcγRIIIa/b (clone 3G8)-PE (used at 1:100 dilution), anti-CD11b-PE/Cy7, anti-human FcγRIIb (clone 2B6)-Dylight650 (used at 10 μg ml−1), and anti-Gr-1-AlexaFluor 700; (vii) for the characterization of dendritic cell populations after antibody treatment: anti-CD103-FITC, anti-Ly6C-PerCP/Cy5.5, anti-NK1.1-AlexaFluor647, anti-CD45-AlexaFluor700, anti-CD11c-eFluor506, anti-CD86-BrilliantViolet605, anti-SiglecF-SuperBright645, anti-Ly6G-BrilliantViolet711, anti-CD11b-BrilliantViolet785, anti-CD40-PE, anti-MHCII-PE/eFluor610, and anti-CD80-PE/Cy7; (viii) for the characterization of T cell populations after antibody treatment: anti-CD4-AlexaFluor488, anti-CD3e-PerCP/Cy5.5, anti-NK1.1-AlexaFluor647, anti-CD45-AlexaFluor700, anti-CD44-BrilliantViolet421, anti-CD62L-BrilliantViolet510, anti-CD25-BrilliantViolet605, anti-CD27-BrilliantViolet650, anti-CD8-BrilliantViolet711, anti-CD11a-PE, anti-CCR7-PE/eFluor610, and anti-CD69-PE/Cy7. For experiments assessing FcγR expression, relevant isotype control antibodies were used and included: mouse IgG1 isotype control-Dylight650 (used at 10 μg ml−1), mouse IgG2b kappa isotype control-FITC (used at 1:50 dilution), mouse IgG1 kappa isotype control-PE (used at 1:100 dilution), mouse IgG1 kappa isotype control-BrilliantViolet605 (used at 1:100 dilution). Cell counts were determined using CountBright absolute counting beads (ThermoFisher). Samples were collected on an Attune NxT flow cytometer (ThermoFisher) using Attune NxT software v3.1.2 and analysed using FlowJo (v10.6) software. For cluster analysis, dendritic cells (defined as Live/Lin+/CD45+/CD11c+/MHCII+) and T cells (defined as Live/CD45+/NK1.1-/CD3+) from individual mice were downsampled using the Downsample v.3.2 plugin (3,000 (dendritic cells) or 6,000 (T cells) events/mouse; 12,000 (dendritic cells) or 24,000 (T cells)/ treatment condition) and concatenated. Cells were clustered and visualized using UMAP reduction (UMAP plugin v2.2) and populations were identified by KNN density estimation (X-shift, v.1.3)35.
Analysis of FcγR expression on human T cells
Leukocyte packs (buffy coats) were purchased from the New York Blood Center, mononuclear cells were isolated by Ficoll gradient centrifugation and stained with the following fluorochrome-conjugated anti-human antibodies: anti-CD14-BrilliantViolet510, anti-CD19-BrilliantViolet510, anti-human FcγRI-BrilliantViolet605 (clone 10.1), anti-CD8-BrilliantViolet650, anti-CD3-BrilliantViolet711, anti-CD11b-BrilliantViolet785, anti-human FcγRIIa-FITC (clone IV.3) (used at 1:50 dilution), anti-CD4-PerCP-Cy5.5, anti-human FcγRIIIa/b-PE (clone 3G8), anti-CD56-PE/Cy7, anti-human FcγRIIb-Dylight650 (clone 2B6) (used at 10 μg ml−1), anti-CD41-AlexaFluor700, and anti-CD42b-AlexaFluor700. The following isotype controls were used: mouse IgG1 isotype control-Dylight650 (used at 10 μg ml−1), mouse IgG2b kappa isotype control-FITC (used at 1:50 dilution), mouse IgG1 kappa isotype control-PE (used at 1:100 dilution), mouse IgG1 kappa isotype control-BrilliantViolet605 (used at 1:100 dilution). Samples were collected on an Attune NxT flow cytometer (ThermoFisher) using Attune NxT software v3.1.2 and analysed using FlowJo (v.10.6) software.
