
Abstract
Oxidative stress is involved in the pathogenesis of hypertension and hypertensive organ damage. Our previous study suggested that stroke-prone spontaneously hypertensive rats (SHRSP) exhibited greater oxidative stress than SHR and that the stroke incidence was significantly greater in SHRSP than SHR. Therefore, we hypothesized that oxidative stress was responsible for the stroke susceptibility in SHRSP. The present study constructed Prdx2 (a gene coding an antioxidative enzyme)-knockout (KO) SHR to examine whether Prdx2 knockout would make SHR more vulnerable to hypertensive organ damage, including stroke. Prdx2-KO SHR were created using CRISPR/CAS9 for genome editing. Eight-week-old male SHR and Prdx2-KO SHR were fed 1% NaCl for 2 months to induce blood pressure (BP) changes and stroke occurrence. The baseline BP was significantly greater in KO SHR, and this difference disappeared after salt loading. The life span of KO SHR was significantly reduced compared to that of SHR despite no differences in BP under salt-loading. However, no stroke was observed in KO SHR. The severity of hypertensive renal and cardiac injuries did not differ significantly between the two strains, but oxidative stress, evaluated using urinary isoprostane excretion and DHE staining, was greater in KO SHR. These results indicated that the Prdx2-depletion caused a shorter life span and modest BP increase in SHR via increased oxidative stress. The pathophysiological roles of oxidative stress in this model should be clarified in future studies.
Similar content being viewed by others

Introduction
Reactive oxygen species (ROS) are generated from different sources as a byproduct or signaling molecule, and imbalance between ROS production and removal may lead to oxidative stress [1, 2]. Oxidative stress may play a role in cardiovascular diseases, including hypertension and hypertensive organ damage, in humans and animal models [3, 4]. Detailed pathophysiological effects of oxidative stress on the cardiovascular system were primarily investigated in animal models, including the spontaneously hypertensive rats (SHR) and stroke-prone SHR (SHRSP) [1, 2].
Salt-induced increases of oxidative stress are generally observed in SHR and SHRSP. However, we found that oxidative stress level was greater in SHRSP than SHR [5]. This observation suggested that oxidative stress played a key role in the pathogenesis of salt-induced cerebrovascular complications in SHRSP [6]. The present study hypothesized that the chronic increase in oxidative stress in SHR accelerated cerebrovascular events in SHR, which were otherwise resistant to stroke [5]. We constructed peroxiredoxin2 (Prdx2)-knockout (KO) SHR using CRISPR/CAS9 genome-editing technology [7].
Humans possess various antioxidant systems to protect our bodies from oxidative stress [8]. Peroxiredoxins (PRDXs) are a group of proteins with potent antioxidant activity that degrade H2O2, lipid hydroperoxides, and peroxynitrite [9]. PRDX1 and 2 are the most abundant of the six isoforms in vivo. PRDX2 is more susceptible to hyperoxidation [10], and it removes H2O2 quickly. PRDX2 exhibited a protective role in vascular remodeling [11], and a recent report demonstrated that Prdx2 deficiency accelerated atherosclerosis in ApoE-KO mice [12]. These observations suggest Prdx2 as a good target for the chronic increase in oxidative stress in SHR.
The present study constructed Prdx2-KO SHR and evaluated the effects of the Prdx2 depletion on blood pressure (BP), stroke, and hypertensive organ damage in SHR.
Methods
Construction of Prdx2-KO SHR
Construction of the KO SHR was performed in Kyoto University.
