
Abstract
Exceptional magmatic events coincided with the largest mass extinctions throughout Earth’s history. Extensive degassing from organic-rich sediments intruded by magmas is a possible driver of the catastrophic environmental changes, which triggered the biotic crises. One of Earth’s largest magmatic events is represented by the Central Atlantic Magmatic Province, which was synchronous with the end-Triassic mass extinction. Here, we show direct evidence for the presence in basaltic magmas of methane, generated or remobilized from the host sedimentary sequence during the emplacement of this Large Igneous Province. Abundant methane-rich fluid inclusions were entrapped within quartz at the end of magmatic crystallization in voluminous (about 1.0 × 106 km3) intrusions in Brazilian Amazonia, indicating a massive (about 7.2 × 103 Gt) fluxing of methane. These micrometre-sized imperfections in quartz crystals attest an extensive release of methane from magma–sediment interaction, which likely contributed to the global climate changes responsible for the end-Triassic mass extinction.
Similar content being viewed by others


Introduction
The geological record shows that massive and rapid inputs of greenhouse gases, such as CO2 and CH4, into the atmosphere–hydrosphere system caused sudden increases of Earth’s surface temperature, leading to major environmental changes and severe biotic crises1,2,3. The largest mass extinction events, such as those at the Permian–Triassic boundary and at the end-Triassic, were synchronous with rapid and voluminous magmatism generating Large Igneous Provinces (LIPs). The emplacement of LIPs involved > 1 × 106 km3 of mainly basaltic magmas, via eruptions or shallow intrusions, often within sedimentary basins rich in organic and inorganic carbon4. LIPs contributed to global climate forcing through both volcanic emissions (e.g., direct release of mainly inorganic carbon from erupted magmas)5 and thermogenic degassing (e.g., release of organic and inorganic carbon from the intruded sedimentary rocks, due to contact metamorphism)6. Significant input of 13C-depleted organic carbon into the surface system during mass extinction events is inferred by the sharp negative carbon-isotope shifts preserved in the sedimentary record7. The Central Atlantic Magmatic Province (CAMP)8 is one of Earth’s most voluminous LIPs, with > 3 × 106 km3 of basaltic magmas emplaced synchronously with the end-Triassic mass extinction (ca. 201.6–201.3 Ma)9,10,11. Direct geological evidence for abundant volcanic CO2 emissions was recently identified within CAMP basaltic lava flows12. Conversely, direct geological evidence for degassing from intruded and heated organic-rich sedimentary rocks, hypothesized and modelled for the CAMP13,14, is still lacking.
Here, we investigate and quantify CH4 preserved within micrometre-sized fluid inclusions (FIs), hosted in shallow basaltic intrusions from the CAMP in Brazilian Amazonia. These findings indicate that CAMP magmatism generated or remobilized large amounts of CH4, which likely contributed to the end-Triassic climatic and biotic crisis.
Results
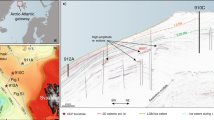
The investigated magmatic rocks were sampled in northern Brazil, where about 1 × 106 km3 of CAMP basaltic magmas, stacked at shallow depths within the Amazonas and Solimões Basins13,15,16, formed some of Earth’s largest magmatic sills. The analysed samples are from several sills emplaced at about 1–4 km depth (on average corresponding to ca. 50 MPa) in the intracratonic Amazonas Basin17,18, and outcropping over > 500 km distances in the State of Pará, northern Brazil (Fig. 1; Supplementary Fig. 1; Supplementary Data 1). These CAMP sills display U–Pb zircon ages ranging from 201.525 ± 0.065 to 201.348 ± 0.034 Ma11,19, overlapping in time with the end-Triassic interval of climatic and environmental changes, carbon cycle disruption and biotic crisis10,20. According to geophysical data and borehole investigations, CAMP basaltic magmas intruded the Amazonas and Solimões Basins at different levels, forming 3–7 sills with a cumulative thickness of up to 1 km18,21. The CAMP sills intruded Devonian to Carboniferous sedimentary formations of continental and marine origin, including voluminous organic-rich shales and evaporites22,23. During CAMP emplacement both the Amazonas and Solimões Basins had a high potential for hydrocarbons, which were either newly generated or remobilized by the thermal anomaly associated with the intrusion of sills13,17,22. The primary hydrocarbon generation was related to the burial of organic matter within the subsiding basin from the Late Carboniferous to the Early Triassic, with a peak during the Late Permian, whereas the secondary hydrocarbon generation was related to the thermal anomaly induced by the CAMP magmatic intrusions at the end-Triassic17,22,24. Evidence for secondary thermal cracking of source rock organic matter after normal sediment diagenesis and for unconventional CH4 generation through the oxidative dissolution of siderite in the Solimões Basin25 highlights the key role of CAMP activity in these hydrocarbon systems.

The geological map of CAMP at about 201 Ma is modified after ref. 16 and references therein, and the geological maps of Monte Alegre, Medicilândia and Placas (Amazonas Basin) are modified after ref. 23. The CAMP sills, intruding Devonian to Carboniferous sedimentary formations and locally named Penatecaua Diabase, outcrop as km-scale elongated intrusive bodies, approximately displaying an E–W orientation23. Among the sedimentary formations and groups intruded by sampled sills, the Urupadi Group includes the Maecuru Formation (Lontra Member; delta fan siliciclastic rocks) and the Ererê Formation (tidal plain siliciclastic rocks). The Curuá Group includes the Barreirinha Formation (mainly black shales deposited in an anoxic marine setting), the Curiri Formation (glacial siliciclastic rocks), the Oriximiná Formation (fluvio–deltaic siliciclastic rocks, with a glacial contribution) and the Faro Formation (fluvio–deltaic siliciclastic rocks), and is locally represented by the undifferentiated Curuá Group (black shales and other siliciclastic rocks of marine, glacial and fluvio–deltaic origin). The Tapajós Group includes the Monte Alegre Formation (siliciclastic and carbonate rocks from fluvio–aeolian and marine environments), the Itaituba Formation (siliciclastic rocks with carbonate intercalations from shallow marine and intertidal plain environments) and the Nova Olinda Formation (siliciclastic rocks at the base, evaporites and limestone lenses at the top, from fluvio–lacustrine and hypersaline restricted marine environments). During CAMP activity hydrocarbons were newly generated or remobilized from pre-existing pools in both Amazonas and Solimões Basins22. The source rocks are mainly represented by marine black shales constituting the lower part of the Barreirinha Formation17,22,24. The reservoir rocks are mainly represented by neritic–deltaic sandstones, siltstones and shales of the Maecuru and Ererê Formations, by glacio–marine diamictites, shales and siltstones of the Curiri Formation, by aeolian sandstones, lacustrine and interdune shales of the Monte Alegre Formation and by lenticular sandstones of the Nova Olinda Formation17,22.
