
Abstract
Pulsatile hemodynamics are associated with brain small perivascular spaces (SPVS). It is unknown whether the stiffness of intermediary arteries connecting the aorta and brain modifies this association. Participants from the Northern Manhattan Study were assessed for SPVS (defined as ≤3 mm T1 voids) and white matter hyperintensity volume (WMH) using MRI. Middle (MCA) and anterior cerebral arterial (ACA) diameters (measured on time-of-flight MRA) and CCA strain (assessed by ultrasound) were used as surrogates of stiffness. Brachial and aortic pulse pressure (PP) and aortic augmentation index (Aix, assessed by applanation tonometry) were used as markers of pulsatility. We tested whether stiffness in intermediary arteries modifies the association between extracranial pulsatility with SPVS and WMH. We found that among 941 participants (mean age 71 ± 9 years, 60% women, 66% Hispanic), the right MCA/ACA diameter was associated with right anterior SPVS (B = 0.177, P = 0.002). Brachial PP was associated with right anterior SPVS (B = 0.003, P = 0.02), and the effect size was bigger with right MCA/ACA diameter in the upper tertile (P = 0.001 for the interaction). The association between right CCA strain and ipsilateral SPVS was modified by MCA/ACA diameter, with the largest effect size in those with ipsilateral MCA/ACA diameter in the upper tertile (P = 0.001 for the interaction). Similar dose-effects and statistical interactions were replicated using aortic AIx or aortic PP. We found no evidence of effect modification between pulsatile measures and WMH by stiffness measures. In summary, pulsatile hemodynamics relate to brain SPVS, and the association is the strongest among individuals with dilated brain arteries.
Similar content being viewed by others


Introduction
Arterial stiffness is inherent to aging, and it has been associated with increased risk of cardiac and cerebrovascular events [1]. The aorta exhibits the earliest pathological correlates of stiffness and subsequently, it progresses centrifugally [2,3,4]. As aortic arterial stiffness progresses, the pulse wave velocity increases [5]. The pulsatility that reaches end organs as the pulse wave propagates through the vascular system depends on the diameter and geometrical configuration of the large arteries distal to the aorta [6, 7], their residual elasticity [8], and the resistance to flow (determined primarily by the arteriolar density of the organ supplied) [9, 10]. Organs with low resistance to flow, such as the brain or the kidneys, may be especially susceptible to aberrant pulsatile hemodynamics. In fact, there is a growing body of literature relating surrogates of arterial stiffness with imaging biomarkers of cerebrovascular disease such as dilated perivascular spaces (PVS), and white matter hyperintensities (WMH) [11, 12] as well as to stroke, dementia, and amyloid deposition [13,14,15]. We have previously reported that systemic pulsatile hemodynamics are associated with PVS and not WMH. Dilatation of the PVS sourrounding brain capillaries and arterioles may reflect increase endothelial hemodynamic stress and inflammation [16], whereas WMH are more typically associated with brain blood barrier dysruption and hypoperfusion [17, 18].
Brain arterial dilatation is an imaging biomarker of vascular risk [19, 20]. Extreme brain arterial luminal dilatation, usually defined as >2 standard deviations above the mean arterial diameter of a given artery, is associated with stroke and vascular death [21, 22]. Pathologically, progressive luminal dilatation of the brain arteries relates to non-atherosclerotic aging, and it is accompanied by elastin loss, disruption of the internal elastic lamina, relatively thickning of the media, and the arterial wall [23]. Consequently, brain arterial aging is likely to cause arterial stiffness by decreasing the elastic modulus of the arterial wall. It is unlikely, however, that brain arterial aging occurs independent of arterial changes in the systemic circulation. In this context, only rarely do the brain arteries exhibit changes not noted in the more proximal large arteries [24, 25]. Consequently, brain arterial dilatation may be a marker of widespread systemic arterial stiffness. In this same context, the same inference may be applicable to common carotid artery (CCA) stiffness. Therefore, we hypothesize that widespread arterial stiffness, as evidenced by greater CCA strain and/or brain arterial dilatation, may magnify the effects of pulsatile hemodynamics in the brain parenchyma by facilitating pulse wave propagation.