Depletion of Prdx2 was performed in SHR/Izm using CRISPR/Cas9 genome editing, as described previously [7]. Briefly, a target sequence in a gRNA was selected to edit the rat Prdx2 gene from the rat genome sequence using the CRISPR Design Tool (http://crispr.mit.edu/). The target sequence selected was CCGGCAACGCGCACATCGGA from +8 to +27 of the Prdx2 gene. A double-stranded DNA, including the T7 promoter, the target sequence above and the gRNA tail, was chemically synthesized, and mRNA was obtained using in vitro transcription and a commercial kit (MEGA shortscript T7 Transcription Kit, Life Technologies, Carlsbad, CA, USA). Cas9 mRNA was obtained using in vitro transcription and the modified hCas9 plasmid [7] as a template with mMESSAGE mMACHINE T7 Ultra Kit (Life Technologies). Cas9 mRNA (100 ng/mL) and gRNA (50 ng/mL) was microinjected into the male-pronuclei of embryos of SHR, and the embryos at the two-cell stage were transferred to pseudo-pregnant females.
Forty-one pups were obtained in total, and screening for deletion/insertion was performed using direct sequencing of PCR products of the target region [primers; CCTTGTACTGGGAGGGTGAA (forward), GGAAGAGGAGAGCGGAAGAG (reverse)]. All the procedures described above were performed in Kyoto University.
Two heterozygous pups with 7-bp and 6-bp deletions were ultimately obtained and backcrossed with SHR. The two strains of homozygous Prdx2-KO SHRs were established in Shimane University. The present study used Prdx2-KO SHR with the 7-bp deletion in which we expected complete depletion of PRDX2 expression because of a frameshift change. The location of the deletion is shown in Fig. 1a.

Construction of Prdx2-knockout SHR. a The target sequence in the Prdx2 gene for genome editing by CRISPR/CAS9. b Protein expression of PRDX2 in the kidney cortex and medulla, and the brainstem of the Prdx2-knockout SHR (KO) and SHR. c mRNA expression of Prdx2 in the kidney cortex and medulla, and the brainstem in SHR (n = 3) and the Prdx2-KO SHR (n = 3). †p < 0.05 vs. SHR
Prdx2-KO SHRs were deposited to the National BioResource Project-Rat under #0814 (SHR-Prdx2em1Izm) and #0815 (SHR-Prdx2em2Izm) for the KO rats with the 7-bp and 6-bp deletions, respectively. The local committee for Animal Research in Kyoto University approved the procedure of genome editing using CRISPR/Cas9.
Animal procedures
SHR/Izm was provided by the Disease Model Cooperative Research Association (Kyoto, Japan). Eight-week-old male SHR/Izm and Prdx2-KO SHR/Izm were used in this study. Rats were fed a stroke-permissive (SP) diet (Funabashi Farm Co., Ltd., Chiba, Japan). BP was measured, and salt-loading was initiated using 1% NaCl in drinking water for 8 weeks until the age of 16 weeks. Control rats received plain water for 8 weeks. Food and water intake was monitored daily, and body weight (BW) and BP were measured using the tail-cuff method every 2 weeks during the experimental period. Twenty-four hours urine, serum, and organs (kidneys, brain, and heart) were collected in some experiments after 8 weeks of salt-loading for biochemical, gene expression, and histological studies, respectively (see below). BP measurements were performed using a radiotelemetry system. Briefly, a telemetry transducer (DSI, St. Paul, MN) was implanted in the abdominal cavity under anesthesia with 1% isoflurane at 11 weeks of age. Animals recovered for 1 week, and baseline BP was monitored for 1 week. Salt-loading was initiated, and changes in BP were monitored for an additional 4 weeks. BP was measured every 10 min, and the average between 11:00 and 13:00 and between 23:00 and 1:00 represented BPs during the light and dark phases, respectively.
The local committee of animal research in Shimane University approved all animal procedures, except genome editing using CRISPR/CAS9.