Our in situ analytical data (see Methods) show that multi-phase FIs, entrapped in minerals of the sill-forming magmatic rocks, represent direct evidence of volatiles mobilized and degassed during magma emplacement (Figs. 2, 3; Supplementary Figs. 2, 3).

a Microgabbro with early-magmatic plagioclase (Pl) and clinopyroxene (Cpx), and interstitial late-magmatic quartz (Qz; sample RP125). b Late-magmatic quartz, brown hornblende (Hbl) and red biotite (Bt; sample RP125). c Graphic texture between late-magmatic quartz and alkali feldspar (Qz+Afs; sample RP130). Transmitted, crossed polarized light (a, c) and transmitted, plane polarized light (b).

a H2O-rich FI, containing 3 phases (i.e., liquid + vapour + solid phases), and CH4-rich FIs, containing 2 phases (i.e., liquid + vapour phases), hosted in late-magmatic quartz (Qz; sample RP128). b, c CH4-rich FI, hosted in late-magmatic quartz (samples RP125 and RP128), along with the corresponding hyperspectral Raman map, where the vapour phase is CH4 (green phase) and the liquid phase is H2O with ca. 10 wt.% salinity (blue phase). Transmitted, plane polarized light.
Magmatic rocks
The sill-forming magmatic rocks, geochemically classified as basalts and basaltic andesites, consist of microgabbros (dolerites), mainly composed of plagioclase, clinopyroxene, Fe–Ti oxides, olivine (when quartz is absent) and interstitial micrographic intergrowths of quartz and alkali feldspar, along with amphibole, biotite and apatite15,23,26,27,28. Three different crystallization stages are identified on the basis of microstructural position and reciprocal equilibria of mineral phases (Fig. 2; Supplementary Note 1). Early-magmatic minerals (70–90 vol.%), such as plagioclase, clinopyroxene and oxides, plus rare olivine in quartz-free samples, constitute the main crystal framework of the sills (Fig. 2a). Late-magmatic minerals (10–30 vol.%), mainly hydrous silicates, feldspars and quartz, often occupy interstitial domains (Fig. 2b). Hydrothermal minerals, like carbonate, quartz and sulfides among others, generally replace the magmatic minerals or crystallize in veins crosscutting the microgabbros (Supplementary Fig. 4). Quartz is the main host mineral phase for FIs and crystallizes during both late-magmatic and hydrothermal stages. Late-magmatic quartz occurs interstitially in single crystals or aggregates, with eu- to an-hedral crystal habit, or in irregular intergrowths with alkali feldspar as graphic textures (Fig. 2c). It represents on average 5 vol.% (up to 13 vol.%) of the investigated microgabbros, as assessed by microscope observations and consistent with calculated normative compositions (Supplementary Data 2).
Fluid inclusions
Primary FIs (i.e., those entrapped during crystallization of the host mineral) are abundant within late-magmatic and hydrothermal quartz (Fig. 3; Supplementary Note 2). These FIs are either vapour- or liquid-rich, measure 1–50 μm in their maximum dimension and are randomly distributed within all investigated crystals (Fig. 3a). In the coexisting vapour- and liquid-rich FIs hosted by late-magmatic quartz, the liquid phase is H2O and the vapour phase is CH4, which has a measured density of about 0.02 g/cm3 (Figs. 3b, c, 4; Supplementary Data 3; Source Data 1, 2). Halite (NaCl) is the only identified solid phase occurring as cubic crystals in some liquid-rich FIs, which have a salinity of > 26 wt.%, calculated after ref. 29. In the mainly liquid-rich FIs hosted by hydrothermal quartz, both liquid and vapour phases are H2O.

Raman spectrum of gaseous CH4 and liquid H2O within a liquid-rich FI (sample RP125), acquired at ambient temperature by confocal Raman microspectroscopy (uppermost Raman spectrum). Gaseous CH4 corresponds to the intense and sharp Raman band at ca. 2917 cm−1, and liquid H2O corresponds to the broad Raman band ranging from 3000 to 3700 cm−1. Raman spectrum of methane clathrate, water ice and hydrohalite (solid phases formed in the H2O–NaCl–CH4 system under freezing conditions) within a liquid-rich FI (sample RP116), acquired at −190 °C by confocal Raman microspectroscopy combined with microthermometry (lowermost Raman spectrum). Methane clathrate corresponds to the sharp Raman band at ca. 2904 cm−1 and to the weak Raman band at ca. 2916 cm−1, water ice corresponds to the intense Raman band at ca. 3100 cm−1 and to the weak Raman band at ca. 3223 cm−1, and hydrohalite corresponds to several Raman bands at ca. 3303, 3325, 3405, 3423, 3436 and 3540 cm−1.
Geothermobarometry
Quartz crystallization and FI entrapment occurred during the late-magmatic to hydrothermal stages (Supplementary Note 3). The crystallization temperatures of quartz, calculated from its titanium content30, are mainly in the range 700–600 °C for the late-magmatic quartz, with generally higher values at crystal cores, and mainly in the range 500–400 °C for the hydrothermal quartz (Fig. 5; Supplementary Fig. 5; Supplementary Data 4, 5).