Methods
We used data from the Northern Manhattan Study (NOMAS) cohort to test our hypothesis. NOMAS represents a multiethnic stroke-free and population-based study that has followed participants since 1993 [26]. In 2003, surviving participants who remained stroke-free were invited to undergo a brain MRI, which also included time-of-flight MRA. Age, sex, and ethnicity were captured by self-report. Hypertension, diabetes, and hypercholesterolemia were determined by self-report and/or evidence of medication use related to these vascular risk factors, and/or objective evidence of increased blood pressure (systolic blood pressure[SBP] > 140 or diastolic blood pressure [DBP] >90), fasting glucose ≥ 126 mg/dl, or total cholesterol ≥240 mg/dl [11]. Cardiac disease was defined by self-report of prior coronary artery disease, angina, myocardial infarction, or bypass surgery. Smoking referred to current smoking at the time of the MRI.
We used a pocket aneroid sphygmomanometer for manual brachial blood pressure (BP) measurements during the MRI visit. The brachial BP was taken twice (>1 h apart), first within the first hour of arrival to the Neurological Institute at Columbia University Medical Center and second after 10 min of rest. We averaged both brachial BP measurements to obtain the MRI visit average brachial DBP and SBP. Brachial pulse pressure (PP) was calculated by subtracting the DBP from SBP and brachial mean arterial pressure (MAP) was calculated as (SBP + (2∗DBP))/3.
MRI measurements
The brain MRI was acquired in a 1.5-T MRI system (Philips Medical Systems). The time-of-flight MRA was acquired with a field of view 15 cm, 1 mm effective slice thickness, acquisition matrix interpolated to 256 × 228 matrix, flip angle 25°, TR 20 ms and TE 2.7 ms and the FLAIR images with a multisection turbo spin-echo mode with an FOV of 250 mm, rectangular FOV of 80%, acquisition matrix of 192 × 133 scaled to 256 × 256 in reconstruction, 3-mm section thickness with no gap, a TE of 144 ms, a TR of 5500 ms, an inversion recovery delay of 1900 ms, and a flip angle of 90°.
Brain arterial diameters were obtained using in-house software with automated vessel centerline tracking, which yielded the luminal diameter, with good to excellent reliability [21]. Briefly, we measured the diameter of brain large arteries proximal to the circle of Willis. Using the Blom method [27], we transformed the anterior (ACA) and middle cerebral arteries (MCA) diameters to a normal distribution because of their naturally different size, to then sum and average both diameters. We used the transformed ACA/MCA mean diameter continuously and categorized arbitrarily into tertiles, with a higher tertile or a larger diameter interpreted as indicative of greater brain arterial stiffness.
Brain small PVS (SPVS) were rated using a semi-quantitative scale that separately considers 14 anatomical regions of the brain with a score of 0–2 per region (possible score range: 0–28). Small PVS were rated combining T1 and FLAIR images, and were defined as T1 hypodensities of <3 mm in effective diameter with no associated FLAIR hyperintensity. This method has good inter- and intra-rater reliabilities [28]. To relate the transformed ACA/MCA mean diameter with the ipsilateral SPVS score, we included only SPVS in the anterior basal ganglia and the frontal and parietal lobes (possible score range of 0–6), given that these areas are predominantly supplied by the combination of the ACA and the MCA blood flow. We also noted the presence or absence of the anterior communicating artery (acomm), which when present, communicates both ACAs.
