
Abstract
Oculocutaneous albinism type 1 (OCA1) is caused by pathogenic variants in the TYR (tyrosinase) gene which encodes the critical and rate-limiting enzyme in melanin synthesis. It is the most common OCA subtype found in Caucasians, accounting for ~50% of cases worldwide. The apparent ‘missing heritability’ in OCA is well described, with ~25–30% of clinically diagnosed individuals lacking two clearly pathogenic variants. Here we undertook empowered genetic studies in an extensive multigenerational Amish family, alongside a review of previously published literature, a retrospective analysis of in-house datasets, and tyrosinase activity studies. Together this provides irrefutable evidence of the pathogenicity of two common TYR variants, p.(Ser192Tyr) and p.(Arg402Gln) when inherited in cis alongside a pathogenic TYR variant in trans. We also show that homozygosity for the p.(Ser192Tyr)/p.(Arg402Gln) TYR haplotype results in a very mild, but fully penetrant, albinism phenotype. Together these data underscore the importance of including the TYR p.(Ser192Tyr)/p.(Arg402Gln) in cis haplotype as a pathogenic allele causative of OCA, which would likely increase molecular diagnoses in this missing heritability albinism cohort by 25–50%.
Similar content being viewed by others

Introduction
Oculocutaneous albinism (OCA) refers to a group of genetically and clinically heterogeneous disorders characterised by abnormal melanin synthesis, resulting in decreased or absent pigmentation of eyes, skin and hair.
Ocular features are present in individuals with OCA and are characteristic of the disease. These include photophobia, nystagmus, foveal hypoplasia, iris transillumination and abnormal decussation of nerve fibres at the optic chiasm resulting in crossed asymmetry on visual evoked potential testing1. These ocular features may, however, be variable with no single defining characteristic found to be present in every individual with OCA2. The cutaneous phenotype may also vary, ranging from total absence to near-normal levels of pigmentation, and can be difficult to evaluate, particularly in individuals with a lightly pigmented ethnic background3,4. As such, OCA can be difficult to distinguish clinically from several other ocular disorders with overlapping phenotypical features, such as GPR143-associated X-linked ocular albinism, where the hypopigmentation is limited to the eye1, FRMD7-associated X-linked idiopathic congenital nystagmus5, SLC38A8-associated foveal hypoplasia (also known as FHONDA; foveal hypoplasia, optic nerve decussation defects and anterior segment dysgenesis)6, and dominant PAX6-related ocular developmental disorders7.
OCA1, associated with TYR gene variants, is the most common OCA subtype found in Caucasians accounting for ~50% of cases worldwide8,9. TYR encodes the enzyme tyrosinase, which is the critical and rate-limiting enzyme in the biosynthesis of melanin in follicular and epidermal melanocytes in hair and skin, as well as in uveal melanocytes in the iris, ciliary body and choroid, and retinal pigment epithelium cells in the eye10. Disease-associated variants in the TYR gene cause complete or partial OCA1 depending on their impact on the residual activity of the encoded mutant tyrosinase enzyme11. TYR gene variants that result in a severe reduction or complete abolition of enzyme activity are associated with OCA1A, characterised by an almost complete absence of hair, skin and eye pigmentation10,11. Hypomorphic TYR variants in which mutant tyrosinase possess residual catalytic activity are associated with OCA1B, where affected individuals present with a milder phenotype with reduced levels of pigmentation10,11.
The apparent missing heritability in OCA is well described, with ~25–30% of clinically affected individuals lacking two clearly pathogenic sequence alterations within the same OCA gene; this proportion is higher in individuals with a partial OCA phenotype11,12. Several hypotheses have been proposed to explain this missing heritability, including variants in the promoter or other regulatory elements, as well as epistatic or synergistic interactions between known genes11,13. Two TYR sequence variants [NM_000372.4:c.575 C > A; p.(Ser192Tyr) or S192Y and c.1205 G > A; p.(Arg402Gln) or R402Q], previously described as non-pathogenic polymorphisms due to their frequency in the general population (25 and 18% respectively), have been found to be enriched in cohorts of OCA patients with only one identified TYR pathogenic variant8,11,14,15,16,17,18,19,20,21,22, leading to suggestions that these variants may in fact account for some of this missing heritability8,9,14,15,18,23,24,25,26,27, although this has, however, been disputed by others17,19,28. We and others have hypothesised that these variants may be pathogenic only when present in cis and inherited in bi-allelic fashion with a second deleterious TYR variant for tyrosinase activity to be sufficiently reduced to a level that will cause an OCA phenotype13,27,29. However, due to the high frequency of the p.(Ser192Tyr) and p.(Arg402Gln) variants in the general population, and the often small family sizes common to modern European populations, in many cases it has not always been possible to obtain informative allele segregation to phase gene variants and prove inheritance of a cis p.(Ser192Tyr)/p.(Arg402Gln) haplotype in trans with the pathogenic TYR alteration in all affected individuals27,30. This remaining uncertainty in clinical interpretation of this haplotype limits its routine reporting in diagnostic testing. This has important diagnostic implications; designating the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype as pathogenic could substantially increase the diagnostic yield by ~25–50% in albinism patient cohorts with missing heritability13. This also further supports the hypothesis that the prevalence of OCA1, commonly quoted as ~1 in 40,00010, likely represents a substantial underestimation, particularly amongst Caucasian populations with fair pigmentation31. In this study, we present extensive genetic data stemming from our investigation of a large multigenerational extended Amish family, alongside functional studies, a review of genotyped UK based albinism cohorts and a review of existing literature to provide strong evidence to support pathogenicity of the TYR p.(Ser192Tyr)/(Arg402Gln) in cis haplotype and its contribution to OCA1B in European populations.
Results
Clinical findings in an extended Amish family
We initially investigated a large multigenerational extended Old Order Amish family of Ohio ancestry residing in Wisconsin (USA) with 9 affected individuals all exhibiting nystagmus and variable levels of hair and skin hypopigmentation (Fig. 1; family 4). On the basis of a detailed medical history, assessment of skin and hair pigmentation, and ophthalmic investigations in selected affected individuals, a diagnosis of likely mild OCA was made in all affected individuals. We subsequently recruited two additional Amish families with a total of four affected individuals with a similar clinical phenotype (Fig. 1; families 2 and 3). In addition, a further Amish family with a single affected individual with OCA was recruited to the study (Fig. 1; family 1). This individual displayed clinical features consistent with a complete OCA phenotype, including pale skin and white/blonde hair and eyelashes, nystagmus, iris transillumination defects and foveal hypoplasia. Affected individuals were not noted to bruise or bleed easily, although specific haematological investigations were not performed. Clinical findings for all affected individuals are summarised in Table 1.