Statistical analysis
Results from multiple experiments are presented as mean ± s.e.m. One- or two-way ANOVA was used to test for differences in the mean values of quantitative variables, and where statistically significant effects were found, post hoc analysis using Bonferroni (adjusted for multiple comparisons) test was performed. Two-tailed t-test was used to test for differences in datasets with two groups. Statistical differences between survival rates were analysed by comparing Kaplan–Meier curves using the log-rank (Mantel–Cox) test. Data were analysed with GraphPad Prism v.8.4 software (GraphPad) and P < 0.05 were considered to be statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Source data are provided with this paper.
References
Bournazos, S., Wang, T. T., Dahan, R., Maamary, J. & Ravetch, J. V. Signaling by antibodies: recent progress. Annu. Rev. Immunol. 35, 285–311 (2017).
Lu, C. L. et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 352, 1001–1004 (2016).
Bournazos, S., DiLillo, D. J. & Ravetch, J. V. The role of Fc-FcγR interactions in IgG-mediated microbial neutralization. J. Exp. Med. 212, 1361–1369 (2015).
Bournazos, S. et al. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 158, 1243–1253 (2014).
DiLillo, D. J., Palese, P., Wilson, P. C. & Ravetch, J. V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest. 126, 605–610 (2016).
DiLillo, D. J., Tan, G. S., Palese, P. & Ravetch, J. V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat. Med. 20, 143–151 (2014).
Kallewaard, N. L. et al. Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell 166, 596–608 (2016).
Corti, D. et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 (2011).
Bournazos, S. IgG Fc receptors: evolutionary considerations. Curr. Top. Microbiol. Immunol. 423, 1–11 (2019).
Smith, P., DiLillo, D. J., Bournazos, S., Li, F. & Ravetch, J. V. Mouse model recapitulating human Fcγ receptor structural and functional diversity. Proc. Natl Acad. Sci. USA 109, 6181–6186 (2012).
Richards, J. O. et al. Optimization of antibody binding to FcγRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 7, 2517–2527 (2008).
DiLillo, D. J. & Ravetch, J. V. Differential Fc-receptor engagement drives an anti-tumor vaccinal effect. Cell 161, 1035–1045 (2015).
Weitzenfeld, P., Bournazos, S. & Ravetch, J. V. Antibodies targeting sialyl Lewis A mediate tumor clearance through distinct effector pathways. J. Clin. Invest. 129, 3952–3962 (2019).
Lazar, G. A. et al. Engineered antibody Fc variants with enhanced effector function. Proc. Natl Acad. Sci. USA 103, 4005–4010 (2006).
Ferrara, C. et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcγRIII and antibodies lacking core fucose. Proc. Natl Acad. Sci. USA 108, 12669–12674 (2011).
Mimoto, F. et al. Engineered antibody Fc variant with selectively enhanced FcγRIIb binding over both FcγRIIa(R131) and FcγRIIa(H131). Protein Eng. Des. Sel. 26, 589–598 (2013).
Wrammert, J. et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 208, 181–193 (2011).
Eisenbarth, S. C. Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol. 19, 89–103 (2019).
Badia, R. et al. CD32 expression is associated to T-cell activation and is not a marker of the HIV-1 reservoir. Nat. Commun. 9, 2739 (2018).
Martin, G. E. et al. CD32-expressing CD4 T cells are phenotypically diverse and can contain proviral HIV DNA. Front. Immunol. 9, 928 (2018).
Descours, B. et al. CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature 543, 564–567 (2017).
Wang, T. T. et al. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science 355, 395–398 (2017).
Monsalvo, A. C. et al. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. 17, 195–199 (2011).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Zalevsky, J. et al. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 28, 157–159 (2010).
Goede, V. et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N. Engl. J. Med. 370, 1101–1110 (2014).
Nishimura, Y. et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature 543, 559–563 (2017).
Niessl, J. et al. Combination anti-HIV-1 antibody therapy is associated with increased virus-specific T cell immunity. Nat. Med. 26, 222–227 (2020).
Petkova, S. B. et al. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int. Immunol. 18, 1759–1769 (2006).
Roopenian, D. C., Christianson, G. J. & Sproule, T. J. Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol. Biol. 602, 93–104 (2010).
Borghi, S. et al. FcRn, but not FcγRs, drives maternal-fetal transplacental transport of human IgG antibodies. Proc. Natl Acad. Sci. USA 117, 12943–12951 (2020).
Okeley, N. M. et al. Development of orally active inhibitors of protein and cellular fucosylation. Proc. Natl Acad. Sci. USA 110, 5404–5409 (2013).