Volumetric WMH distribution across 14 brain regions (brainstem, cerebellum, and bilateral frontal, occipital, temporal, and parietal lobes, and bilateral anterior and posterior periventricular white matter, defined as within 1 cm of the lateral ventricles) was determined separately for each hemisphere. The brain atlas was individually projected on to each FLAIR image using the inverse transform calculated in the first step. The volume voxels overlapping with the total WMH mask from the second step provided regional WMH volumes. For the purpose on this analysis, we only used the WMH volume downstream from the ACA/MCA arterial territory, i.e., frontal lobe (subcortical and periventricular as well as subcortical parietal lobe excluding the posterior wall of the lateral ventricles and posterior caps, as these may relate to posterior cerebral arteries).
Common carotid artery measurements
We performed a high-resolution carotid ultrasound using a GE LOGIQ 700 system (GE Healthcare, Milwaukee, WI) equipped with a multifrequency 9/13-MHz linear array transducer. We collected doppler measurements only on the right common carotid (CCA). The right CCA was imaged in transverse (short axis) and longitudinal planes (anterior, lateral, and posterior views) with standardized scanning and reading protocols, as previously described [12]. A real-time digital clip of the CCA was recorded for 10 s. We measured the CCA intraluminal systolic and diastolic diameters off-line with IMAGE-Pro analysis software. We traced the best-visualized intima-media boundaries from up to 10 cardiac cycles on the M mode ultrasound and computed the systolic (SDIAM) and diastolic (DDIAM) intraluminal CCA diameters that were then averaged. We defined CCA strain as equal to (SDIAM-DDIAM)/DDIAM [12]. We used CCA strain as a surrogate measure of carotid stiffness.
Aortic measurements
Pulse wave analysis of the radial artery by applanation tonometry was performed using a commercially available device (SphygmoCor, Pulse Wave Analysis System, AtCor Medical) in a subsample of the NOMAS cohort [29]. Briefly, central SBP, DBP, MAP, and PP were calculated from the radial pulse wave by a validated generalized transfer function [30]. The aortic augmented pressure from the reflected wave was measured as the difference between central SBP and the pressure at the onset of the reflected wave from the lower body. The aortic augmentation index (AIx), an index of wave reflection, was calculated as the ratio between the augmented pressure and central PP expressed as percent.
Statistical analyses
We used Chi-squared and student’s t-tests to assess differences between the sample used for this analysis and the whole NOMAS MRI sample. The outcome measures for this analysis were the lateralized SPVS score and WMH. First, we evaluated the association between brain arterial diameters with ipsilateral SPVS score and WMH and used the contralateral SPVS score and WMH as control, stratified by the presence or absence of the acomm. This stratification is justified by the previously noted interrelationship between brain arterial diameters on the contralateral hemisphere depending on whether an acomm was present or not [31]. All subsequent models were restricted to right-sided anterior SPVS score, WMH partial volume, transformed MCA/ACA mean diameter, and CCA strain. The main exposures were brachial and aortic PP and aortic AIx, and the two possible effect modifiers were brain arterial diameters and right CCA strain. We used Pearson’s correlation to assess the unadjusted correlation between exposures and effect modifiers. We used general linear models with an identity link to assess the relationship between exposure (brachial and aortic PP and aortic AIx) and outcome (right anterior SPVS and right WMH partial volume) as well as to stratify analyses by effect modifiers (tertiles of right transformed MCA/ACA mean diameter and right CCA strain). A P value <0.05 was considered statistically significant. The analyses were carried out with SAS version 9.4 (SAS Institute, Cary, NC).
Results
Sample description
The sample included 941 NOMAS participants (mean age 71 ± 9 years, 60% women, 66% Hispanic) with concomitant (i.e., within one year) carotid ultrasound and brain MRA while only 434 participants had also concomitant aortic measures. There were no significant differences between the NOMAS participants included in this study and the rest of the MRI cohort, with the exception of dyslipidemia (Table 1).