a Calculated temperature ranges of both late-magmatic and hydrothermal quartz (for all the analysed samples) in box-and-whisker plots. In box-and-whisker plots, the middle line of the box indicates the median, the bottom and top edges of the box show the lower and upper quartiles respectively, the whiskers extend to the minimum and maximum data that are not outliers, and the outliers are plotted individually. For each temperature datum, the average total uncertainty is ±40 °C for late-magmatic quartz and ±32 °C for hydrothermal quartz. Note that the data displaying below-detection-limit values or > 15 % analytical errors for Ti or displaying > 5000 ppm values for Al are not plotted. b Temperature transect (α–β; panels c and d) of late-magmatic quartz crystals (sample RP136), based on TitaniQ thermobarometry30. For each calculated temperature, the error bar represents the total uncertainty, which is given by the sum of internal (depending on analytical measurements) and external (depending on both crystallization pressure and TiO2 activity relative to rutile saturation) uncertainties. c, d Backscattered-electron (BSE) and greyscale cathodoluminescence (CL) images of the analysed late-magmatic quartz (Qz) crystals, displaying normal growth zonation, with high-Ti (CL-brighter) cores and low-Ti (CL-darker) rims.
Magma–sediment interaction
Primitive CAMP basalts of both low-Ti (TiO2 < 2.0 wt.%) and high-Ti (TiO2 > 2.0 wt.%) types contain olivine31,32 and are thus not compatible with the presence of quartz. Hence, significant enrichment in SiO2 is required to achieve quartz saturation and crystallization in the Amazonian CAMP sills15 (Fig. 6a, b). Starting from typical low-Ti33 and high-Ti31 CAMP basalts with 0.5 wt.% H2O12 (Supplementary Data 2), Rhyolite-MELTS modelling34 (see Methods) suggests that the observed, relatively abundant quartz crystallization (ca. 5 vol.%) requires at least 10 wt.% assimilation of silica-rich rocks such as shales (Supplementary Data 6). Shales, which are present in the intruded Paleozoic sedimentary sequence (Fig. 1; Supplementary Fig. 1), would dehydrate from ca. 500 °C and melt from ca. 700 °C35,36, and their assimilation would also enhance zircon saturation, consistently with the relative abundance of zircon among the late-magmatic minerals of quartz-bearing basaltic rocks in the Amazonian CAMP sills11,19. Crustal assimilation is particularly relevant for the high-Ti magmas (> 50 % of the investigated samples; Supplementary Data 2), which would not crystallize any magmatic quartz without contamination (Supplementary Data 6). According to our petrologic modelling, the contaminated basaltic magma contains relatively large amounts of H2O (ca. 0.9 wt.%), suggesting that the entire volume of Amazonian CAMP sills released about 2.7 × 104 Gt H2O.

a, b Magmatic crystallization started in rising magmas within the deep transcrustal magmatic system, below the basins, and continued within the sills. One anatectic melt phase and two immiscible fluid phases from the heated organic-rich sedimentary rocks migrated within the sills during the late-magmatic crystallization. Hydrothermal alteration and vein formation occurred after the magmatic crystallization. c Coexisting fluid phases, occasionally entrapped as FIs mainly in crystallizing quartz, are shown at entrapment conditions of 550 °C and 50 MPa (modified after ref. 37). Two immiscible fluid phases, dominated by H2O+NaCl and H2O+CH4 respectively (small red stars), coexist along tie-lines for the average composition of a bulk fluid system (large red star) in the highlighted 2-phase field. When the FIs cooled to ambient temperature, CH4 bubbles nucleated and NaCl crystals precipitated.
Methane and halite in aqueous fluids
In addition to H2O, the amounts of CH4 and NaCl in the Amazonian CAMP sills during late-magmatic quartz crystallization are estimated using our microscopy and microspectroscopy data (see Methods). These data combined with experimental phase equilibria in the ternary H2O–NaCl–CH4 system37, at a pressure and temperature consistent with those of late-magmatic quartz crystallization, suggest that in the Amazonian CAMP sills the H2O/CH4 molar ratio was about 3.3 and the H2O/NaCl molar ratio was about 2.6 (Fig. 6c). Therefore, the total amount of CH4 (7.2 × 103 Gt; 4.5 × 1017 mol) and NaCl (3.4 × 104 Gt; 5.8 × 1017 mol) for all Amazonian CAMP sills during late-magmatic quartz crystallization hinges on the estimated amount of H2O. Potentially, similar or even larger amounts of fluid phases were also mobilized from the intruded sedimentary sequence before the crystallization of late-magmatic quartz, but were not preserved by FIs in other mineral phases within the sills.
Discussion
The rapid and pulsed intrusion of about 1 × 106 km3 CAMP basaltic magmas in Amazonia likely produced widespread and prolonged heating within the shallow sedimentary basins11,19. The magmatic interaction with sedimentary rocks such as shales and evaporites may account for the excess chlorine observed in the analysed FIs and biotites from Amazonian CAMP sills28. The formation of thermogenic CH4 requires relatively low temperatures, from ca. 60 to 300 °C38, and may occur within the metamorphic aureoles, which extend up to 250% of sills thickness39. Massive amounts of fluids in the H2O–NaCl–CH4 system were likely released during heating of the intruded Paleozoic sedimentary sequence up to its melting temperature (ca. 700 °C; Fig. 6a). Favoured by temperature and pressure gradients, H2O–NaCl–CH4 fluids migrated after being generated through devolatilization reactions and organic cracking within the organic-rich sedimentary basins28,39,40. Furthermore, CH4 already present within Paleozoic reservoir rocks at 201 Ma was remobilized due to CAMP intrusions, which likely induced additional maturation of organic matter, forming new CH4 within the Amazonian sedimentary basins17,22.
A dominant deep magmatic origin of CH4 is not likely since its maximum solubility in basaltic magmas at low oxygen fugacity is very low (< 0.1 wt.%)41 and melt inclusions from CAMP basaltic rocks indicate exsolution of magmatic carbon mainly as CO2 at greater depths, before the magmas reached the shallow sedimentary basins12. Similarly, the lack of graphite and native iron in the analysed microgabbros and the presence of H2O–NaCl–CH4 fluids in equilibrium with these rocks rule out direct magmatic assimilation of organic matter to form CH442.