He, W., Mullarkey, C. E. & Miller, M. S. Measuring the neutralization potency of influenza A virus hemagglutinin stalk/stem-binding antibodies in polyclonal preparations by microneutralization assay. Methods 90, 95–100 (2015).
Kaufmann, L. et al. An optimized hemagglutination inhibition (HI) assay to quantify influenza-specific antibody titers. J. Vis. Exp. 130, 55833 (2017).
Samusik, N., Good, Z., Spitzer, M. H., Davis, K. L. & Nolan, G. P. Automated mapping of phenotype space with single-cell data. Nat. Methods 13, 493–496 (2016).
Dahan, R. et al. Therapeutic activity of agonistic, human anti-CD40 monoclonal antibodies requires selective FcγR engagement. Cancer Cell 29, 820–831 (2016).
Acknowledgements
We thank R. Francis for technical assistance in antibody cloning and expression, in vitro experiments, flow cytometry, and in vivo influenza challenge studies, M. Ye and E. Lam for maintaining the mouse colonies and for assisting with in vitro and in vivo experiments, P. Smith for maintaining the humanized mouse strains and all the members of the Laboratory of the Molecular Genetics and Immunology for discussions. We also thank S. Jaconi, K. Culap, E. Cameroni and B. Guarino for providing the FY1 monoclonal antibody Fc variants and for technical assistance in characterizing the in vitro ADCC/ADCP activity of Fc variants. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (R01AI129795 and R01AI145870). We acknowledge support from the Rockefeller University and Vir Biotechnology Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Contributions
S.B. designed the study, performed experiments, analysed data, and wrote the manuscript; D.C. and H.W.V. provided intellectual input; J.V.R. designed the study and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
S.B. and J.V.R. are inventors on a patent (WO2019125846A1) describing the GAALIE variant and its use in therapeutic monoclonal antibodies; D.C. and H.W.V. are employees of Vir Biotechnology Inc. and may hold shares in Vir Biotechnology Inc.; J.V.R. is a member of the scientific advisory board and a consultant of Vir Biotechnology Inc.
Additional information
Peer review information Nature thanks George Georgiou and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Characterization of the FcγR binding profile and Fc effector activity of Fc domain variants.
Fc domain variants with differential FcγR binding affinity were generated through the introduction of amino acid substitutions at the hinge proximal region of the CH2 domain of human IgG1. a, The positions of the mutated residues for the different Fc domain variants are highlighted. b, The affinity of these human IgG1 Fc variants for the different human FcγRs was assessed by surface plasmon resonance and the dissociation constant (Kd) (in M) is presented. The following references are cited in the table: refs. 13,36. c, d, HPLC analysis of Fc domain variants using size-exclusion chromatography (SEC) columns was performed to determine whether mutations at the Fc domain are associated with increased antibody aggregation. The SEC profiles (c: overlay; d: individual Fc variants) and the abundance (percentage) of monomeric IgG is presented for the different Fc variants. e, Fc variants for the anti-NP monoclonal antibody 3B62 were generated and their binding (monomeric for FcγRI or as IgG immune complexes using NP-BSA for all other FcγRs) to immobilized human FcγRI, FcγRIIa, FcγRIIb and FcγRIIIa was assessed by ELISA. Results are from one experiment performed in duplicates. The Fc effector activity of anti-influenza monoclonal antibody FY1 Fc variants was assessed in vitro using FcγRIIa-expressing (f: n = 2 independent experiments for wild type, n = 1 for other groups, i: n = 2 independent experiments) and FcγRIIIa-expressing (g (F158 allele), j (V158 allele); n = 1 for each variant, except for GAALIE–LS (n = 2 independent experiments)) NFAT reporter cell lines. Fc variants with enhanced affinity for FcγRIIa or FcγRIIIa demonstrated increased capacity to induce NFAT reporter activation. h, Similarly, FY1 Fc variants engineered for FcγRIIIa binding exhibited improved primary human natural killer cell-mediated ADCC activity against HA-expressing cells. Results are the mean from two independent experiments using different natural killer cell donors.
Extended Data Fig. 2 Anti-HA and NA monoclonal antibody titration studies to determine the optimal dose required for protection against mouse influenza infection.