a Pedigree diagram showing segregation of TYR variants p.(Ser192Tyr), p.(Arg402Gln) and p.(Met252Arg) (highlighted in red). The two disease-causing haplotypes are shaded; the p.(Met252Arg) haplotype in blue, and the p.(Ser192Tyr)/p.(Arg402Gln) in cis haplotype in yellow. b Sequence chromatograms showing TYR c.575 C > A; p.(Ser192Tyr), c.755 T > G; p.(Met252Arg) and c.1205 G > A; p.(Arg402Gln) variants in heterozygous form. Schematic localisation of TYR p.(Ser192Tyr), p.(Met252Arg) and p.(Arg402Gln) variants within the catalytic tyrosinase domain of the TYR polypeptide. The p.(Ser192Tyr) and p.(Arg402Gln) variants are located at or near the copper-containing catalytic binding sites (the red diamonds denote the histidine residues that bind to copper atoms and hence structurally coordinate the positions of the metal-binding sites). Conservation of TYR p.(Ser192Tyr), p.(Met252Arg) and p.(Arg402Gln) variants across species. c Tyrosinase activity in wild-type, p.(Ser192Tyr)/S192Y mutant, p.(Arg402Gln)/R402Q mutant and double-mutant HEK293 cells. The absorbance of dopachrome, a product synthesised by the transformation of L-DOPA by tyrosinase was quantified as a measure of tyrosinase activity in wild-type and TYR-mutant cell lines. Cumulative production of dopachrome (top row) was quantified from the start of L-DOPA treatment (0 min) to 180 min. Statistical differences between cell lines were analysed at 180 min (bottom row). Data are shown as mean ± SEM and statistically significant differences between groups are indicated by asterisks (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001); ns not significant.
Genetic findings in Amish families (families 1–4)
Exome sequencing was initially performed in two affected individuals in family 4 (individuals IX:9 and IX:22) for targeted evaluation using the “Albinism or congenital nystagmus v1.0” PanelApp virtual gene panel (41 genes). Subsequently, variants predicted to have a functional consequence (including copy number variants) located genome-wide were identified and filtered according to allele frequency (gnomAD minor allele frequency (MAF) of <0.01). This identified only a single plausible candidate disease variant in both individuals, a heterozygous TYR missense variant (GRCh38) chr11:g.89178708 T > G; NM_000372.4:c.755 T > G; p.(Met252Arg) or M252R. The p.Met252 amino acid residue is located in the catalytic domain of the tyrosinase protein and is conserved across a variety of vertebrate species (Fig. 1b). This variant was absent in gnomAD and Genome Project population databases, although it was present in an Amish control exome dataset (allele frequency 0.0023) in heterozygous form only. In silico analysis of the p.(Met252Arg) variant using SIFT, PolyPhen-2 and PROVEAN predicted the variant to be deleterious, possibly damaging and deleterious. This variant has been reported in the compound heterozygous form [with a previously reported p.(Arg217Trp) variant] in a single individual with OCA22 and is considered to be likely pathogenic. Exome sequencing did not identify any additional candidate single nucleotide or structural disease variants in any OCA-associated genes.
To explore this apparent missing heritability, targeted dideoxy sequencing of all coding regions and intron-exon junctions of the TYR gene was performed in these two individuals. This confirmed the presence of the p.(Met252Arg) variant and also identified a further two TYR missense variants (GRCh38) chr11:g.89178528 C > A; NM_000372.4:c.575 C > A; p.(Ser192Tyr) (S192Y) and (GRCh38) chr11:g.89284793 G > A; NM_000372.4:c.1205 G > A; p.(Arg402Gln) (R402Q) in the same two individuals, excluded from the exome sequencing analysis based on individual population frequencies of 0.25 and 0.18, respectively. Segregation of all three TYR variants in all Amish families (families 1–4) is shown in Fig. 1, which demonstrates that the p.(Ser192Tyr)/p.(Arg402Gln) variants were linked in cis and inherited in a compound heterozygous fashion with p.(Met252Arg) (which itself occurs in cis with p.(Arg402Gln)) in all affected individuals except for a single affected individual with OCA, found to be homozygous for p.(Met252Arg) through targeted dideoxy sequencing. Individuals compound heterozygous for TYR p.(Met252Arg) and p.(Ser192Tyr)/p.(Arg402Gln) alleles displayed clinical features suggestive of partial albinism with variable skin and hair depigmentation, while the individual homozygous for the TYR p.(Met252Arg) variant displayed features of classical OCA including nystagmus, iris transillumination defects, a depigmented fundus and foveal hypoplasia (Table 1). Notably, individuals carrying the TYR p.(Met252Arg) variant on one allele and only the p.(Arg402Gln) or the p.(Ser192Tyr) variant on the other allele were apparently unaffected with no clinical features of OCA (individuals VIII:9, IX:2, IX:21, X:6, X:8, IX:1 and IX:4; Fig. 1a).
Additive temperature-sensitive effects of p.(Ser192Tyr) (S192Y) and p.(Arg402Gln) (R402Q) variants on TYR enzymatic activity
The TYR p.(Arg402Gln) variant alone has previously been proposed to contribute to OCA when inherited in trans with a pathogenic TYR variant9,14,15,16,22,23,25,26,32. Our pedigree analysis, however, appears to dispute this, with five individuals compound heterozygous for the pathogenic TYR p.(Met252Arg) variant as well as the p.(Arg402Gln) variant and yet showing no clinical features of OCA (individuals VIII:9, IX:2, IX:21, X:6 and X:8; Fig. 1a). At the same time, 13 individuals who were compound heterozygous for TYR p.(Met252Arg) and p.(Ser192Tyr)/p.(Arg402Gln) alleles all displayed clinical features of partial albinism, suggesting an additive impact of the p.(Ser192Tyr) and p.(Arg402Gln) variants on tyrosinase function. To investigate this further, we designed functional experiments to study and quantify the effects of the p.(Ser192Tyr) and p.(Arg402Gln) variants both independently and in combination compared to wild-type tyrosinase enzyme.