From source or reservoir rocks, CH4-bearing fluids migrated into the crystallizing gabbroic sills, with records of their presence entrapped by FIs, before potentially reaching the atmosphere13. Decompression and cracking induced by contraction of the cooling magma bodies may have promoted the migration of fluids towards the sills28, favouring their partial entrapment within the crystallizing quartz during the late-magmatic stage, consistent with CH4 stability up to 1200 °C in both C–O–H and C–O–H–S systems42,43.
The large amount of CH4 (4.5 × 1017 mol) estimated from the FI analysis is consistent with the emissions of carbon (2.0 × 1018 mol) modelled for the contact metamorphism induced by CAMP intrusions within the Amazonian basins13. The release of carbon species due to thermogenic degassing from carbon-rich sedimentary sequences is also suggested by the presence of similar CH4-bearing FIs and hydrothermal vents from the North Atlantic LIP6,44, likely responsible for the Palaeocene–Eocene Thermal Maximum45, and was thermodynamically modelled for several LIPs46,47. A modern analogue of the Amazonian CAMP magma–sediment interaction, even if on a much smaller scale, is represented by the Lusi mud-eruption (NE Java, Indonesia). Lusi is a sediment-hosted hydrothermal system, where shallow magmatic intrusions within a sedimentary sequence affect source rocks and/or hydrocarbon reservoirs, degassing large amounts of both volcanic and thermogenic carbon species into the atmosphere48.
The direct evidence for magma–sediment interaction recorded by FIs of the investigated CAMP sills indicates that large amounts of CH4 were generated or remobilized due to the end-Triassic magmatism. A minimal part of this volatile load was entrapped within quartz-hosted FIs. The majority was either reintroduced into the sedimentary host rocks, forming exploitable CH4 reservoirs in structural and lithological traps17, or discharged into the surface system during CAMP activity11, possibly through hydrothermal vents and pipes like those detected in other LIPs6,49. Methane is among the highest impact greenhouse gases50 and chlorine, which is mostly dissolved in H2O associated with CH4, can induce ozone depletion51. In addition to the CO2 exsolved from CAMP magmas in the deep transcrustal magmatic system12, the CH4 released from the shallow sedimentary basins likely contributed to the end-Triassic global warming of up to 4 °C52, forcing the global climate changes and triggering the mass extinction. The injection of sedimentary organic CH4, displaying a 13C-depleted isotopic signature, into the exogenic carbon cycle may have also produced the large negative carbon-isotope shifts in the sedimentary record14. Our observations highlight the role of LIP magmatism and its interactions with rocks and fluids within sedimentary basins in controlling global climate changes and the evolution of life on Earth, as well as in enhancing hydrocarbon maturation and remobilization.
Methods
Sample selection and preparation
The microgabbros analysed for this study are from CAMP sills intruded in the Amazonas Basin (Fig. 1). Mainly based on the variability in mineralogical composition and hydrothermal alteration, 20 out of a suite of 50 intrusive rock samples were selected from the outcrops near the villages of Monte Alegre, Medicilândia and Placas in the State of Pará, northern Brazil (Fig. 1; Supplementary Fig. 1; Supplementary Data 1). These outcrops mainly occur along road cuts and in quarries, and are generally scattered and not continuous, due to the flat topography and dense vegetation. Within the Paleozoic sequence of the Amazonas Basin, the sills sampled at Monte Alegre, north of the Amazonas River, intruded both the Curuá Group (Devonian–Carboniferous) and the Tapajós Group (Carboniferous)23. The sill sampled at Medicilândia, south of the Amazonas River, intruded between the Urupadi Group (Devonian) and the Curuá Group (Devonian–Carboniferous)23. Lastly, the sill sampled at Placas, south of the Amazonas River, intruded the Tapajós Group (Carboniferous)23. From the screened microgabbros, the 10 most representative FI-bearing samples were characterized in detail by optical microscopy and confocal Raman microspectroscopy (Supplementary Data 1). Glue-free double-polished thick (about 100 μm) sections were employed for in situ confocal Raman microspectroscopy, in order to avoid contamination and signal interferences from any artificial carbon-bearing compounds.
X-ray fluorescence (XRF) spectrometry
This analytical technique was employed to determine the whole-rock chemical composition (major elements) of the most representative FI-bearing samples (Supplementary Data 1). The analyses were conducted at the Department of Geosciences, University of Padova, using a WDS Philips PW2400 sequential spectrometer. The analytical uncertainty is < 3% for all major elements.
Microthermometry
This analytical technique was employed to investigate the unexposed liquid-rich FIs through the phase transition temperatures, mainly of last-melting and homogenization, and to determine additional phases, in combination with confocal Raman microspectroscopy, under freezing conditions (e.g., hydrohalite; Supplementary Data 1). The analyses were conducted at the Lithosphere Fluid Research Lab and at the Research and Industrial Relations Center of the Faculty of Science, Eötvös Loránd University of Budapest, using a LINKAM THMS600 heating–freezing stage, mounted on a NIKON Eclipse E600 petrographic microscope. The investigated FIs were cooled down to −190 °C using liquid N2, and then heated until homogenization was reached, potentially up to 600 °C. A NIKON × 50 objective was used to observe the phase transitions.