Given the differential epitope specificities and in vitro neutralization potency of the selected anti-influenza monoclonal antibodies, titration studies were performed to assess the capacity of these antibodies to protect mice against lethal influenza infection. Anti-HA and NA antibodies (a, FI6v3 (n = 5 mice per group for 8 mg kg−1 group, n = 6 mice per group for all other groups); b, FY1 (n = 6 mice per group for 4 and 2 mg kg−1, n = 5 mice per group for 1 and 0.5 mg kg−1, n = 4 mice per group for PBS); c, 4G05 (n = 7 mice per group for 0.5 mg kg−1 group, n = 6 mice per group for all other groups); d, 1A01 (n = 4 mice per group for 1 mg kg−1 group, n = 5 mice per group for all other groups); e, 3C05 (n = 5 mice per group for 20 and 5 mg kg−1, n = 4 mice per group for 10 mg kg−1 and PBS groups)). All antibodies were expressed as human IgG1 and administered intraperitoneally (for FI6v3 and FY1) or intravenously (for 4G05, 1A01, and 3C05) at the dose indicated to mice (C57BL/6) 4 h before lethal challenge with influenza (five mLD50; H1N1 PR8 for a, b; H1N1 Neth/09 for c–e). Survival was monitored for 14 days.
Extended Data Fig. 3 In vitro characterization of the antigenic specificity, neutralization potency, and HAI activity of Fc domain variants of anti-influenza monoclonal antibodies.
a–l, To study the role of Fc–FcγR interactions in the antibody-mediated protection against influenza infection, Fc domain variants with differential FcγR affinities (Fig. 1b) were generated for antibodies that target distinct epitopes on influenza antigens (Fig. 1a). These antibodies include FI6v3 (a–c) and FY1 (d–f), which both recognize the stalk region of influenza HA and exhibit broad and potent neutralizing activity against group 1 and 2 influenza strains, 4G05 (g–i), which is a pan-H1 monoclonal antibody against the globular head of HA and exhibits potent neutralizing and HAI activity, 1A01 (j–l), which is a pan-H1 anti-globular head HA antibody with no neutralizing or HAI activity, and 3C05 (m–o), a broadly (pan-H1) protective antibody against NA. To confirm that changes in the Fc domain have no effect on the antigenic activity and Fab-mediated functions of these antibodies, Fc domain variants were characterized by ELISA (a, d, g, j, m; n = 1 experiment performed in duplicates) to assess their specificity against purified HA (H1N1 PR8 strain for FI6v3 and FY1; Cal/09 strain for 4G05, 1A01 and 3C05), by microneutralization assay (b, e, h, k and n; c, f, i, l and o for IC50 values; n = 2 independent experiments; data were fitted with nonlinear regression analysis (four-parameter) to calculate IC50 values) to evaluate their neutralizing activity against H1N1 (H1N1 PR8 strain for FI6v3 and FY1; Neth/09 strain for 4G05, 1A01, and 3C05), and by HAI assay (H1N1 PR8 strain for FI6v3 and FY1; Neth/09 strain for 4G05, 1A01, and 3C05; n = 2 independent experiments) to determine their HAI titre (c, f, i, l and o).
Extended Data Fig. 4 In vivo half-life of Fc domain variants.
a, Fc variants of an anti-HIV antibody (3BNC117) were administered (intravenously; 100 μg) to FcγR-humanized mice and antibody serum levels were determined by ELISA at various time points after antibody administration. n = 4 mice per group in two independent experiments. Data are mean ± s.e.m. b–f, To ensure that the observed differences in the protective activity of Fc variants of anti-HA and NA antibodies (Figs. 1 and 2) were not due to differential in vivo antibody half-lives, serum was obtained from influenza-infected mice (day 3 after infection) and analysed by ELISA to quantify antibody levels. (b: n = 9 mice per group for GAALIE, n = 10 mice per group for all other groups; c: n = 10 mice per group for wild type, n = 8 mice per group for all other groups ; d: n = 10 mice per group; e: n = 6 mice per group for GRLR, n = 8 mice per group for all other groups; f: n = 12 mice per group for wild type and GA; n = 10 mice per group for GRLR and ALIE; n = 9 mice per group for GAALIE). g, To ensure that the observed differences in the protective activity of Fc variants of FY1 antibodies (Fig. 3e, f, Extended Data Fig. 10d, e) were not due to differential in vivo antibody half-lives, serum was obtained from influenza-infected mice (day 3 after infection) and analysed by ELISA to quantify FY1 antibody levels. n = 12 mice per group for wild type/isotype, GAALIE/isotype, and GAALIE/anti-CD8 groups, n = 11 mice per group for wild-type/anti-CD8, n = 8 mice per group for GA/isotype, GA/anti-CD4, and GA/anti-CD4 groups, n = 6 mice per group for wild-type/anti-CD4 group. h, Titration studies were performed in a mouse model of antibody-mediated prophylaxis of influenza infection (Fig. 4d) and serum was obtained at the time of virus challenge and analysed by ELISA to quantify FY1 antibody levels. n = 10 mice per group for LS 0.1 mg kg−1 dose, n = 8 for GAALIE–LS at 1.6 and 0.4 mg kg−1 doses, n = 9 mice per group for all other groups.