Correlation between exposure variables
Brachial PP correlated with aortic PP (R = 0.53, P < 0.001) and brachial MAP (R = 0.42, P < 0.001) and to a lesser extent with aortic MAP (R = 0.16, P = 0.001) or Aix (R = 0.21, P = 0.001). Aortic PP correlated with Aix (R = 0.48, P < 0.001) and aortic MAP (R = 0.46, P < 0.001) and to a lesser extent with brachial MAP (R = 0.29, P = 0.001). Aortic AIx collated better with aortic MAP (R = 0.27, P = 0.001) than with brachial MAP (R = 0.09, P = 0.02).
Transformed MCA/ACA mean diameter correlations with SPVS
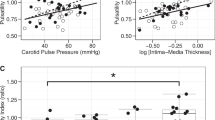
The transformed right MCA/ACA mean diameter was associated with ipsilateral SPVS score (B = 0.177, P = 0.002, Table 2) and this association persisted after adjusting for demographic and vascular risk factors (B = 0.207, P < 0.001, Fig. 1). A similar association was noted between the transformed left MCA/ACA mean diameter with ipsilateral SPVS score but it did not reach statistical significance (B = 0.056, P = 0.31). The presence of an acomm modified the relationship between anterior brain arterial diameters and the contralateral SPVS score. The right MCA/ACA mean diameter related to the left SPVS score only in the presence of an acomm (B = 0.309, P = 0.001) whereas the transformed left MCA/ACA mean diameter related positively to the right anterior SPVS score in the presence of acomm (B = 0.132, P = 0.08) but negatively in the absence of acomm (B = −0.018, P = 0.82). The transformed right MCA/ACA mean diameter associated negatively with ipsilateral WMH volume (B = −0.259 ± 0.104, P = 0.002) but this association no longer persisted after adjusting for demographic and vascular risk factors. There were no other significant association between brain arterial diameters with ipsilateral or contralateral WMH volumes in adjusted models.

In panel a, the right anterior and middle cerebral arteries seem dilated, possibly in relationship to compensatory changes given the absence of a contralateral first segment of the anterior cerebral artery. The brain MRI shows evidence of dilated perivascular spaces in the basal ganglia, more demarcated in the right caudate and anterior putamen regions, both which are perfused through penetrating arteries arising from the ipsilateral first segments of the anterior and middle cerebral arteries. On the contralateral basal ganglia, there are dilated perivascular spaces which appear smaller and less well demarcated
Effect modification of the association between pulsatile hemodynamics with SPVS score and WMH by ipsilateral brain arterial diameters and carotid stiffness
Brachial PP was associated with right anterior SPVS score (B = 0.007, P = 0.04) independent of demographic and clinical factors described in Table 1. The associations of brachial PP with the right anterior SPVS score was greatest among participants with a transformed right MCA/ACA mean diameter in the upper tertile compared with those in the mid or lower tertiles (P = 0.001 for the trend). An additional effect modification was noted between brachial PP and right anterior SPVS score with right CCA strain. The joint effect of brachial PP and transformed right MCA/ACA mean diameter on the right anterior SPVS score was higher among participants with carotid stiffness in the mid or high tertile compared with the lower tertile (P < 0.001 for the trend). Conversely, the effects of carotid stiffness were only significant among participants with a transformed average right MCA/ACA diameter in the upper tertile (B = 0.001, P = 0.001) compared with the mid or the lowest tertile (P = 0.001 for the trend). Substituting brachial PP with aortic AIx or aortic PP in the subsample with available aortic measures reproduced the effect modification of brain arterial diameters with the ipsilateral downstream SPVS score (Table 3).
Using the adjusted model from Tables 3 or 4, brachial (B = 0.036 ± 0.009, P < 0.001) or aortic MAP (0.026 ± 0.012, P = 0.04) but not brachial PP (B = −0.010 ± 0.007, P = 0.12) or aortic PP (−0.003 ± 0.010, P = 0.74) were associated with right WMH volume. Aix was associated with WMH volume (0.040 ± 0.016, P = 0.01), however. Using the same models as for SPVS we did not find effect modifications by carotid strain or brain arterial diameter in the association between any of three measures of pulsatile hemodynamics with WMH volume.