Confocal Raman microspectroscopy
This analytical technique was employed to characterize the fluid and solid phases of unexposed FIs (Supplementary Data 1). The analyses were conducted at the Research and Industrial Relations Center of the Faculty of Science, Eötvös Loránd University of Budapest, using a HORIBA Jobin Yvon LabRAM HR 800 Raman microspectrometer. Both spot and areal analyses were carried out below the sample surface for all the phases within unexposed FIs. Spot Raman analysis allowed to acquire the spectra of fluid and solid phases, and to characterize them through their spectral features. Areal Raman analysis (i.e., Raman mapping) allowed to reconstruct and image the spatial distribution of the same phases within unexposed FIs. A frequency doubled Nd–YAG green laser with a 532 nm excitation wavelength was employed, displaying 120 mW at the source and 23 mW on the sample surface, and a He–Ne red laser with a 633 nm excitation wavelength was employed exclusively to analyse H2O within calcite-hosted FIs. An OLYMPUS × 100 (N.A. = 0.9) objective was used to focus the laser on the analysed sites. Raman spectra acquisition was conducted in both single- and multi-window settings, from ambient temperature down to −190 °C, combined with a LINKAM THMS600 microthermometric stage. For spot Raman analysis, a 100 μm confocal hole, a 1800 grooves/mm optical grating, 2–3 accumulations and a 30–60 s exposition time were employed. For areal Raman analysis, a 100 μm confocal hole, a 600 grooves/mm optical grating, 1–20 accumulations and a 1–30 s exposition time were employed. The investigated spectra range from 70 to 4000 cm−1, depending on the spectral region of interest for each analysed phase. The spectral resolution of measurements varies from 0.8 to 3.0 cm−1 for the spot spectra, and from 2.4 to 3.0 cm−1 for the maps. Spot Raman data were processed through LabSpec 5 software, and areal Raman data were processed through both LabSpec 5 and LabSpec 6 softwares.
Electron microprobe (EMP) analysis
This analytical technique was employed to measure the Ti and Al concentrations in both late-magmatic and hydrothermal quartz (Supplementary Data 1). The analyses were conducted at the Department of Mineral Sciences, Smithsonian Institution, Washington DC, using a JEOL 8530F hyperprobe. The beam current was set at 200 nA, the accelerating voltage at 15 kV, and the spot diameter at 10 μm. The Ti concentrations were measured using 3 PETL spectrometers, and the Al concentrations using 2 TAP spectrometers, for 300 s on the peak and 15 s on the background, yielding a detection limit of 6–8 ppm for Ti and 4–6 ppm for Al. As standards, ilmenite was used for Ti and corundum for Al.
Cathodoluminescence (CL) analysis
This analytical technique was employed to distinguish late-magmatic and hydrothermal quartz (Supplementary Data 1). Colour CL images were acquired at the C.N.R., Institute of Geosciences and Georesources, Padova, using a Cold Cathode Luminescence 8200 mk3. The beam current was set at 220 µA and the accelerating voltage at 20 kV. Greyscale CL images were acquired at the Department of Mineral Sciences, Smithsonian Institution, Washington DC, using a JEOL panchromatic cathodoluminescence detector. The beam current was set at 20 nA and the accelerating voltage at 15 kV.
Petrologic modelling of magma crystallization
The equilibrium crystallization of CAMP sills was modelled in open and closed system, using Rhyolite-MELTS (code version 1.0.2)34. We started from typical low-Ti (sample AN133)33 and high-Ti (sample M13)31 CAMP basalts with 0.5 wt.% H2O12 in the initial magma, and considered an average shale with 5.0 wt.% H2O53 as the assimilated crustal material (Supplementary Data 2, 6). The initial temperature of the assimilated shale was set at 100 °C, and its amount was set at 10 wt.% for low-Ti magmas and at 20 wt.% for high-Ti magmas. The oxygen fugacity was set at the FMQ buffer, and the pressure generally at 50 MPa, corresponding to the average intrusion depth of the investigated CAMP sills. Our petrologic modelling suggests that at least 10 wt.% crustal assimilation is necessary to reproduce the observed amount of quartz within the Amazonian CAMP sills. Furthermore, high-Ti CAMP basalts do not reach quartz saturation without shale assimilation and up to 20 wt.% crustal assimilation is necessary to reproduce the same observed amount of quartz.
Quantification of H2O, NaCl and CH4
During the late-magmatic stage, at ca. 900–550 °C and 50 MPa, the system was likely formed by at least one exsolved fluid phase (H2O–NaCl–CH4) coexisting with a silicate melt, from which quartz, alkali feldspar and hydrous mineral phases, such as amphibole and biotite, crystallized. Within late-magmatic quartz, liquid- and vapour-rich FIs are associated together without any distinctive distribution between crystal cores, mantles and rims, and thus constitute a peculiar FI assemblage, that is typical of shallow, silicic, magmatic/hydrothermal environments54. This FI assemblage reveals that two different, immiscible fluid phases were present during the late-magmatic stage, and were occasionally trapped into crystallizing quartz as FIs. In respect of Roedder’s rules54, the liquid-rich FIs likely trapped a single homogeneous H2O+NaCl-dominated fluid phase, containing minor amounts of CH4 (i.e., high-density fluid phase), whereas the vapour-rich FIs likely trapped a single homogeneous H2O+CH4-dominated fluid phase, containing minor amounts of NaCl (i.e., low-density fluid phase). Considering the complex H2O–NaCl–CH4 ternary system (Fig. 6c), broad regions of immiscibility are present at pressures and temperatures consistent with the entrapment conditions of the investigated FIs37,55,56,57. For any bulk composition that lies in the largest 2-phase field at these conditions, one high-density fluid phase and one low-density fluid phase coexist55,56. The high-density fluid phase is enriched in H2O and NaCl (i.e., H2O+NaCl-dominated fluid) and corresponds to our 2 or 3 phase-bearing, liquid-rich FIs at ambient temperature. The low-density fluid phase is enriched in H2O and CH4 (i.e., H2O+CH4-dominated fluid) and corresponds to our vapour-rich FIs at ambient temperature. The presence of NaCl and the low pressure of crystallization promote fluid immiscibility, and the 1-phase field, depending on both pressure and temperature, is generally very restricted37.
The FIs hosted in late-magmatic quartz may contain H2O+NaCl+CH4, H2O+CH4 or pure CH4. Therefore, CH4 is the only component always present during the late-magmatic stage. However, this variability in the chemical composition of the investigated FIs reveals a continuous compositional evolution of the fluid phases throughout the whole temperature range for the crystallization of late-magmatic quartz. For instance, the addition of further exsolved fluids from rising magmatic recharges, as well as of brines from intruded sedimentary rocks, and the crystallization of hydrous mineral phases along with quartz may progressively modify the composition of the complex H2O–NaCl–CH4 system.