Figure 1c shows the DOPA-oxidase activity for all tyrosinase mutants analysed from 0 min to 180 min at 31 °C and 37 °C. At 37 °C, a slight decrease in DOPA-oxidase activity of the p.(Ser192Tyr) mutants was observed, and an almost total loss of DOPA-oxidase activity in the p.(Arg402Gln) mutants and p.(Ser192Tyr)/p.(Arg402Gln) double mutants. At 31 °C, the loss of tyrosinase activity caused by all of the TYR-mutants was reduced but still significant when compared to wildtype. For all the TYR mutant cell lines, the p.(Ser192Tyr)/p.(Arg402Gln) double mutants showed the most reduced tyrosinase activity, followed by p.(Arg402Gln) mutant, with the p.(Ser192Tyr) mutant least affected. There was a statistically significant difference between all three mutant groups, indicative of a cumulative effect of both p.(Ser192Tyr) and p.(Arg402Gln) mutations on tyrosinase activity.
Enrichment of the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype in OCA and control cohorts
Interrogation of a clinical cohort of 161 affected individuals with nystagmus and/or albinism (Southampton cohort) (including individuals previously reported by Norman et al. and O’Gorman et al.13,27) identified 71 individuals with two pathogenic or likely pathogenic variants (molecularly diagnosed including TYR, OCA2, GPR143 and PAX6 genes), 51 individuals carrying only a single likely disease-associated TYR variant with no candidate pathogenic variants identified in other OCA genes (missing heritability), and 39 individuals with no disease-associated TYR variants. All patients were sequenced using either the “Albinism or congenital nystagmus v1.0” PanelApp gene panel (41 genes) (https://panelapp.genomicsengland.co.uk/panels/) or a broader research panel as previously described13,27. Copy number analysis was not performed. Of these, 2 of the 71 individuals in the molecularly diagnosed group and 49 of the 51 individuals in the missing heritability group were found to have a genotype consistent with the presence of the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype (i.e. individuals who were homozygous or heterozygous for both these variants) (Table 2); this information was unavailable for the 39 molecularly undiagnosed individuals in this clinical cohort. A review of seven published OCA cohorts with missing heritability (i.e. individuals in whom only a single pathogenic TYR variant has been identified), together with our study cohort, found that approximately half of all affected individuals (50.7%) had a genotype consistent with the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype (Table 2). This is markedly enriched compared to molecularly diagnosed OCA cohorts (2.0%), as well as a control cohort of Amish individuals with no OCA diagnoses (16.9%; Pearson’s Chi-squared test, p < 2.2e-16). These findings strongly suggest that the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype contributes to the OCA phenotype.
Prevalence of TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype in OCA cohorts with missing heritability
Forty-nine affected individuals in our (Southampton and Salisbury) study cohorts were identified as carrying only a single pathogenic or likely pathogenic TYR variant as well as harbouring homozygous or heterozygous TYR p.(Ser192Tyr) and p.(Arg402Gln) variants; of these, familial segregation was performed in 41 individuals and their parents to assess the phase of the variants. In 23 individuals, this confirmed that the TYR p.(Ser192Tyr) and p.(Arg402Gln) variants were inherited in cis, and this haplotype was in trans to the previously identified pathogenic or likely pathogenic TYR variant (Table 3). For the remaining 18 cases, definitive segregation was not possible. Notably, no case was identified in which segregation showed that p.(Ser192Tyr) and p.(Arg402Gln) were not in trans with the pathogenic or likely pathogenic variant.
In five of the seven published OCA cohorts with missing heritability reviewed (Table 2), it was possible to determine the cis/trans phase of the TYR p.(Ser192Tyr) and p.(Arg402Gln) variants in a proportion of individuals reported17,20,22,30,31 (Table 3); in the remaining individuals this was not possible due to familial samples being unavailable for segregation analysis, or uninformative segregation results (owing to the high allele frequency of the p.(Ser192Tyr) and p.(Arg402Gln) TYR variants in the general population). For the remaining two studies of OCA cohorts with missing heritability, the cis/trans phase of the TYR p.(Ser192Tyr) and p.(Arg402Gln) variants could not be determined from the reported genotypes8,15. There were 41 OCA individuals with missing heritability from these five studies in whom the p.(Ser192Tyr)/p.(Arg402Gln) haplotype was possible, and where the cis/trans phase of the TYR p.(Ser192Tyr) and p.(Arg402Gln) variants could also be determined. In accordance with the findings from our local research cohorts, together with this additional informative cohort derived from five published studies, the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype segregated in trans with the pathogenic TYR variant in all 64 cases (amounting to 25.5% of total missing heritability cases) (Table 3). Taken together with the findings in Table 2, this suggests that the p.(Ser192Tyr)/p.(Arg402Gln) haplotype completes the molecular diagnosis in ~25–50% of OCA individuals with missing heritability.
Discussion
The pathogenicity of TYR p.(Ser192Tyr) and p.(Arg402Gln) variants and their contribution to the OCA phenotype, either in isolation or when linked in cis, has been heavily debated in many studies8,9,14,15,17,18,19,23,24,25,26,27,28. As such, these TYR variants are variably reported by clinical testing laboratories and potentially excluded, even when shown to be in cis. Here our genomic and functional data, initiated by our search for the cause of OCA in a number of Amish families, provide irrefutably strong evidence that the TYR p.(Ser192Tyr) and p.(Arg402Gln) variants are pathogenic when in cis. The increased frequency of the TYR p.(Met252Arg) variant in the Amish community, likely due to founder effects and endogamy, together with the large family sizes typical within the community, permitted empowered cosegregation studies able to determine the haplotype, phasing and inheritance of the common p.(Ser192Tyr) and p.(Arg402Gln) TYR variants together with the p.(Met252Arg) variant in a large number of related individuals.