Extended Data Fig. 5 Evaluation of the role of FcγRIIa and the contribution of neutrophils in the antibody-mediated protection against influenza infection.
a, b, To confirm the dependence of FcγRIIa engagement in driving the protective activity of the GA variant, mice expressing the human FcγRIIa transgene on an FcγRnull background (hFcγRIIa+; gating strategy (a) and representative flow cytometry histograms (b) of FcγRIIa expression in lung-resident leukocytes) or deficient for all classes of FcγRs (hFcγRIIa−) were administered with GA variants of FY1 (intraperitoneally 2 mg kg−1) (n = 7 mice per group for FcγRIIa+, n = 6 mice per group for FcγRIIa− in two independent experiments) or PBS (n = 4 mice per group for FcγRIIa+, n = 5 mice per group for FcγRIIa− in two independent experiments) 4 h before lethal challenge with influenza (H1N1; PR8, 5 mLD50). c, d, Weight loss (c) (mean ± s.e.m.) and survival (d) were monitored for 14 days and compared by two-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) (c: *P = 0.02 §P = 0.03, ¶P = 0.006, ^P < 0.0001, #P = 0.0002, ^^P = 0.0006, **P = 0.001, ##P = 0.002 versus GA-treated FcγRIIa−) and log-rank (Mantel–Cox) test, respectively (d: *P = 0.0006 versus GA-treated FcγRIIa−). e, f, FcγR-humanized mice were administered with GA variants of FI6v3 (intraperitoneally 4 mg kg−1) before challenge with PR8 (as described in c, d). To block the ligand binding activity of FcγRIIa, recombinant anti-FcγRIIa (clone IV.3) expressed as human IgG1 GRLR variant to abrogate FcγR binding or isotype control (anti-hapten (NP) monoclonal antibody; clone 3C13) was administered intranasally (80 μg) to mice 1 day after virus challenge (n = 7 mice per group for GA/IV.3-treated group, n = 5 mice per group for all other groups in two independent experiments) and weight loss (e) (mean ± s.e.m.) and survival (f) were monitored. g, To establish the efficiency of antibody-mediated neutrophil depletion, FcγR-humanized mice (n = 3 mice per group in one experiment) were injected intravenously with 150 μg anti-mouse Gr-1 monoclonal antibody (clone RB6-8C5) or isotype control (clone LTF-2). The abundance of neutrophils (Ly6G+) in peripheral blood was determined 2 days after antibody administration by flow cytometry. **P = 0.0053, two-sided unpaired t-test. h, i, To assess the contribution of neutrophils to the protective activity of FcγRIIa-enhanced variants of anti-influenza antibodies, GA variants of the anti-HA stalk antibody FY1 were administered intraperitoneally (2 mg kg−1) to FcγR-humanized mice (n = 6 mice per group, except for PBS/Gr-1 treated (n = 5 mice per group) in two independent experiments) 4 h before lethal challenge with PR8. Isotype or anti-mouse Gr-1 monoclonal antibodies (150 μg intravenously) were administered on day 1 after infection and weight loss (h) (mean ± s.e.m.) and survival (i) were recorded.