Quantitative real time PCR
Measurement of mRNA was performed as described previously [13]. In brief, rats were sacrificed under deep anesthesia (inhalational isoflurane), and the kidneys and brain were dissected out immediately. Total RNA was isolated using Sepasol-RNA I Super G (Nakalai Tesque, Kyoto, Japan) according to the manufacturer’s instructions. Complementary DNA was synthesized using PrimeScript RT reagent Kit with gDNA Eraser (Takara Bio, Shiga, Japan). Quantitative reverse transcription PCR (RT-PCR) was performed using the real-time PCR system 7300 or StepOne Plus (Applied Biosystems, Foster City, CA, USA) and SYBR Premix EX-TaqII (Takara Bio, Shiga, Japan). The quantity of mRNA was standardized to β-actin mRNA. The primers used in the experiments are listed in Supplementary Table 1.
Western blot analysis of PRDX2 protein
Kidney cortex, medulla, and brainstem were dissected from rats under deep anesthesia and homogenized in RIPA buffer (Nakalai Tesque, Kyoto, Japan). The same volume of 50 mM Tris–HCl buffer (pH 6.8) containing 10% glycerol, 2% sodium dodecyl sulfate, 0.01% bromophenol blue, and 5% 2-mercaptoethanol was added to the homogenate, which was mixed and boiled at 100 °C for 5 min. Each aliquot of 30 µg protein was loaded on a 9% polyacrylamide gel for electrophoresis at a constant current of 15 mA for 2 h at room temperature. Separated proteins were electrically transferred to a membrane (Immobilon-P, Millipore, Billerica, MA, USA) previously treated with 100% methanol. Membranes were blocked with 5% skim milk dissolved in 20 mM Tris–HCl buffer (pH 7.5) containing 137 mM NaCl and 0.05% Tween 20 and incubated with a rabbit anti-PRDX2 antibody (Cell Signaling, #46855, 1:1000, Danvers, MA, USA) or mouse anti-β-actin antibody (Sigma-Aldrich, AC-15, 1:5000, St. Louis, MO) for 16 h at 4 °C. Membranes were incubated with an anti-rabbit IgG antibody conjugated with peroxidase (1:5000) or an anti-mouse IgG antibody conjugated with peroxidase (1:5000). All antibodies used in this experiment were diluted in 1% skim milk dissolved in 20 mM Tris–HCl buffer (pH 7.5) containing 137 mM NaCl and 0.05% Tween 20. Membranes were washed and developed using Image Quant LAS 4000 (GE Healthcare UK Ltd., Buckinghamshire, England) and the Clarity Western ECL Substrate (Bio-Rad, Hercules, CA, USA) to enhance chemiluminescence signals.
Biochemical assays
Urine was collected for 24 h in a metabolic cage and centrifuged at 3000 rpm for 10 min at 4 °C. The supernatant was collected and stored at −30 °C. Urinary isoprostane was measured using an ELISA kit (Nikken Seil Co., Ltd., Shizuoka, Japan) following the manufacturer’s protocol. Urinary protein was determined in 24-h urine samples using the Protein Assay Bicinchoninate Kit (Nakalai Tesque, Kyoto, Japan). Blood samples were collected and centrifuged at 3000 rpm for 5 min at 4 °C for serum collection and stored at −80 °C until analysis. Levels of blood urea nitrogen (BUN) and serum creatinine were measured using a chemical analyzer (SPOTCHEM EZ sp-4430, Arkray, Kyoto, Japan).
Dihydroethidium (DHE) staining
Superoxide level in tissue was evaluated using the DHE staining method [14]. In brief, cryosections (5 μm) of kidney and brainstem were stained with DHE (10 μmol/L) (Sigma Aldrich, St. Louis, MO, USA) for 30 min at room temperature in the dark. Photographs were obtained using the DS-Ri1-U2 microscope (Nikon, Tokyo, Japan). Photographs were analyzed, and fluorescence intensity was calculated in NIH Image J (ver 1.46r).