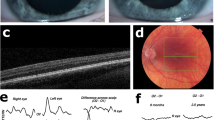
Both Jagirdar et al. and our group have previously proposed that both TYR p.(Ser192Tyr) and p.(Arg402Gln) variants, acting in cis, may have an additive effect producing a greater reduction in enzyme activity compared to each variant individually27,29. Both p.(Ser192Tyr) and p.(Arg402Gln) variants are common in Caucasian populations with allele frequencies of 36% and 27%, respectively (gnomAD v2.1.1), and would thus normally be considered benign. Indeed, our study demonstrates that inheritance of either variant individually in compound heterozygous form with the deleterious p.(Met252Arg) variant is insufficient to result in an OCA phenotype (individuals VIII:9, IX:1, IX:2, IX:4, X:6 and X:8; Fig. 1a). The p.(Ser192Tyr) and p.(Arg402Gln) variants are believed to have arisen independently on different ancestral haplotypes33, and their combined presence in cis on a recombinant haplotype is relatively rare, predicted to be between 1.1% to 1.9% in European populations27,29,31. Our studies here, alongside other previous studies8,13,17,22,27,30,31, provide strong support to show that the TYR p.(Ser192Tyr)/p.(Arg402Gln) the haplotype is enriched in Caucasian OCA cohorts with missing heritability (Table 2), and contributes to an OCA1B diagnosis when inherited in trans with a second deleterious TYR variant, particularly in individuals with lower pigmentary backgrounds, who may be more susceptible to the damaging effects of hypomorphic variants16,34. However, given the number of apparently unaffected individuals homozygous for the p.(Ser192Tyr)/p.(Arg402Gln) haplotype reported in the literature (Supplementary Table 1)29,30,31, the penetrance of the p.(Ser192Tyr)/p.(Arg402Gln) haplotype might appear to be incomplete, confounding the argument that it is a pathogenic allele. The apparently reduced penetrance of the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype may relate to the modifying effects of sequence variants in genes encoding other melanosomal proteins35,36,37,38,39, although other genetic and molecular studies would be required to confirm this. However, we propose that individuals homozygous for the hypomorphic TYR p.(Ser192Tyr)/p.(Arg402Gln) allele may have instead a consistent but mild phenotype, which is easily missed by incomplete phenotyping. In support of this, our studies identified five individuals with a clinical diagnosis of ‘possible hypomorphic’ OCA who were homozygous for TYR p.(Ser192Tyr)/p.(Arg402Gln), with no other known or likely TYR or other OCA gene-associated variants identified (Supplementary Table 1). All were noted to have foveal hypoplasia on OCT investigation but most had very mild, if any, other OCA features. Additionally, an apparently unaffected relative in our study was also identified as homozygous for TYR p.(Ser192Tyr)/p.(Arg402Gln). Despite the absence of nystagmus or any other pigmentary phenotype in this unaffected individual and visual acuities of 0.1 and 0.08 LogMAR (right and left eye respectively), the further detailed clinical investigation identified very mild iris transillumination and significant foveal hypoplasia (Supplementary Fig. 1). A review of the literature identified a further seven affected individuals in two studies with detailed clinical phenotyping available30,31 (Supplementary Table 1). Foveal hypoplasia, as well as iris transillumination, was documented in all thirteen individuals homozygous for both TYR p.(Ser192Tyr) and p.(Arg402Gln) (Supplementary Table 1). It, therefore, seems possible that individuals homozygous for the hypomorphic p.(Ser192Tyr)/p.(Arg402Gln) TYR allele have such a mild phenotype that they can easily go unidentified and unreported due to minimal effects on visual function or clear features of albinism; further phenotypic studies in large genomic population cohorts may be able to further clarify this potential association.
The TYR p.(Arg402Gln) variant is located near the copper-containing catalytic binding site CuB, and functional studies have shown that this amino acid alteration results in an enzyme with decreased thermal stability, disrupted copper-binding and reduced catalytic activity, thought to be mediated by decreased protein stability resulting in increased retention of the mutant tyrosinase protein as an unprocessed and misfolded glycoform in the endoplasmic reticulum (ER)29,40,41,42,43,44,45,46. The TYR p.(Ser192Tyr) variant is located within the copper-containing catalytic binding site CuA, and has been shown to reduce tyrosinase enzymatic activity and melanocyte pigment production independent of the p.(Arg402Gln) variant29,47,48. Genome-wide association studies have identified associations with skin, hair and eye pigmentation for both p.(Ser192Tyr) and p.(Arg402Gln) variants49,50,51,52,53, suggesting these TYR variants have a role in normal pigmentary variation, and that the double-variant p.(Ser192Tyr)/p.(Arg402Gln) haplotype appears to show an additive effect on these pigmentary phenotypes compared to each variant individually29. It is difficult however from the literature review alone to quantify the functional effects of the p.(Ser192Tyr) and p.(Arg402Gln) TYR variants both independently and in combination, compared to wild-type tyrosinase enzyme. This issue arises from the historical use of the human TYR expression construct pcTYR containing the p.(Ser192Tyr) variant to study the effects of “wildtype” tyrosinase activity42,54. Computational approaches to TYR functional activity, based on protein flexibility and dynamic properties, suggest that the p.(Ser192Tyr) and p.(Arg402Gln) variants both result in a TYR protein that is less stable and has reduced enzyme activity compared to a wild-type molecule; the combined effect of having both changes together in a single TYR molecule, however, has not been previously investigated55. Our study now shows for the first time a thermosensitive additive decrease in enzymatic function of the double-variant p.(Ser192Tyr)/p.(Arg402Gln) TYR protein compared to each variant acting individually (Fig. 1c), lending further support to the pathogenicity of the p.(Ser192Tyr)/p.(Arg402Gln) haplotype. Homology modelling of tyrosinase protein structure does not appear to show a direct interaction between the 192 and 402 amino acid residues31, and therefore this additional reduction in enzyme function in the double-mutant protein may instead be mediated by a combination of increased ER retention of the misfolded mutant protein [caused by p.(Arg402Gln) reducing protein stability] and reduced enzyme activity of any released mutant protein [possibly resulting from steric hindrance effects of p.(Ser192Tyr) affecting the CuA binding site]47, as proposed by Gronskov et al.31.
Subcellular localisation studies have determined that disease-associated TYR variants commonly result in near-absolute and irreversible ER retention of the mutant protein. The p.(Arg402Gln) variant, however, results in a thermosensitive tyrosinase protein that is retained in the ER at higher temperatures but is able to partially exit the ER at lower, more permissive temperatures40,44,46. Homozygosity for the p.(Ser192Tyr)/p.(Arg402Gln) haplotype may therefore still permit sufficient quantities of mutant tyrosinase to reach the inner surface of the melanosomal membrane, where the mutant protein is still able to participate in protein–protein interactions with other melanosomal proteins involved in melanogenesis, such as TYRP1 and TYRP256,57, resulting in a less severe functional impact and a milder pigmentary phenotype that may not always be clinically significant. This thermosensitivity of the double-variant mutant TYR protein also provides a compelling explanation for our discovery of a consistent foveal hypoplasia phenotype in individuals who are homozygous for both p.(Ser192Tyr)/p.(Arg402Gln) TYR variants, as higher temperatures within the developing eye may result in a larger impact of these variants on tyrosinase function14, while lower temperatures at the skin and extremities instead result in greater preservation of mutant protein function and a milder and more variable pigmentary phenotype.