Discussion
There is a strong theoretical argument that the status of the intermediary arteries between the aorta and the brain may modify the effects of central arterial stiffness with cerebrovascular disease. In this analysis, we provide evidence that brain arterial dilation, a marker of brain arterial stiffness, is associated with brain SPVS and not with WMH. Furthermore, brachial or aortic PP and aortic AIx association with brains SPVS is modified by the status of the intermediary arteries, confirming out initial hypothesis. We did not find an association between brachial or aortic PP with WMH volume independent of the association with MAP, a finding consistent with what we previously reported [11]. We did find association between Aix and WMH volume, but there was no effect modification by stiffness in the intermediary arteries (Table 4). These findings support the hypothesis that measures of stiffness in the intermediary arteries connecting the aorta to the brain magnify the effects of pulsatile hemodynamics as it relates to SPVS and not WMH volume. The evidence provided here also supports the argument that SPVS and WMH, although both traditionally referred as imaging biomarkers of small artery disease, may have distinct physiopathology. Dilated brain arteries are associated with downstream arteriolar dilatation [32], which in addition to the decreased impedance to flow may expossse brain capillaries to systemic hemodynamics from which they should be protected in physiological circumstances via cerebral autoregulation [33].
Brain SPVS are biomarkers of vascular risk and death, and in particular of myocardial infarction [28]. The strong association between SPVS and brachial or aortic PP as compared with brachial or aortic MAP [11], the association of SPVS with myocardial infarction [28], and the association between SPVS with brain arterial dilatation presented here suggest that brain SPVS may represent end-organ damage related to arterial stiffness. Although other studies have found an association between measures of arterial stiffness and WMH [15], it is not clear whether the effects of arterial stiffness would remain significant after adjusting for MAP. As we have demonstrated before, non-atherosclerotic brain arterial aging consists of brain arterial dilatation and elastin loss, and it relates to Alzheimer dementia independent of brain large artery atherosclerosis and brain infarctions [23]. Brain arterial dilatation in the anterior circulation relates to carotid intima-media thickness and carotid plaques, both of which presumably decrease compliance of the carotid artery [31]. Because brain SPVS have been related to Alzheimer dementia independent of amyloid deposition [34], and central measures of arterial stiffness have been related to poorer cognitive performance and dementia [13], brain PVS and their underlying pathophysiology most likely related to arterial stiffness may represent a vascular contribution to Alzheimer dementia. There is conflicting data on whether the contribution may be amyloid mediated or not [15, 34], but it is plausible that strategies to curb arterial stiffness or maintain arterial elasticity may alter cerebrovascular health and consequently modify its contribution to Alzheimer dementia.
Atherosclerosis is perhaps the most studied arterial disease, but as we have pointed out in brain arteries, more than half of the brain arteries in individuals over the age of 80 have no pathological evidence of cholesterol deposition, either loosely arranged or concentrated in a necrotic core [35]. The idea that atherosclerosis is not the only arterial phenotype that relates to cerebrovascular health is also supported by studies of non-cerebral arteries. For example, the prevalence of moderate carotid stenosis is ~4% in the general population and the prevalence of severe stenosis is even lower [36]. Although aortic plaques are overall more prevalent in 50 year or older adults than carotid plaques (38% of the women and 41% of the men), atherosclerotic plaques in the thoracic aorta are much less prevalent (~6%) and all together, the majority of old adults do not have atherosclerotic plaques [37, 38]. Absence of atherosclerosis does not indicate absence of arterial disease, however. For example, in the cervical carotid, intima-media thickness results from diffuse intima thickening [39], which may be the result of compensatory process to chronic blood flow and not necessarily related to carotid plaques and or atherosclerosis [40, 41]. Diffuse intima thickening and elastin loss accompanies non-atherosclerotic aging of brain arteries and the aorta [2, 23]. As a counterargument to the idea that non-atherosclerotic arterial aging is distinct from atherosclerosis, it may be argued that intima thickening is a predisposing lesion to develop atherosclerosis and that a given arterial segment with intima thickening will eventually develop atherosclerosis. It is fair to say, however, that loss of elastic properties of the artery and wall thickening suffice to explain increased arterial stiffness [5, 42, 43], and the end-organ damage related to stiffness may precede any possible future development of atherosclerosis.