The total volume of ca. 1.0 × 106 km3 for CAMP sills in Amazonia16 corresponds to a total mass of ca. 3.0 × 106 Gt, assuming a density of 3.0 g/cm3 for gabbroic rocks58. Our petrologic modelling suggests that at least 2.7 × 105 Gt of shales may have been assimilated during the emplacement of the Amazonian CAMP sills. According to our Rhyolite-MELTS modelling, the H2O dissolved in the melt at quartz saturation is about 2.2 wt.%, consistently with experimental data on granitic systems59,60, and the H2O exsolved from the crystallizing sills is about 0.9 wt.%. Therefore, the total mass of H2O released from the Amazonian CAMP sills is about 2.7 × 104 Gt, equivalent to 1.5 × 1018 mol. However, the higher amount of hydrous mineral phases and the lower crystallization temperatures of quartz in the investigated microgabbros compared to our modelling results, suggest that the late-magmatic stage was likely richer in H2O, thus not derived exclusively from the magmatic (i.e., juvenile and assimilated) component considered in our petrologic modelling, but possibly also from sedimentary and metamorphic components originated in the host sedimentary sequence. Similarly, the occurrence of pure CH4-bearing FIs hosted in late-magmatic quartz likely represents CH4 fluxes from the metamorphic aureoles into the crystallizing sills.
Experimental phase equilibria in the ternary H2O–NaCl–CH4 system at 550 °C and 50 MPa37 indicate that the investigated FI assemblage represents a fluid system in a 2-phase field for pressure and temperature consistent with the entrapment conditions (Fig. 6c). In order to quantify the amount of CH4, we observed that within the vapour-rich FIs at ambient temperature the gaseous CH4 occupies most commonly about 95 vol.% of the whole FI and coexists with liquid H2O displaying ca. 10 wt.% salinity. Given an average density of ca. 0.02 g/cm3 for gaseous CH4 and of ca. 1.07 g/cm3 for liquid H2O with 10 wt.% salinity61, the H2O/CH4 molar ratio is generally about 2.2 in the vapour-rich FIs. The bulk fluid system is considered to have the average composition between those of the coexisting low- and high-density fluid phases, forming the vapour- and liquid-rich FIs respectively (Fig. 6c). Therefore, the H2O/CH4 molar ratio was about 3.3 in the bulk fluid system, and the total amount of carbon, in the form of CH4, within the sills during late-magmatic quartz crystallization was approximately 4.5 × 1017 mol (7.2 × 103 Gt CH4). Similarly, the H2O/NaCl molar ratio was about 2.6 in the bulk fluid system, and the total amount of chlorine, in the form of NaCl, within the sills during late-magmatic quartz crystallization was approximately 5.8 × 1017 mol (3.4 × 104 Gt NaCl).
Data availability
All data generated in this study are provided in the Supplementary Information (Supplementary Notes 1–3, Supplementary Figs. 1–5 and Supplementary References), in the Supplementary Data 1–6 and in the Source Data 1, 2. Source data are provided with this paper.
References
Wignall, P. B. The Worst of Times: How Life on Earth Survived Eighty Million Years of Extinctions (Princeton University Press, 2015).
Bond, D. P. G. & Grasby, S. E. On the causes of mass extinctions. Palaeogeogr. Palaeoclimatol. Palaeoecol. 478, 3–29 (2017).
Ernst, R. E. & Youbi, N. How Large Igneous Provinces affect global climate, sometimes cause mass extinctions, and represent natural markers in the geological record. Palaeogeogr. Palaeoclimatol. Palaeoecol. 478, 30–52 (2017).
Ganino, C. & Arndt, N. T. Climate changes caused by degassing of sediments during the emplacement of large igneous provinces. Geology 37, 323–326 (2009).
Black, B. A. & Gibson, S. A. Deep Carbon and the Life Cycle of Large Igneous Provinces. Elements 15, 319–324 (2019).
Svensen, H. et al. Release of methane from a volcanic basin as a mechanism for initial Eocene global warming. Nature 429, 542–545 (2004).
Schobben, M., van de Schootbrugge, B. & Wignall, P. B. Interpreting the Carbon Isotope Record of Mass Extinctions. Elements 15, 331–337 (2019).
Marzoli, A. et al. Extensive 200-million-year-old continental flood basalts of the Central Atlantic Magmatic Province. Science 284, 5414 (1999).
Marzoli, A. et al. Synchrony of the Central Atlantic magmatic province and the Triassic–Jurassic boundary climatic and biotic crisis. Geology 32, 973–976 (2004).
Schoene, B., Guex, J., Bartolini, A., Schaltegger, U. & Blackburn, T. J. Correlating the end-Triassic mass extinction and flood basalt volcanism at the 100 ka level. Geology 38, 387–390 (2010).
Davies, J. H. F. L. et al. End-Triassic mass extinction started by intrusive CAMP activity. Nat. Commun. 8, 15596 (2017).
Capriolo, M. et al. Deep CO2 in the end-Triassic Central Atlantic Magmatic Province. Nat. Commun. 11, 1670 (2020).
Heimdal, T. H. et al. Large-scale sill emplacement in Brazil as a trigger for the end-Triassic crisis. Sci. Rep. 8, 141 (2018).
Heimdal, T. H., Jones, M. T. & Svensen, H. H. Thermogenic carbon release from the Central Atlantic magmatic province caused major end-Triassic carbon cycle perturbations. Proc. Natl Acad. Sci. USA 117, 11968–11974 (2020).
De Min, A. et al. in The Central Atlantic Magmatic Province. Insights from fragments of Pangea Vol. 136 (eds Hames, W. E., McHone, J. G., Renne, P. R. & Ruppel, C.) (AGU Geophysical Monograph, 2003).