Together, our studies define the genotype, biochemical and phenotype correlation of the p.(Met252Arg) and p.(Ser192Tyr)/p.(Arg402Gln) TYR variants and collectively demonstrate that the in cis p.(Ser192Tyr)/p.(Arg402Gln) allele is pathogenic. As such, the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype should be included as a pathogenic allele in future and retrospective genetic diagnoses of OCA, supporting the idea for a review of all previously undiagnosed OCA cases where these variants have been excluded. Reporting of the p.(Ser192Tyr)/p.(Arg402Gln) genotype in individuals in whom only a single deleterious TYR variant has been identified could permit a 25–50% uplift in confirmatory molecular diagnoses (when the phase has been determined) in this diagnostically challenging patient group (Tables 2, 3). Additionally, for patients with an albinism phenotype but no apparent variants in albinism genes, consideration of these variants when identified in cis as a pathogenic allele in its own right may also help provide clinical direction. For example, in individuals heterozygous for this allele, alternative diagnoses such as syndromic albinism might be considered less likely as they would be considered ‘at least a carrier of a pathogenic OCA1B allele’, and genomic data may be re-examined in a targeted fashion to search for further non-coding splice or structural variants in the TYR gene. In individuals with a very mild albinism phenotype or isolated foveal hypoplasia, identification of this pathogenic allele in homozygous form may provide the molecular diagnosis, ending their diagnostic odyssey. It will be crucially important to accurately determine the phase of these common variants, and due to the high frequencies of these variants alone in the population which can limit informative phase studies in relatives, consideration should perhaps be given to the use of amplicon-based long-read sequencing technologies that allow haplotype phasing in the genomic workup of such patients58. Achieving an accurate molecular diagnosis will bring about important benefits in affected individuals and their families, allowing accurate prognostic information and family counselling to be provided, avoiding the need for further invasive investigations to confirm the clinical diagnosis or rule out syndromic forms of the disease or masquerading conditions, and has important therapeutic implications, given the emerging therapies currently under development and in clinical trials for OCA59,60.
Methods
Ethics statement
This study was approved by the institutional review board of all participating institutions (University of Arizona IRB—1000000050, Akron Children’s Hospital IRB—project number 986876–3, South Central—Hampshire A Research Ethics Committee—IRAS:174564), and all participating individuals were recruited with written informed consent.
Patient ascertainment and clinical phenotyping
Affected individuals and unaffected family members from four Ohio and Wisconsin Amish families with a common Ohio ancestry were recruited to this study (Fig. 1). Medical history was taken in all recruited family members, as well as detailed phenotyping of skin and hair pigmentation, particularly in the context of familial pigmentary background. A diagnosis of nystagmus was established in all affected individuals, and further ophthalmic investigations including electroretinography and optical coherence tomography (OCT) were performed in selected individuals. Blood/buccal samples were obtained with informed consent.
Molecular genetic analysis
Participating individuals had either peripheral venous blood samples taken in EDTA containing vacutainer tubes or buccal cell collection using the ORAcollect® for paediatrics kit (DNA Genotek). Genomic DNA extraction was performed using either the ReliaPrepTM kit (Blood gDNA Miniprep System, Promega) for venous blood samples or the Xtreme DNA kit (Isohelix) for buccal samples, according to the manufacturer’s protocol. Exome sequencing (whole-exome sequencing, Exeter laboratory for individual IX:9 and Illumina TruSightTM One clinical exome sequencing panel, Southampton laboratory for individual IX:22) was performed as previously described27,61. The whole-exome sequencing sample was prepared using Agilent Sureselect Whole Exome v6 targeting, while the TruSightTM One panel provides targeted sequencing for 4813 genes associated with clinical phenotypes and captures most of the coding regions of genes responsible for OCA subtypes 1–4 & 6 (TYR, OCA2, TYRP1, SLC45A2 and SLC24A5, respectively), the ocular albinism gene (GPR143), all syndromic albinism genes and PAX6. Next-generation sequencing analysis (NextSeq500: Illumina) involved: read alignment (BWA-MEM (v0.7.12), mate-pairs fixed and duplicates removed (Picard v1.129), InDel realignment/base quality recalibration (GATK v3.4–46), single-nucleotide variant (SNV)/InDel detection (GATK HaplotypeCaller), annotation (Alamut v1.4.4), and read depth (GATK DepthOfCoverage). Additional filtering was performed using virtual gene panel analysis of exome data using the “Albinism or congenital nystagmus v1.0” PanelApp gene panel (41 genes) (https://panelapp.genomicsengland.co.uk/panels/), with variants prioritised by call quality, frequency in control datasets (Genome Aggregation Database; gnomAD v2.1.1 and 1000 Genomes Project) and predicted functional consequence13,27. Primers were designed with Primer3 web software to cover all five coding exons and associated intron-exon junctions in TYR. As the 3′ region encompassing coding exons 4 and 5 of TYR shares high homology with a pseudogene, TYRL62, locus-specific amplification primers were designed for TYR exons 4 and 5 to prevent co-amplification of TYR and TYRL and subsequent misinterpretation of results. Dideoxy sequencing products were sequenced by Source BioScience Lifesciences (https://www.sourcebioscience.com/). Primer sequences and polymerase chain reaction conditions are listed in supplementary Table 3. The TYR c.755 T > G; p.(Met252Arg) variant and c.[575 C > A;1205 G > A]; p.[Ser192Tyr;Arg402Gln] variants-in-cis haplotype were submitted to ClinVar (www.ncbi.nlm.nih.gov/clinvar, accession numbers SCV001984755 and SCV001984756).
Establishment of Tyr mutant cell lines
The plasmid vector p3XFLAG-CMV-14 containing TYR cDNA was purchased from Addgene (Massachusetts, USA) and was initially deposited by Ruth Halaban63. Upon arrival, sequencing revealed the p.(Ser192Tyr) (c.C575A) common population variant to be present. Site-directed mutagenesis was used to create the wild-type sequence (c.575 C, p.192Ser) as well as the p.(Arg402Gln) variant. The primers used for each variant inserted through site-directed mutagenesis are listed in Supplementary Table 2. Site-directed mutagenesis was carried out using the non-strand displacing activity of Pfu DNA polymerase to incorporate and extend the mutagenic primers. The reaction mixture contained Phusion Pfu Polymerase and its buffer, forward and reverse primers (0.5 µM), dNTPs (200 µM) and the cDNA template. PCRs were performed in a total volume of 50 µl. Touch-down PCR conditions were set at 98 °C for 30 sec followed by 30 cycles of 98 °C for 10 sec, 45–72 °C for 10–30 sec and 72 °C for 15–30 sec, and a final extension step of 72 °C for 5–10 min. The PCR product was treated with DpnI to digest the methylated parental DNA.