Extended Data Fig. 6 Abundance and FcγR expression profile of leukocyte populations in the lungs of influenza-infected FcγR-humanized mice at different time points afteer infection.
a–f, To determine the abundance and FcγR expression profile of lung resident and infiltrated leukocytes during the course of influenza infection, cohorts of FcγR-humanized mice were infected (intranasally with H1N1 PR8; 5 mLD50) and euthanized at different time points after infection (day 0 to 6). Lungs were homogenized and analysed by flow cytometry (a: gating strategy) to determine the frequency (b, c) and FcγR expression profile (f) of innate effector leukocytes. n = 3 and n = 4 mice per time point for day 0–2 and day 3–6 time points, respectively. Influenza infection was associated with the recruitment of natural killer (NK) cells, neutrophils and monocytes, whereas the number of alveolar macrophages was reduced at the later stages of infection. Owing to the high degree of sequence similarity between FcγRIIa and FcγRIIb, expression of these FcγRs was assessed using antibody clones (d: IV.3 for FcγRIIa; e: 2B6 for FcγRIIb) that exhibit high specificity, as assessed by ELISA using recombinant FcγRs (n = 1 experiment performed in duplicates). Analysis of the FcγR expression profile (MFI) revealed that influenza infection had no effect on the levels of FcγRs expressed by the various leukocyte types. With the exception of natural killer cells, which expressed only FcγRIIIa, most innate effector leukocytes co-expressed multiple FcγRs, including the activating FcγRs, FcγRIIa and FcγRIIIa/b, as well as the inhibitory FcγRIIb. n = 3 and n = 4 mice per time point for day 0–2 and day 3–6 time points, respectively.
Extended Data Fig. 7 Treatment of FcγR-humanized mice with GAALIE variants of anti-HA antibodies is associated with increased frequency of activated dendritic cells.
a–f, To determine the abundance and FcγR expression profile of dendritic cell subsets during the course of influenza infection, cohorts of FcγR-humanized mice were infected (intranasally with H1N1 PR8; 5 mLD50) and euthanized at different time points after infection (day 0 to 6). Lungs were homogenized and analysed by flow cytometry (a: gating strategy) to determine the frequency (b) and FcγR expression profile (c: representative flow cytometry overlay; d–f: MFI) of the three major dendritic cell subsets identified: cDC1, cDC2 and tipDCs. Influenza infection was not associated with any major changes in the number of lung-resident cDC1 and cDC2, whereas tipDCs were almost absent at baseline, but their number increased markedly after infection. cDC1 and cDC2 expressed FcγRIIa and FcγRIIb, but they were negative for FcγRIIIa. By contrast, tipDCs expressed FcγRIIa and FcγRIIIa, along with the inhibitory FcγRIIb. Owing to the very low number of tipDCs at baseline, FcγR expression (MFI) was omitted. n = 4 mice per time point assessed. g–i, To investigate the effect of enhanced FcγRIIa engagement by GAALIE variants on the maturation status of dendritic cells, FcγR-humanized mice were treated with Fc domain variants of the anti-HA stalk antibody FI6v3, exhibiting differential FcγR affinity—wild type IgG1 (baseline FcγR affinity), GRLR (diminished binding to all classes of FcγRs), and GAALIE (increased FcγRIIa and FcγRIIIa affinity). Fc domain variants were administered intraperitoneally (3 mg kg−1) to FcγR-humanized mice (n = 4 mice per treatment group in two independent experiments) 4 h before lethal challenge with H1N1 (PR8; 5 mLD50). Mice were euthanized on day 4 and lung-resident dendritic cells were analysed by flow cytometry. The abundance of mature (defined as CD80highCD86high) cDC1 (g), cDC2 (h), and tipDCs (i) was compared between mice treated with the various Fc domain variants of FI6v3. Representative flow cytometry plots from data presented in Fig. 3a. In contrast to cDC1 and cDC2, no differences were observed in the maturation status of tipDCs among mice treated with different FI6v3 Fc variants (one-way ANOVA). j, k, In vitro differentiated monocyte-derived dendritic cells (n = 4 peripheral blood mononuclear cell donors performed in two independent experiments) were stimulated overnight with IgG immune complexes (anti-NP–NP-BSA immune complexes (j) and heat-aggregated IgG complexes (k)). The abundance (percentage) of mature dendritic cells (defined as CD80highCD86high) was assessed by multicolour flow cytometry and compared against the corresponding wild-type-treated group by one-way ANOVA (Bonferroni post hoc analysis adjusted for multiple comparisons) *P = 0.0417, **P = 0.0134, ***P < 0.0001, ^P = 0.0049. l–n, Cluster analysis of dendritic cell populations present in the lungs of influenza-infected mice treated with Fc domain variants of anti-HA monoclonal antibodies. Mice (n = 4 mice per group in two independent experiments) were treated as described in g–i, euthanized on day 4 after infection and dendritic cells (Lin+MHCII+CD11c+) were analysed by multicolour flow cytometry. Data were dimensionally reduced and visualized using the UMAP algorithm. l, UMAP plots of dendritic cells in mice treated with the different Fc domain variants are presented. Populations were identified by X-shift using KNN density estimation and assigned IDs (A-J). m, The abundance of the various dendritic cell clusters in the different treatment groups was plotted and populations that are enriched or reduced in GAALIE-treated mice were identified. n, Histogram plots of the expression of CD80, CD86, CD40 and MHCII in dendritic cell populations that are enriched (red, A; and orange, H) or reduced (cyan, D; purple, E; and blue, F) in GAALIE-treated mice. GAALIE treatment was associated with the enrichment of dendritic cell populations characterized by high levels of CD86 and CD40 expression. Results are from four mice per treatment condition in two independent experiments.