The results of these analyses should be considered in the setting of their limitations. It is less certain whether this result would be applicable to other populations. We do not have carotid-femoral pulse wave velocity, considered the gold standard for central arterial stiffness. As the ejection fraction decreases with age so does the aortic AIx while the pulse wave velocity increases exponentially after age 60 [5]. Based on this, the aortic AIx may be a less reliable biomarker of arterial stiffness in populations over 60 years, which is a limitation of this study. The fact that we were able to demonstrate aortic AIx as an effect modifier of the association between brain PVS and brain arterial diameters suggests that with an even more precise measure of central arterial stiffness such as carotid-femoral pulse wave velocity, the association may be even stronger.
Perspectives
In summary, we confirmed the hypothesis that brain arterial dilatation is associated with brain SPVS. Greater brain arterial dilatation and/or carotid stiffness magnify the association between pulsatile hemodynamics with brain SPVS. Discovering ways to modify arterial stiffness or preserve arterial elasticity may help offset the effects of systemic arterial disease in the brain parenchyma and possibly in its function.
References
Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, et al. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121:505–11.
Schlatmann TJ, Becker AE. Histologic changes in the normal aging aorta: implications for dissecting aortic aneurysm. Am J Cardiol. 1977;39:13–20.
Mitchell GF, Lacourcière Y, Ouellet JP, Izzo JL Jr., Neutel J, Kerwin LJ, et al. Determinants of elevated pulse pressure in middle-aged and older subjects with uncomplicated systolic hypertension: the role of proximal aortic diameter and the aortic pressure-flow relationship. Circulation. 2003;108:1592–8.
Sollberg LA, McGarry PA, Moossy J, Strong JP, Tejada C, Loken AC. Severity of atherosclerosis in cerebral arteries, coronary arteries, and aortas. Ann N Y Acad Sci. 1968;149:956–73.
Mitchell GF. Arterial stiffness and wave reflection: biomarkers of cardiovascular risk. Artery Res. 2009;3:56–64.
Boyajian RA, Schwend RB, Wolfe MM, Bickerton RE, Otis SM. Measurement of anterior and posterior circulation flow contributions to cerebral blood flow. J Neuroimaging. 1995;5:1–3.
Scuteri A, Chen CH, Yin FC, Chih-Tai T, Spurgeon HA, Lakatta EG. Functional correlates of central arterial geometric phenotypes. Hypertension. 2001;38:1471–5.
Gosling RG, Budge MM. Terminology for describing the elastic behavior of arteries. Hypertension. 2003;41:1180–2.
Marmarou A, Takagi H, Shulman K. Biomechanics of brain edema and effects on local cerebral blood flow. Adv Neurol. 1980;28:345–58.
Wang JJ, Parker KH. Wave propagation in a model of the arterial circulation. J Biomech. 2004;37:457–70.
Gutierrez J, Elkind MS, Cheung K, Rundek T, Sacco RL, Wright CB. Pulsatile and steady components of blood pressure and subclinical cerebrovascular disease: the Northern Manhattan Study. J Hypertens. 2015;33:2115–22.
Rundek T, Della-Morte D, Gardener H, Dong C, Markert MS, Gutierrez J, et al. Relationship between carotid arterial properties and cerebral white matter hyperintensities. Neurology. 2017;88:2036–42.