Histopathological examination
The left kidney and heart of each rat were preserved in 10% formalin. Sliced sections (5-μm thick) of the maximal cut surface of the kidney and middle of heart ventricle were stained using hematoxylin & eosin (HE) or the Azan method. Glomeruli were categorized into three groups according to the severity of glomerulosclerosis in HE sections (see Supplementary Fig. 1). Approximately 100–200 glomeruli were examined in each section (~400–600 glomeruli were examined from three rats per group), and the prevalence of sclerotic glomeruli was compared between experimental groups using the χ2 test. Analyses were performed on ‘completely’ sclerotic glomeruli or total (completely+partially) sclerotic glomeruli (Fig. 6a). The area of fibrotic regions in the kidney and the left cardiac ventricle of Azan sections (regions stained blue in the sections, see Supplementary Fig. 1) were measured on digital images using NIH Image J, and a relative fibrotic area (%) was calculated as fibrotic area/total area × 100.
Statistics
All values are expressed as the means ± SD. p < 0.05 was considered statistically significant.
Results
Depletion of PRDX2 expression
The kidney and brainstem of the Prdx2-KO SHR and SHR were analyzed for PRDX2 protein expression. We confirmed the depletion of PRDX2 protein in KO SHR (Fig. 1b). RT-PCR analysis indicated that no mRNA expression of Prdx2 was detected in KO SHR (Fig. 1c). The Prdx2 mRNA with the deletion may be unstable and rapidly degraded.
Prdx2 deficiency increases oxidative stress
Urinary excretion of isoprostane was increased under salt-loading in the Prdx2-KO and control SHR compared to water-treated rats (Fig. 2a), which indicates that salt intake per se increased oxidative stress in both strains. As expected, oxidative stress, as estimated by urinary isoprostane, was greater in the Prdx2-KO SHR than the control SHR under baseline and salt-loaded conditions (Fig. 2a). DHE staining also revealed that the level of oxidative stress in the kidney was greater in the Prdx2-KO SHR than SHR (Fig. 2b, c). Increased oxidative stress levels play a role in sympatho-excitation mechanisms in the brainstem (reviewed in ref. [2]). Oxidative stress tended to be greater in KO SHR, but did not reach a significant level.

Effect of Prdx2 depletion on oxidative stress. a Urinary isoprostane level measured in SHR and Prdx2-KO SHR (KO) fed plain water (Control) and 1% salt water (1% NaCl). Five rats were used in each group. b Levels of oxidative stress measured using DHE staining. Representative photographs are shown for each set of treatment. The scale bar indicates 25 μm. c Quantitative analyses of DHE fluorescence intensity in SHR (n = 4) and Prdx2-KO SHR (n = 4). †p < 0.05 vs. salt-loaded SHR and *p < 0.05 vs. control. DHE dihydroethidium
Effects of Prdx2 deficiency on BP
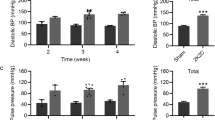
BP measurements using the tail-cuff were significantly greater in KO SHR than SHR at 10 weeks of age (170 ± 12 and 152 ± 12 mmHg, respectively, Fig. 3a) under the baseline condition (i.e., without salt-loading). Salt-loading significantly increased BP in both strains, but this difference between the two strains disappeared (Fig. 3a). Telemetry measurements revealed a small but significant difference in baseline BP, but no significant difference was observed under salt-loading, which was consistent with the tail-cuff method results (Fig. 3b, c).