Purified mutated tyrosinase PCR products were employed to transform NEB® 5-alpha Competent E. coli (High Efficiency; New England Biolabs, UK) via heat shock method. Briefly, 50 µl of thawed cells were kept on ice and combined with ~100 ng of plasmid DNA and incubated for 30 min. The cell-DNA mixture was heat-shocked at 42 °C for 30 sec and then placed on ice for 5 min. Cells were given S.O.C medium and incubated for an hour in a shaking incubator before being plated on ampicillin selection (100 ug/ml) LB agar plates. After overnight incubation at 37 °C, single ampicillin-resistant colonies were picked and grown in LB broth for approximately 16 h, at which point the cells were pelleted by centrifugation and the DNA extracted. When the stocks were diminished, competent cells were produced through treatment with CaCl2 and subsequently transformed using the heat shock method described above.
Cell culture conditions
Human Embryonic Kidney 293 Freestyle (HEK293F) cells (Invitrogen, California, USA) were cultured in Freestyle culture medium (Invitrogen, California, USA) at 37 °C in a shaking incubator at 125 rpm with 8% CO2. When cells reached a density of 1 × 106 cells/ml, they were transfected with 30 µg of plasmids containing the p.(Arg402Gln) or p.(Ser192Tyr) mutations or co-transfected with both plasmids. The lipid-based reagent, 293fectin (60 µl) (ThermoFisher, UK), was diluted in Opti-MEM (ThermoFisher, UK) and incubated at room temperature for 5 mins. DNA and 293fectin were combined, gently mixed and incubated at room temperature for 30 mins before adding to cells. Then, cells were incubated in 6 wells plates for 72 h at 31 °C or 37 °C to reach 90% confluency, and the enzymatic activity assays were performed.
Enzymatic activity assays
The DOPA-oxidase activity was assessed in the different mutants. First, transfected cells from the different mutant clones were treated with L-DOPA, and the DOPA-oxidase activity was measured as the accumulation of the downstream product, dopachrome, following the manufacturer’s protocol. Briefly, cells cultured in six-well plates were lysed in NP40 Cell Lysis Buffer (ThermoFisher, UK) containing 1 mM phenylmethylsulfonyl fluoride (PMSF) (in DMSO with a final concentration of 1%) and 1X protease and phosphatase inhibitor (Halt™ Phosphatase Inhibitor Cocktail, Thermo Fisher Scientific, UK), and protein concentration was measured by BCA assay (Thermo Scientific™ Pierce™ BCA Protein Assay Kit). Samples were then diluted into 4 µg/µl, and 50 µl or 30 µl sample aliquots were used for the DOPA assays. After adding the volume of the samples to 96-well plates, 150 µl of a phosphate buffer with L-DOPA 1 mM was added to the wells. Enzymatic activity was recorded as the absorbance of dopachrome at 492 nm from the start of L-dopa treatment (0 min) and at 30 min intervals thereafter for a total of 180 min at both 31 °C and 37 °C. Assays were routinely performed in triplicate and the results are presented as the means of the independent assays ± standard error.
Statistics
Results of enzymatic activity at 180 min were normalised to wild-type, with the values for wild-type taken to be 100% of the expected enzymatic activity. One-way ANOVA was performed followed by a Sidak’s post-hoc test. A probability level of at least p < 0.05 was considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Evaluating the prevalence of TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype in OCA and control cohorts
A clinical cohort of affected individuals with nystagmus and/or albinism was retrospectively ascertained through the Southampton (161 individuals) and Salisbury (131 individuals) research databases. All individuals had been referred from a regional paediatric nystagmus clinic. Next-generation sequencing (Illumina TruSight One clinical exome sequencing panel), alignment and filtering were performed as previously described13,27. The genomic data were interrogated to ascertain the frequency of the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype in this cohort. A literature review was also performed to evaluate the reported prevalence of the TYR p.(Ser192Tyr)/p.(Arg402Gln) haplotype in additional published OCA cohorts. This was compared against an in-house exome database of Amish individuals unaffected by OCA. Statistical analysis was performed using an established software package (R Core Team 2015; R Foundation for Statistical Computing, Vienna, Austria)64.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The genetic variants investigated are deposited in ClinVar (accession codes SCV001984755 and SCV001984756). While the in-house Amish exome database is not publicly accessible due to the informed consent restrictions, de-identified information may be accessible and requested from corresponding authors A.H.C. (a.h.crosby@exeter.ac.uk) and E.L.B. (E.Baple@exeter.ac.uk).
References
McKay, B. S. Pigmentation and vision: is GPR143 in control? J. Neurosci. Res. 97, 77–87 (2019).
Kruijt, C. C. et al. The phenotypic spectrum of albinism. Ophthalmology 125, 1953–1960 (2018).
Sjöström, A., Kraemer, M., Ohlsson, J. & Villarreal, G. Subnormal visual acuity syndromes (SVAS): albinism in Swedish 12-13-year-old children. Doc. Ophthalmol. 103, 35–46 (2001).
Sjöström, A. et al. Subnormal visual acuity (svas) and albinism in mexican 12–13-year-old children. Doc. Ophthalmol. 108, 9–15 (2004).
Thomas, M. G., Maconachie, G., Hisaund, M. & Gottlob, I. FRMD7-related infantile nystagmus. GeneReviews. https://www.ncbi.nlm.nih.gov/books/NBK3822/ (2009).
Poulter, J. A. et al. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am. J. Hum. Genet. 93, 1143–1150 (2013).
Lima Cunha, D., Arno, G., Corton, M. & Moosajee, M. The spectrum of PAX6 mutations and genotype-phenotype correlations in the eye. Genes (Basel) https://doi.org/10.3390/genes10121050 (2019).
Hutton, S. M. & Spritz, R. A. Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 is the most prevalent OCA type. J. Invest. Dermatol. 128, 2442–2450 (2008a).