Mitchell GF, van Buchem MA, Sigurdsson S, Gotal JD, Jonsdottir MK, Kjartansson O, et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility–Reykjavik study. Brain. 2011;134:3398–407.
Tsao CW, Seshadri S, Beiser AS, Westwood AJ, Decarli C, Au R, et al. Relations of arterial stiffness and endothelial function to brain aging in the community. Neurology. 2013;81:984–91.
Hughes TM, Kuller LH, Barinas-Mitchell EJ, Mackey RH, McDade EM, Klunk WE, et al. Pulse wave velocity is associated with beta-amyloid deposition in the brains of very elderly adults. Neurology. 2013;81:1711–8.
Wuerfel J, Haertle M, Waiczies H, Tysiak E, Bechmann I, Wernecke KD, et al. Perivascular spaces–MRI marker of inflammatory activity in the brain? Brain. 2008;131:2332–40.
Young VG, Halliday GM, Kril JJ. Neuropathologic correlates of white matter hyperintensities. Neurology. 2008;71:804–11.
Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, et al. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry. 2011;82:126–35.
Gutierrez J, Cheung K, Bagci A, Rundek T, Alperin N, Sacco RL, et al. Brain arterial diameters as a risk factor for vascular events. J Am Heart Assoc. 2015;4:e002289.
Tanaka M, Sakaguchi M, Miwa K, Okazaki S, Furukado S, Yagita Y, et al. Basilar artery diameter is an independent predictor of incident cardiovascular events. Arterioscler Thromb Vasc Biol. 2013;33:2240–4.
Gutierrez J, Bagci A, Gardener H, Rundek T, Ekind MS, Alperin N, et al. Dolichoectasia diagnostic methods in a multi-ethnic, stroke-free cohort: results from the northern Manhattan study. J Neuroimaging. 2014;24:226–31.
Passero SG, Rossi S. Natural history of vertebrobasilar dolichoectasia. Neurology. 2008;70:66–72.
Gutierrez J, Honig L, Elkind MS, Mohr JP, Goldman J, Dwork AJ, et al. Brain arterial aging and its relationship to Alzheimer dementia. Neurology. 2016;86:1507–15.
Sorbara R. Étude comparative du vieillissement des artères du polygone de Willis. Toulouse: Université Paul-Sabatier; 1972.
Bouissou H, Emery M, Sorbara R. Age related changes of the middle cerebral artery and a comparison with the radial and coronary artery. Angiology. 1975;26:257–68.
Sacco RL, Kargman DE, Gu Q, Zamanillo MC. Race-ethnicity and determinants of intracranial atherosclerotic cerebral infarction. The Northern Manhattan Stroke Study. Stroke. 1995;26:14–20.
Altman DG. Practical statistics for medical research. CRC press, London; 1990.
Gutierrez J, Elkind MS, Dong C, Di Tullio M, Rundek T, Sacco RL, et al. Brain perivascular spaces as biomarkers of vascular risk: results from the Northern Manhattan Study. AJNR Am J Neuroradiol. 2017;38:862–7.
Russo C, Jin Z, Palmieri V, Homma S, Rundek T, Elkind MS, et al. Arterial stiffness and wave reflection: sex differences and relationship with left ventricular diastolic function. Hypertension. 2012;60:362–8.
Chen CH, Nevo E, Fetics B, Pak PH, Yin FC, Maughan WL, et al. Estimation of central aortic pressure waveform by mathematical transformation of radial tonometry pressure. Valid Gen Transf Funct Circ. 1997;95:1827–36.
Gutierrez J, Elkind MS, Gomez-Schneider M, DeRosa JT, Cheung K, Bagci A, et al. Compensatory intracranial arterial dilatation in extracranial carotid atherosclerosis: The Northern Manhattan Study. Int J Stroke. 2015;10:843–8.
Gutierrez J, Murray J, Chon C, Morgello S. Relationship between brain large artery characteristics and their downstream arterioles. J Neurovirol. 2018;24:106–12.