Effects of Prdx2 depletion on BP change. a SBP measured using the tail-cuff method in SHR and Prdx2-KO SHR. Open triangles and circles for SHR and Prdx2-KO SHR-fed plain water (control), respectively, and closed triangles and circles are for SHR and the Prdx2-KO SHR under 1% salt loading, respectively. Eleven rats were used for each group. †p < 0.05 vs. control SHR, *p < 0.05 vs. control. b SBP in SHR (n = 8) and the Prdx2-KO SHR (n = 5) measured using telemetry. Open and closed circles are for SHR and the Prdx2-KO SHR, respectively. c Averaged SBP during light and dark phases using the telemetry method. SBP during light and dark phases was averaged for 7 days of the baseline period before salt loading (shown as ‘baseline’) or during salt loading (shown as ‘1% NaCl’). Data of the last 7 days were used for the salt-loaded period. †p < 0.05 vs. baseline SHR. *p < 0.05 vs. the baseline SBP
Prdx2 deficiency reduces life span
The life span of Prdx2-KO SHR was significantly reduced compared to SHR under salt-loading (median of the life span; 52 and 81 days, respectively, p = 0.049 using the log-rank test, Fig. 4a). However, no symptoms of stroke (i.e., paralysis, convulsion, and akinesia) were observed during the experimental period, and no edema or hemorrhage was found in the brain using MRI examination or macroscopic observation of dissected brains (Fig. 4b). Therefore, KO SHR and SHR were not likely to die from cerebral stroke in this experiment.

Effect of Prdx2 depletion on life span. a Survival of SHR (n = 16) and Prdx2-KO SHR (n = 16) was examined until 90 days of salt loading. The life span was significantly reduced in Prdx2-KO SHR compared to SHR (p = 0.049 by log-rank test). b Representative observation of the brain from deceased Prdx2-KO SHR. No changes suggest hemorrhage or infarction on MRI (upper panels) or macroscopic inspection (lower panels)
Effects of Prdx2 deficiency on hypertensive organ damage
Salt-loading significantly increased the kidney weight, but it did not differ between the two strains with or without salt-loading (Fig. 6a). Salt-loading also increased urinary protein excretion and BUN level, but no significant differences in urinary protein, BUN, and serum creatinine were found between the two strains (Fig. 5a, b). Histological evaluations of glomerular sclerosis revealed no significant differences in the prevalence of complete sclerotic glomeruli under salt loading. However, we found a significant difference in the prevalence of total (completely+partially) sclerotic glomeruli between the two strains (19% and 12% for KO SHR and SHR, respectively, Fig. 6a). The fibrotic area tended to be greater in KO SHR, but did not reach a significant level (Fig. 6a).

Effects of the Prdx2 depletion on urinary protein excretion, serum BUN, serum creatinine, and genetic markers for renal injury. a and b Urinary excretion of protein, serum BUN, and serum creatinine were measured in control and salt-loaded rats. Each group included five rats. c Expression of genetic markers for renal injury [n = 6 except Kim1 (n = 5)]. *p < 0.05 vs. control

Effects of Prdx2 depletion on kidney and heart. a Relative kidney weight (mg/g BW, n = 8) and relative area of renal fibrosis (n = 3) were evaluated in SHR and Prdx2-KO SHR. The incidence of total sclerotic glomeruli [%, (complete+partial) sclerotic glomeruli/total glomeruli] and complete sclerotic glomeruli (%, sclerotic glomeruli/total glomeruli) are shown in parenthesis above the column), *p < 0.05 vs. control. †p < 0.05 vs. salt-loaded SHR. b Relative heart weights (mg/g BW) were evaluated in SHR and Prdx2-KO SHR (n = 8). *p < 0.05 vs. control. Relative area of cardiac fibrosis in SHR and Prdx2-KO SHR (n = 3)
We further examined the expression of several marker genes for renal fibrosis, including α-smooth muscle actin (α-Sma), transforming growth factor-β (Tgf-β), collagen type I alpha 1 chain (Col1a1), and collagen type IV alpha 1 chain (Col4a1), and markers of tubular damage, such as kidney injury molecule 1 (Kim1), using RT-PCR. The expression levels of these markers increased significantly in salt-treated SHR and Prdx2-KO SHR. However, the expression levels were not significantly different between the two strains (Fig. 5c).
Similar results were obtained in the heart. Heart weight increased significantly under salt loading, but no significant difference was found between the two strains either with or without salt loading (Fig. 6b). Microscopic observations revealed that the fibrotic area in the left ventricular wall was not significantly different between the two strains (Fig. 6b).