Rooryck, C. et al. Molecular diagnosis of oculocutaneous albinism: new mutations in the OCA1-4 genes and practical aspects. Pigment Cell Melanoma Res. 21, 583–587 (2008).
Grønskov, K., Ek, J. & Brondum-Nielsen, K. Oculocutaneous albinism. Orphanet J. Rare Dis. 2, 1–8 (2007).
Simeonov, D. R. et al. DNA variations in oculocutaneous albinism: an updated mutation list and current outstanding issues in molecular diagnostics. Hum. Mutat. 34, 827–835 (2013).
King, R. A. et al. Tyrosinase gene mutations in oculocutaneous albinism 1 (OCA1): definition of the phenotype. Hum. Genet. 113, 502–513 (2003).
O’Gorman, L. et al. A small gene sequencing panel realises a high diagnostic rate in patients with congenital nystagmus following basic phenotyping. Sci. Rep. 9, 13229 (2019).
Fukai, K. et al. Autosomal recessive ocular albinism associated with a functionally significant tyrosinase gene polymorphism. Nat. Genet. 9, 92–95 (1995).
Hutton, S. M. & Spritz, R. A. A comprehensive genetic study of autosomal recessive ocular albinism in Caucasian patients. Invest. Ophthalmol. Vis. Sci. 49, 868–872 (2008b).
Chiang, P. W., Spector, E. & Tsai, A. C. Oculocutaneous albinism spectrum. Am. J. Med. Genet. A 149A, 1590–1591 (2009).
Oetting, W. S. et al. The R402Q tyrosinase variant does not cause autosomal recessive ocular albinism. Am. J. Med. Genet. A 149A, 466–469 (2009).
Gargiulo, A. et al. Molecular and clinical characterization of albinism in a large cohort of Italian patients. Invest. Ophthalmol. Vis. Sci. 52, 1281–1289 (2011).
Preising, M. N., Forster, H., Gonser, M. & Lorenz, B. Screening of TYR, OCA2, GPR143, and MC1R in patients with congenital nystagmus, macular hypoplasia, and fundus hypopigmentation indicating albinism. Mol. Vis. 17, 939–948 (2011).
Ghodsinejad Kalahroudi, V. et al. Two novel tyrosinase (TYR) gene mutations with pathogenic impact on oculocutaneous albinism type 1 (OCA1). PLoS ONE 9, e106656 (2014).
Mauri, L. et al. Clinical evaluation and molecular screening of a large consecutive series of albino patients. J. Hum. Genet. 62, 277–290 (2017).
Lasseaux, E. et al. Molecular characterization of a series of 990 index patients with albinism. Pigment Cell Melanoma Res. 31, 466–474 (2018).
Chiang, P.-W., Drautz, J. M., Tsai, A. C.-H., Spector, E. & Clericuzio, C. L. A new hypothesis of OCA1B. Am. J. Med. Genet. Part A 146, 2968–2970 (2008).
Kausar, T., Bhatti, M. A., Ali, M., Shaikh, R. S. & Ahmed, Z. M. OCA5, a novel locus for non-syndromic oculocutaneous albinism, maps to chromosome 4q24. Clin. Genet. 84, 91–93 (2013).
Kubal, A., Dagnelie, G. & Goldberg, M. Ocular albinism with absent foveal pits but without nystagmus, photophobia, or severely reduced vision. J. AAPOS 13, 610–612 (2009).
Thomas, M. G., Maconachie, G. D., Sheth, V., McLean, R. J. & Gottlob, I. Development and clinical utility of a novel diagnostic nystagmus gene panel using targeted next-generation sequencing. Eur. J. Hum. Genet. 25, 725–734 (2017).
Norman, C. S. et al. Identification of a functionally significant tri-allelic genotype in the Tyrosinase gene (TYR) causing hypomorphic oculocutaneous albinism (OCA1B). Sci. Rep. 7, 4415 (2017).
Gronskov, K. et al. Birth prevalence and mutation spectrum in danish patients with autosomal recessive albinism. Invest. Ophthalmol. Vis. Sci. 50, 1058–1064 (2009).
Jagirdar, K. et al. Molecular analysis of common polymorphisms within the human Tyrosinase locus and genetic association with pigmentation traits. Pigment Cell Melanoma Res. 27, 552–564 (2014).
Campbell, P. et al. Clinical and genetic variability in children with partial albinism. Sci. Rep. 9, 16576 (2019).
Gronskov, K. et al. A pathogenic haplotype, common in Europeans, causes autosomal recessive albinism and uncovers missing heritability in OCA1. Sci. Rep. 9, 645 (2019).
Marti, A. et al. Lessons of a day hospital: comprehensive assessment of patients with albinism in a European setting. Pigment Cell Melanoma Res. 31, 318–329 (2018).
Hudjashov, G., Villems, R. & Kivisild, T. Global patterns of diversity and selection in human tyrosinase gene. PLoS ONE 8, e74307 (2013).
Mondal, M., Sengupta, M. & Ray, K. Functional assessment of tyrosinase variants identified in individuals with albinism is essential for unequivocal determination of genotype-to-phenotype correlation. Br. J. Dermatol. 175, 1232–1242 (2016).
King, R. A. et al. MC1R mutations modify the classic phenotype of oculocutaneous albinism type 2 (OCA2). Am. J. Hum. Genet. 73, 638–645 (2003).
Manga, P. et al. Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. Am. J. Hum. Genet. 61, 1095–1101 (1997).
Chiang, P. W., Fulton, A. B., Spector, E. & Hisama, F. M. Synergistic interaction of the OCA2 and OCA3 genes in a family. Am. J. Med. Genet. A 146A, 2427–2430 (2008).
Sajid, Z. et al. Genetic causes of oculocutaneous albinism in Pakistani population. Genes (Basel) https://doi.org/10.3390/genes12040492 (2021).
Wei, A. H., Yang, X. M., Lian, S. & Li, W. Genetic analyses of Chinese patients with digenic oculocutaneous albinism. Chin. Med. J. (Engl.) 126, 226–230 (2013).
Berson, J. F., Frank, D. W., Calvo, P. A., Bieler, B. M. & Marks, M. S. A common temperature-sensitive allelic form of human tyrosinase is retained in the endoplasmic reticulum at the nonpermissive temperature. J. Biol. Chem. 275, 12281–12289 (2000).
Spritz, R. A., Ho, L., Furumura, M. & Hearing, V. J. Jr. Mutational analysis of copper binding by human tyrosinase. J. Invest. Dermatol. 109, 207–212 (1997).