Extended Data Fig. 8 Treatment of FcγR-humanized mice with GAALIE variants of anti-HA stalk antibodies is associated with increased activation of CD8+ and CD4+ T cells.
To investigate whether the observed increase in the frequency of mature dendritic cells in mice treated with GAALIE variants of anti-HA monoclonal antibodies was associated with enhanced T cell responses, the activation status of CD8+ and CD4+ T cells was analysed and compared between mice treated with anti-HA Fc domain variants with differential FcγR affinity (wild-type IgG1, GRLR and GAALIE). Fc domain variants of the anti-HA stalk antibody FI6v3 were administered (intraperitoneally 3 mg kg−1) to FcγR-humanized mice before lethal challenge with H1N1 (PR8; 5 mLD50). Mice (n = 4 mice per group in two independent experiments) were euthanized on day 4 after infection and T cell populations were analysed by multicolour flow cytometry. a, b, The frequency of activated (defined as CD44hiCD69+) CD8+ (a) and CD4+ (b) T cells was compared between mice treated with the various Fc domain variants of FI6v3. Representative flow cytometry plots from data in Fig. 3c. In addition, cluster analysis of T cell populations present in the lungs of influenza-infected mice treated with Fc domain variants of anti-HA monoclonal antibodies was performed. Flow cytometry data were dimensionally reduced and visualized using the UMAP algorithm. c, UMAP plots of T cells in mice treated with the different Fc domain variants are presented. Populations were identified by X-shift using KNN density estimation and assigned IDs (A-M). d, Heat map of the abundance of the various T cell clusters in the different treatment groups. Populations that are enriched or reduced in GAALIE-treated mice were identified. e, Histogram plots of the expression of CD69, CD25, CD44 and CD62L in T cell populations that are enriched (red, G; green, E; orange, J) or reduced (cyan, C; blue, D; magenta, L) in GAALIE-treated mice. GAALIE treatment was associated with the enrichment of T cell populations characterized by high levels of CD69, CD44 and CD25 expression. Results are from four mice per treatment condition.
Extended Data Fig. 9 FcγR expression analysis of T cells.
To determine whether human T cells express human FcγRs, human peripheral blood mononuclear cells were analysed by multicolour flow cytometry. a, Gating strategy for identifying human T cells. Monocytes (CD14+CD11b+) and B cells (CD19+CD11b−) were included as controls for FcγR immunofluorescence staining. b, g, Analysis of FcγR expression revealed that human T cells are negative for FcγR expression (b: representative flow cytometry histogram overlays; g: quantification of FcγR+ cells; n = 3 donors). c–f, h–l, Human FcγR expression on peripheral blood (c, d, h, i), spleen (e, f, j, k), or lung (l) T cells from naive (h, j) or influenza-infected (i, k, l; day 6 after infection (5 mLD50; H1N1 PR8)) FcγR-humanized mice was assessed by multicolour flow cytometry. Gating strategy for identifying T cells in mouse blood (c), spleen or lungs (e). Myeloid cells (CD11b+) and B cells (B220+) were included as controls for FcγR immunofluorescence staining. Analysis of FcγR expression on T cells from naive or influenza-infected FcγR-humanized mice revealed minimal expression of FcγRs on T cell (d, f: representative flow cytometry histogram overlays; h–l: quantification of FcγR+ cells (n = 2 mice per group in one experiment) in the blood (h, i), spleen (j, k) and lungs (l) of naive or influenza-infected mice). Although a small fraction of B cells appears positive for FcγRIIa expression, this is probably due to the cross-reactivity of the anti-FcγRIIa antibody (clone IV.3) tο FcγRIIb (see Extended Data Fig. 6d).