Discussion
The present study demonstrated that depletion of the Prdx2 gene increased baseline BP and shortened the life span of SHR under a salt-loaded condition. However, no evidence of a greater incidence of cerebral stroke was observed in KO SHR compared with the original SHR.
Many enzymes and proteins play important roles to reduce oxidative stress in vivo [1]. The PRDX family is a key system in controlling oxidative stress because it reduces >90% of cellular peroxides [9]. PRDX2 is the most abundant enzyme, and it is expressed ubiquitously in the body, including the vasculature [9, 11]. Therefore, it was reasonable to expect that Prdx2 depletion would increase oxidative stress in vivo. We found that urinary isoprostane excretion and DHE staining of the kidney increased significantly in Prdx2-KO SHR (see Fig. 2). However, stroke was not obvious in the KO rats in the present study. This result suggests that a modest increase in oxidative stress is not sufficient to promote stroke in SHR. SHRs are resistant to stroke even under salt-loaded condition [5]. Salt loading produced a substantial increase in oxidative stress in SHRs (see Fig. 2), but the additional modest increase in oxidative stress following the Prdx2 deletion may not be sufficient to increase stroke susceptibility.
Oxidative stress is generally believed to exert deteriorating effects on cerebral stroke [15], but minor roles of oxidative stress in the pathogenesis of stroke were suggested in humans and model animals [16, 17]. For example, Yao et al. demonstrated that P22phox-depleted congenic SHRSPs did not exhibit reduced infarct volume in the middle-cerebral-artery occlusion model [18]. P22PHOX is an essential subunit of NADPH oxidases, and P22phox-depletion reduced oxidative stress [18]. These observations by Yao et al. support a less important role of oxidative stress in the pathogenesis of cerebral infarction in SHRSPs.
Variation in the levels of oxidative stress in different models may be responsible for the discrepant observations on the effects of oxidative stress, and strict quantitative estimation of oxidative stress may be essential to interpret the discrepancies between studies.
We did not find significant differences in renal fibrosis or proteinuria between our two strains. The prevalence of total (complete+partial) glomerulosclerosis was significantly greater in KO SHR, but the incidence of ‘complete’ sclerosis was not different (see Fig. 6a). We must be prudent in our conclusion that histological damage was greater in KO SHR based on all of the histological data. Genetic markers for fibrosis and tubular damage did not reveal significant differences between the two strains, which is consistent with the histological evaluations. Expression of the genes examined in this study correlated significantly with proteinuria. Therefore, we expected that these genes would be good markers of renal damage (see Supplementary Fig. 2). We concluded that salt loading-induced renal damage did not clearly differ between the two strains. The Prdx2 depletion may produce limited effects on hypertensive renal damage compared to the clear deteriorating effects of salt on renal injury (see Figs. 5 and 6a).
This interpretation also seemed applicable to the heart. The heart weights did not significantly differ between the two strains. Microscopic observations indicated no significant difference in fibrotic areas between the two strains (see Fig. 6b). Salt loading per se elicited clear changes in the heart weight, but the Prdx2 depletion produced a rather minor effect on heart pathology.
In conclusion, we established Prdx2-KO SHR, which exhibited greater oxidative stress, a small but significant increase in baseline BP and a shorter life span under salt loading compared to those of SHR. The pathological mechanisms of the reduced life span were not clear because we could not find clear effects of the Prdx2 depletion on the cerebral, renal, and cardiac pathologies. The pathophysiological pathways responsible for the increased oxidative stress and reduction in the life span in KO SHR must be clarified in future studies.
References
Datla SR, Griendling KK. Reactive oxygen species, NADPH oxidases, and hypertension. Hypertension. 2010;56:325–30.
Hirooka Y, Sagara Y, Kishi T, Sunagawa K. Oxidative stress and central cardiovascular regulation: pathogenesis of hypertension and therapeutic aspects. Circ J. 2010;274:827–35.