Tripathi, R. K., Giebel, L. B., Strunk, K. M. & Spritz, R. A. A polymorphism of the human tyrosinase gene is associated with temperature-sensitive enzymatic activity. Gene Expr. 1, 103–110 (1991).
Tripathi, R. K., Hearing, V. J., Urabe, K., Aroca, P. & Spritz, R. A. Mutational mapping of the catalytic activities of human tyrosinase. J. Biol. Chem. 267, 23707–23712 (1992).
Halaban, R. et al. Endoplasmic reticulum retention is a common defect associated with tyrosinase-negative albinism. Proc. Natl Acad. Sci. USA 97, 5889–5894 (2000).
Dolinska, M. B. et al. Oculocutaneous albinism type 1: link between mutations, tyrosinase conformational stability, and enzymatic activity. Pigment Cell Melanoma Res. 30, 41–52 (2017).
Toyofuku, K., Wada, I., Spritz, R. A. & Hearing, V. J. The molecular basis of oculocutaneous albinism type 1 (OCA1): sorting failure and degradation of mutant tyrosinases results in a lack of pigmentation. Biochem. J. 355, 259–269 (2001).
Chaki, M. et al. Molecular and functional studies of tyrosinase variants among Indian oculocutaneous albinism type 1 patients. J. Invest. Dermatol. 131, 260–262 (2011).
Wei, A. H., Zang, D. J., Zhang, Z., Yang, X. M. & Li, W. Prenatal genotyping of four common oculocutaneous albinism genes in 51 Chinese families. J. Genet. Genomics 42, 279–286 (2015).
Sulem, P. et al. Two newly identified genetic determinants of pigmentation in Europeans. Nat. Genet. 40, 835–837 (2008).
Nan, H., Kraft, P., Hunter, D. J. & Han, J. Genetic variants in pigmentation genes, pigmentary phenotypes, and risk of skin cancer in Caucasians. Int. J. Cancer 125, 909–917 (2009).
Sulem, P. et al. Genetic determinants of hair, eye and skin pigmentation in Europeans. Nat. Genet. 39, 1443–1452 (2007).
Stokowski, R. P. et al. A genomewide association study of skin pigmentation in a South Asian population. Am. J. Hum. Genet. 81, 1119–1132 (2007).
Hu, H. H. et al. Assessment of tyrosinase variants and skin cancer risk in a large cohort of French subjects. J. Dermatol. Sci. 64, 127–133 (2011).
Bouchard, B., Fuller, B. B., Vijayasaradhi, S. & Houghton, A. N. Induction of pigmentation in mouse fibroblasts by expression of human tyrosinase cDNA. J. Exp. Med. 169, 2029–2042 (1989).
K, B. & Purohit, R. Mutational analysis of TYR gene and its structural consequences in OCA1A. Gene 513, 184–195 (2013).
Kobayashi, T. & Hearing, V. J. Direct interaction of tyrosinase with Tyrp1 to form heterodimeric complexes in vivo. J. Cell Sci. 120, 4261–4268 (2007).
Orlow, S. J. et al. High-molecular-weight forms of tyrosinase and the tyrosinase-related proteins: evidence for a melanogenic complex. J. Invest. Dermatol. 103, 196–201 (1994).
van Dijk, E. L., Jaszczyszyn, Y., Naquin, D. & Thermes, C. The third revolution in sequencing technology. Trends Genet. 34, 666–681 (2018).
Adams, D. R. et al. One-year pilot study on the effects of nitisinone on melanin in patients with OCA-1B. JCI Insight https://doi.org/10.1172/jci.insight.124387 (2019).
Lee, H., Scott, J., Griffiths, H., Self, J. E. & Lotery, A. Oral levodopa rescues retinal morphology and visual function in a murine model of human albinism. Pigment Cell Melanoma Res. 32, 657–671 (2019).
Rawlins, L. E. et al. An Amish founder variant consolidates disruption of CEP55 as a cause of hydranencephaly and renal dysplasia. Eur. J. Hum. Genet. 27, 657–662 (2019).
Chaki, M., Mukhopadhyay, A. & Ray, K. Determination of variants in the 3’-region of the tyrosinase gene requires locus specific amplification. Hum. Mutat. 26, 53–58 (2005).
Halaban, R., Cheng, E. & Hebert, D. N. Coexpression of wild-type tyrosinase enhances maturation of temperature-sensitive tyrosinase mutants. J. Invest. Dermatol. 119, 481–488 (2002).
R Core Team. R: A language and environment for statistical computing. (Vienna, Austria, 2013). https://www.r-project.org/.
Acknowledgements
We would like to thank family members for their involvement in this study, and to acknowledge Dr. Caroline Wright for her assistance with this work and helpful comments on the manuscript. The work was supported by University of Exeter Vice Chancellor Scholarship (S.L.), the Gift of Sight Appeal (J.E.S., H.L., J.A.R., A.S.B., C.S.N.), MRC (Proximity to Discovery and Confidence in Concept grants MC_PC_18047, MC_PC_15054, MC_PC_15047 to University of Exeter, E.L.B. and A.H.C., G1001931 to E.L.B. and G1002279 to A.H.C.).
Author information
Authors and Affiliations
Contributions
Clinical data collection, collation and analysis: S.L., J.D., K.B.W., M.A.S., A.V.D., H.E.C., S.H., O.W., E.L.B., A.H.C., J.E.S. Genetic and functional studies and data analysis: S.L., A.S.B., J.S.L., J.A.R., C.S.N., G.V.H., E.L.B., A.H.C., J.E.S. Manuscript writing and revision: S.L., A.S.B., H.L., N.S.T., J.C., D.B., S.E., E.L.B., A.H.C., J.E.S. Design and conception of studies, supervision and coordination: H.L., N.S.T., J.C., D.B., S.E., E.L.B., A.H.C., J.E.S. All authors have read and approve of the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, S., Sanchez-Bretaño, A., Leslie, J.S. et al. Evidence that the Ser192Tyr/Arg402Gln in cis Tyrosinase gene haplotype is a disease-causing allele in oculocutaneous albinism type 1B (OCA1B). npj Genom. Med. 7, 2 (2022). https://doi.org/10.1038/s41525-021-00275-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-021-00275-9
This article is cited by
-
The contribution of common regulatory and protein-coding TYR variants to the genetic architecture of albinism
Nature Communications (2022)