Marzoli, A. et al. in The Late Triassic World. Earth in a Time of Transition Vol. 46 (ed. Tanner, L. H.) (Springer, 2018).
Gonzaga, F. G., Gonçalves, F. T. T. & Coutinho, L. F. C. in Petroleum Systems of South Atlantic Margins Vol. 73 (eds Mello, M. R. & Katz, B. J.) (AAPG Memoir, 2000).
Caputo, M. V. & Soares, E. A. A. Eustatic and tectonic change effects in the reversion of the transcontinental Amazon River drainage system. Braz. J. Geol. 46, 301–328 (2016).
Davies, J. H. F. L. et al. Zircon petrochronology in large igneous provinces reveals upper crustal contamination processes: new U–Pb ages, Hf and O isotopes, and trace elements from the Central Atlantic Magmatic Province (CAMP). Contrib. Mineral. Petrol. 176, 9 (2021).
Lindström, S. et al. Tracing volcanic emissions from the Central Atlantic Magmatic Province in the sedimentary record. Earth-Sci. Rev. 212, 103444 (2021).
Wanderley Filho, J. R., Travassos, W. A. S. & Alves, D. B. O diabásio nas bacias paleozóicas amazônicas - herói ou vilão? B. Geoci. Petrobras 14, 177–184 (2006).
Milani, E. J. & Zalán, P. V. An outline of the geology and petroleum systems of the Paleozoic interior basins of South America. Episodes 22, 199–205 (1999).
Vasquez, M. L., Sousa, C. S. & Carvalho, J. M. A. Mapa Geológico do Estado do Pará (1:1.000.000). Programa Geologia do Brasil (2008).
Silva, M., Mohriak, W., Holanda, W., Reis, D. & Ferreira, J. Analysis of the hydrocarbon maturation windows of the Amazon Basin. 14th International Congress of the Brazilian Geophysical Society (2015).
Milesi, V., Prinzhofer, A., Guyot, F., Benedetti, M. & Rodrigues, R. Contribution of siderite–water interaction for the unconventional generation of hydrocarbon gases in the Solimões basin, north-west Brazil. Mar. Pet. Geol. 71, 168–182 (2016).
Costa, J., Gouvêa Vasconcellos, E. M., de Mesquita Barros, C. E., Cury, L. F. & Virmond Juk, K. F. Petrologia e geoquímica da soleira de Medicilândia, diabásio Penatecaua, PA. Rev. Bras. Geociênc. 42, 754–771 (2012).
Silva, E. F., Pinto, M. B., Peregovich, B. G. & Brenner, W. W. Petrologia do diabásio Penatecaua, soleira de Rurópolis, Pará, Brasil. Comun. Geol. 101, 213–216 (2014).
Heimdal, T. H. et al. Evidence for magma–evaporite interactions during the emplacement of the Central Atlantic Magmatic Province (CAMP) in Brazil. Earth Planet. Sci. Lett. 506, 476–492 (2019).
Cohen-Adad, R. & Lorimer, J. W. (eds.) Alkali Metal and Ammonium Chlorides in Water and Heavy Water (Binary Systems) (Pergamon Press, 1991).
Zhang, C. et al. Ti-in-quartz thermobarometry and TiO2 solubility in rhyolitic melts: new experiments and parametrization. Earth Planet. Sci. Lett. 538, 116213 (2020).
Merle, R. et al. 40Ar/39Ar ages and Sr–Nd–Pb–Os geochemistry of CAMP tholeiites from Western Maranhão basin (NE Brazil). Lithos 122, 137–151 (2011).
Callegaro, S. et al. Upper and lower crust recycling in the source of CAMP basaltic dykes from southeastern North America. Earth Planet. Sci. Lett. 376, 186–199 (2013).
Marzoli, A. et al. The Central Atlantic Magmatic Province (CAMP) in Morocco. J. Petrol. 60, 945–996 (2019).
Gualda, G. A. R., Ghiorso, M. S., Lemons, R. V. & Carley, T. L. Rhyolite-MELTS: a modified calibration of MELTS optimized for silica-rich, fluid-bearing magmatic systems. J. Petrol. 53, 875–890 (2012).
Wyllie, P. J. & Tuttle, O. F. Hydrothermal Melting of Shales. Geol. Mag. 98, 56–66 (1961).
Clark, B. H. & Peacor, D. R. Pyrometamorphism and partial melting of shales during combustion metamorphism: mineralogical, textural, and chemical effects. Contrib. Mineral. Petrol. 112, 558–568 (1992).
Liebscher, A. Experimental studies in model fluid systems. Rev. Mineral. Geochem. 65, 15–47 (2007).
Stolper, D. A. et al. in From Source to Seep: Geochemical Applications in Hydrocarbon Systems Vol. 468 (eds Lawson, M., Formolo, M. J. & Eiler, J. M.) (Geol. Soc. Lond., 2017).
Aarnes, I., Svensen, H., Connolly, J. A. D. & Podladchikov, Y. Y. How contact metamorphism can trigger global climate changes: Modeling gas generation around igneous sills in sedimentary basins. Geochim. Cosmochim. Acta 74, 7179–7195 (2010).
Svensen, H. et al. Sandstone dikes in dolerite sills: Evidence for high-pressure gradients and sediment mobilization during solidification of magmatic sheet intrusions in sedimentary basins. Geosphere 6, 211–224 (2010).
Ardia, P., Hirschmann, M. M., Withers, A. C. & Stanley, B. D. Solubility of CH4 in a synthetic basaltic melt, with applications to atmosphere–magma ocean–core partitioning of volatiles and to the evolution of the Martian atmosphere. Geochim. Cosmochim. Acta 114, 52–71 (2013).
Iacono-Marziano, G. et al. Extremely reducing conditions reached during basaltic intrusion in organic matter-bearing sediments. Earth Planet. Sci. Lett. 357–358, 319–326 (2012).
Kueter, N., Schmidt, M. W., Lilley, M. D. & Bernasconi, S. M. Experimental determination of equilibrium CH4–CO2–CO carbon isotope fractionation factors (300–1200 °C). Earth Planet. Sci. Lett. 506, 64–75 (2019).