Lee RMKW, Dickhout JG, Sandow SL. Vascular structural and functional changes: their association with causality in hypertension: models, remodeling and relevance. Hypertens Res. 2016;40:311.
Banerjee G, Kim HJ, Fox Z, Jager HR, Wilson D, Charidimou A, et al. MRI-visible perivascular space location is associated with Alzheimer’s disease independently of amyloid burden. Brain. 2017;140:1107–16.
Gutierrez J, Elkind MS, Virmani R, Goldman J, Honig L, Morgello S, et al. A pathological perspective on the natural history of cerebral atherosclerosis. Int J Stroke. 2015;10:1074–80.
de Weerd M, Greving JP, de Jong AWF, Buskens E, Bots ML. Prevalence of asymptomatic carotid artery stenosis according to age and sex. Syst Rev Metaregression Anal. 2009;40:1105–13.
Jaffer FA, O’Donnell CJ, Larson MG, Chan SK, Kissinger KV, Kupka MJ, et al. Age and sex distribution of subclinical aortic atherosclerosis. A Magn Reson Imaging Exam Fram Heart Study. 2002;22:849–54.
Maroules CD, Rosero E, Ayers C, Peshock RM, Khera A. Abdominal aortic atherosclerosis at MR imaging is associated with cardiovascular events: The Dallas Heart Study. Radiology. 2013;269:84–91.
Finn AV, Kolodgie FD, Virmani R. Correlation between carotid intimal/medial thickness and atherosclerosis: a point of view from pathology. Arterioscler Thromb Vasc Biol. 2010;30:177–81.
Baroncini LAV, de Castro Sylvestre L, Filho RP. Carotid intima-media thickness and carotid plaque represent different adaptive responses to traditional cardiovascular risk factors. IJC Heart Vasc. 2015;9:48–51.
Rundek T, Gardener H, Della-Morte D, Dong C, Cabral D, Tiozzo E, et al. The relationship between carotid intima-media thickness and carotid plaque in the Northern Manhattan Study. Atherosclerosis . 2015;241:364–70.
Nichols WW, O’Rourke MF, McDonald DA. Special circulations. In: Nichols WW, O’Rourke MF, McDonald DA, editors. McDonald’s blood flow in arteries: theoretical, experimental and clinical principles. 6th edn. London: Hodder Arnold; 2011. p. 755.
O’Rourke MF, Hashimoto J. Mechanical factors in arterial aging: a clinical perspective. J Am Coll Cardiol. 2007;50:1–13.
Funding
The work was supported by National Institutes of Health (5R01NS029993, R37 NS029993, K24 NS 062737-03, 1R01AG057709-).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Jose Gutierrez, MD, MPH: Dr Gutierrez receives compensation for serving as an expert witness in medical litigation. Noam Alperin, PhD: Shareholder in Alperin Noninvasive Diagnostics, Inc. Clinton B Wright, MD, MS: Dr. Wright receives royalties from UpToDate.com for two chapters on vascular dementia. Mitchell SV Elkind, MD, MS: Dr. Elkind receives compensation for providing consultative services for Biotelemetry/Cardionet, BMS-Pfizer Partnership, Boehringer-Ingelheim, and Sanofi-Regeneron Partnership; receives compensation for serving as an expert witness in litigation for BMS-Sanofi (Plavix), Merck/Organon (Nuvaring), and Hi-Tech Pharmaceuticals (dimethylamylamine); serves on the National, Founders Affiliate, and New York City chapter boards of the American Heart Association/American Stroke Association; and receives royalties from UptoDate for chapters related to stroke. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gutierrez, J., DiTullio, M., K Cheung, Y.K. et al. Brain arterial dilatation modifies the association between extracranial pulsatile hemodynamics and brain perivascular spaces: the Northern Manhattan Study. Hypertens Res 42, 1019–1028 (2019). https://doi.org/10.1038/s41440-019-0255-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-019-0255-1