Bayorh MA, Ganafa AA, Socci RR, Silvestrov N, Abukhalaf IK. The role of oxidative stress in salt-induced hypertension. Am J Hypertens. 2004;17:31–36.
Zhao W, Chen SS, Chen Y, Ahokas RA, Sun Y. Kidney fibrosis in hypertensive rats: role of oxidative stress. Am J Nephrol. 2008;28:548–54.
Griffin KA, Churchill PC, Picken M, Webb RC, Kurtz TW, Bidani AK. Differential salt-sensitivity in the pathogenesis of renal damage in SHR and stroke prone SHR. AJH. 2001;14:311–20.
Gandolgor TA, Ohara H, Cui ZH, Hirashima T, Ogawa T, Saar K, et al. Two genomic regions of chromosomes 1 and 18 explain most of the stroke susceptibility under salt loading in stroke-prone spontaneously hypertensive rat/Izm. Hypertension. 2013;62:55–61.
Yoshimi K, Kunihiro Y, Kaneko T, Nagahora H, Voigt B, Mashimo T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat Commun. 2016;7:10431.
Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–52.
Chae HZ, Kim HJ, Kang SW, Rhee SG. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res Clin Pract. 1999;45:101–12.
Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140:517–28.
Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, et al. Regulation of PDGF signaling and vascular remodeling by peroxiredoxin II. Nature. 2005;435:347–53.
Park JG, Yoo JY, Jeong SJ, Choi JH, Lee MR, Lee MN, et al. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein E-deficient mice. Circ Res. 2011;109:739–49.
Ferdaus MZ, Xiao B, Ohara H, Nemoto K, Harada Y, Saar K, et al. Identification of Stim1 as a candidate gene for exaggerated sympathetic response to stress in the stroke-prone spontaneously hypertensive rat. PLoS One. 2014;9:e95091.
Schupp N, Kolkhof P, Queisser N, Gärtner S, Schmid U, Kretschmer A, et al. Mineralocorticoid receptor-mediated DNA damage in kidneys of DOCA-salt hypertensive rats. FASEB J. 2011;25:968–78.
Cherubini A, Ruggiero C, Polidori MC, Mecocci P. Potential markers of oxidative stress in stroke. Free Radic Biol Med. 2005;39:841–52.
Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–70.
Rodrigo R, Fernández-Gajardo R, Gutiérrez R, Matamala JM, Carrasco R, Miranda-Merchak A, et al. Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets. 2013;12:698–714.
Yao H, Ferdaus MZ, Zahid HM, Ohara H, Nakahara T, Nabika T. Focal ischemic injury with complex middle cerebral artery in stroke-prone spontaneously hypertensive rats with loss-of-function in NADPH oxidases. PLoS One. 2015;10:e0138551.
Acknowledgements
The authors thank Satoko Mishima for her great secretarial and pathohistological (preparation of tissue sections and staining) work.
Funding
This work was supported by JSPS KAKENHI 26293086.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Mahal, Z., Fujikawa, K., Matsuo, H. et al. Effects of the Prdx2 depletion on blood pressure and life span in spontaneously hypertensive rats. Hypertens Res 42, 610–617 (2019). https://doi.org/10.1038/s41440-019-0207-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-019-0207-9
Keywords
This article is cited by
-
Cpxm2 as a novel candidate for cardiac hypertrophy and failure in hypertension
Hypertension Research (2022)
-
Implications of CRISPR/Cas9 system in Hypertension and its related diseases
Journal of Human Hypertension (2021)
-
Thiol-based redox-active proteins as cardioprotective therapeutic agents in cardiovascular diseases
Basic Research in Cardiology (2021)
-
Rat models of human diseases and related phenotypes: a systematic inventory of the causative genes
Journal of Biomedical Science (2020)

{kind=link}
{kind=link}