Extended Data Fig. 10 T cell depletion experiments and the effect of FcγRIIa-enhanced variants on anti-influenza IgG responses.
a, b, To establish the efficiency of antibody-mediated CD8+ or CD4+ T cell depletion, FcγR-humanized mice were injected intravenously with 150 μg anti-mouse CD8α monoclonal antibody (clone 2.43) or anti-mouse CD4 (clone GK1.5), or isotype control (clone LTF-2). The abundance of CD8+ (a) or CD4+ (b) T cells in peripheral blood was determined at various time points after antibody administration by flow cytometry. Baseline CD8+ or CD4+ T cell frequencies were determined in blood samples obtained before antibody administration. Results are expressed as the percentage of CD8+CD3+ or CD4+CD3+ T cells (n = 6 for anti-CD8 and anti-CD4-treated groups, n = 7 mice per group for isotype groups in two independent experiments). c–e, To determine the contribution of CD4+ T cells in the protective activity of FcγRIIa-enhanced variants of anti-influenza monoclonal antibodies, CD4+ depletion studies were performed. c, Wild-type or GA variants of the anti-HA stalk antibody FY1 were administered intraperitoneally (2 mg kg−1) to FcγR-humanized mice 4 h before lethal challenge with influenza (5 mLD50; H1N1 PR8). Isotype or anti-mouse CD4 antibodies (150 μg intravenously) were administered on day 3 after infection. Isotype-treated groups: n = 12 mice per group for wild-type and PBS groups, n = 8 mice per group for GA group; anti-CD4-treated groups: n = 6 mice per group for wild type, n = 8 mice/group for GA, n = 4 mice per group for PBS in two independent experiments. d, e, Weight loss (d) (mean ± s.e.m.) and survival (e) were monitored for 14 days. Data for isotype-treated mice are also in Fig. 3e, f. f–m, Wild-type or GAALIE Fc variants for the anti-HA globular head antibody 1A01 were administered (intravenously 4 mg kg−1) to FcγR-humanized mice (n = 7 mice per group for wild-type group, n = 11 mice per group for GAALIE-treated group, n = 5 mice per group for PBS-treated in two independent experiments) 4 h before lethal challenge with influenza (5 mLD50; H1N1 Neth/09). f, g, Weight loss (mean ± s.e.m.) and survival (g) were monitored for 14 days. h–k, IgG responses against HA (h, i) and NP (j, k) from homologous (H1N1; A/California/04/2009) or heterologous (H3N2; A/x31) strains were evaluated in surviving mice. l, Analysis of IgG titres against homologous or heterologous HA or NP revealed no differences among mice previously treated with wild-type or GAALIE Fc variants of 1A01 antibody. m, Similarly, comparable HAI titres were noted among the treatment groups.
Supplementary information
Source data
Rights and permissions
About this article

Cite this article
Bournazos, S., Corti, D., Virgin, H.W. et al. Fc-optimized antibodies elicit CD8 immunity to viral respiratory infection. Nature 588, 485–490 (2020). https://doi.org/10.1038/s41586-020-2838-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2838-z
This article is cited by
-
Monoclonal antibody applications in travel medicine
Tropical Diseases, Travel Medicine and Vaccines (2024)
-
The elusive nature of the oligoclonal bands in multiple sclerosis
Journal of Neurology (2024)
-
Mechanism of glycoform specificity and in vivo protection by an anti-afucosylated IgG nanobody
Nature Communications (2023)
-
Multiple sclerosis plasma IgG aggregates induce complement-dependent neuronal apoptosis
Cell Death & Disease (2023)
-
Human FcγRIIIa activation on splenic macrophages drives dengue pathogenesis in mice
Nature Microbiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