Larsen, R. B., Brooks, C. K. & Bird, D. K. Methane-bearing, aqueous, saline solutions in the Skaergaard intrusion, east Greenland. Contrib. Mineral. Petrol. 112, 428–437 (1992).
Jones, S. M., Hoggett, M., Greene, S. E. & Dunkley Jones, T. Large Igneous Province thermogenic greenhouse gas flux could have initiated Paleocene–Eocene Thermal Maximum climate change. Nat. Commun. 10, 5547 (2019).
Iacono-Marziano, G. et al. Gas emissions due to magma–sediment interactions during flood magmatism at the Siberian Traps: gas dispersion and environmental consequences. Earth Planet. Sci. Lett. 357–358, 308–318 (2012).
Aarnes, I., Planke, S., Trulsvik, M. & Svensen, H. Contact metamorphism and thermogenic gas generation in the Vøring and Møre basins, offshore Norway, during the Paleocene–Eocene thermal maximum. J. Geol. Soc. Lond. 172, 588–598 (2015).
Mazzini, A. 10 years of Lusi eruption: Lessons learned from multidisciplinary studies (LUSI LAB). Mar. Pet. Geol. 90, 1–9 (2018).
Svensen, H. et al. Siberian gas venting and the end-Permian environmental crisis. Earth Planet. Sci. Lett. 277, 490–500 (2009).
Montzka, S. A., Dlugokencky, E. J. & Butler, J. H. Non-CO2 greenhouse gases and climate change. Nature 476, 43–50 (2011).
Black, B. A., Lamarque, J.-F., Shields, C. A., Elkins-Tanton, L. T. & Kiehl, J. T. Acid rain and ozone depletion from pulsed Siberian Traps magmatism. Geology 42, 67–70 (2014).
Landwehrs, J. P., Feulner, G., Hofmann, M. & Petri, S. Climatic fluctuations modeled for carbon and sulfur emissions from end-Triassic volcanism. Earth Planet. Sci. Lett. 537, 116174 (2020).
Ptáček, M. P., Dauphas, N. & Greber, N. D. Chemical evolution of the continental crust from a data-driven inversion of terrigenous sediment compositions. Earth Planet. Sci. Lett. 539, 116090 (2020).
Bodnar, R. J. in Fluid Inclusions: Analysis and Interpretation Vol. 32 (eds Samson, I., Anderson, A. & Marshall, D.) (Mineral. Assoc. Canada, 2003).
Lamb, W. M., Popp, R. K. & Boockoff, L. A. The determination of phase relations in the CH4–H2O–NaCl system at 1 kbar, 400 to 600 °C using synthetic fluid inclusions. Geochim. Cosmochim. Acta 60, 1885–1897 (1996).
Lamb, W. M., McShane, C. J. & Popp, R. K. Phase relations in the CH4–H2O–NaCl system at 2 kbar, 300 to 600 °C as determined using synthetic fluid inclusions. Geochim. Cosmochim. Acta 66, 3971–3986 (2002).
Duan, Z., Møller, N. & Weare, J. H. Equations of State for the NaCl–H2O–CH4 System and the NaCl–H2O–CO2–CH4 System: Phase Equilibria and Volumetric Properties above 573 K. Geochim. Cosmochim. Acta 67, 671–680 (2003).
Emerson, D. W. Notes on Mass Properties of Rocks – Density, Porosity, Permeability. Explor. Geophys. 21, 209–216 (1990).
Johannes, W. & Holtz, F. Petrogenesis and Experimental Petrology of Granitic Rocks Vol. 22 (Springer, 1996).
Baker, D. R. & Alletti, M. Fluid saturation and volatile partitioning between melts and hydrous fluids in crustal magmatic systems: The contribution of experimental measurements and solubility models. Earth-Sci. Rev. 114, 298–324 (2012).
Simion, A. I., Grigoraş, C.-G., Roşu, A.-M. & Gavrilӑ, L. Mathematical modelling of density and viscosity of NaCl aqueous solutions. J. Agroaliment. Process. Technol. 21, 41–52 (2015).
Acknowledgements
This study was supported by the following collaborative research projects: PRIN 20178LPCP (Italy) to A.M. and A.D.M., TKP2020-IKA-05 (Hungarian Ministry of Human Capacities) to L.E.A. and Cs.Sz., BART_SID19_01 (Italy) to O.B., Young Research Talents Project 301096 MAPLES (Research Council of Norway) to S.C., and NERC Large Grant NE/N018559/1 (UK) to R.J.N. The authors thank A. Sansone (University of Padova) for sample selection, L. Tauro (University of Padova) for sample preparation, and G. Bellieni for valuable assistance during sampling.
Author information
Authors and Affiliations
Contributions
M.C. and A.M. devised the project. A.M., M.E., E.M.G.V. and A.D.M. carried out the field work. M.C., A.M., L.E.A. and M.R.A. collected the analytical data. M.C., A.M., L.E.A., M.R.A., O.B., S.C., J.D.C., M.E., E.M.G.V., A.D.M., R.J.N. and Cs.Sz. contributed to data interpretation and writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Frances Deegan, Sofie Lindström and Lawrence Percival for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Capriolo, M., Marzoli, A., Aradi, L.E. et al. Massive methane fluxing from magma–sediment interaction in the end-Triassic Central Atlantic Magmatic Province. Nat Commun 12, 5534 (2021). https://doi.org/10.1038/s41467-021-25510-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25510-w
This article is cited by
-
New U–Pb geochronology for the Central Atlantic Magmatic Province, critical reevaluation of high-precision ages and their impact on the end-Triassic extinction event
Scientific Reports (2023)
-
Butterfly effect of shallow-ocean deoxygenation on past marine biodiversity
Nature Geoscience (2023)
-
Mercury evidence for combustion of organic-rich sediments during the end-Triassic crisis
Nature Communications (2022)
-
High temperature methane emissions from Large Igneous Provinces as contributors to late Permian mass extinctions
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
