
Abstract
The detection of distribution patterns of genetic diversity of plant and animal species has contributed to the understanding of biodiversity and evolutionary history of the Atlantic Forest. We used microsatellite markers to access the genetic diversity and structure of 26 populations and 527 adult individuals of Euterpe edulis, a native palm which is an important food resource for fauna and is intensively exploited due to economic reasons. We found high genetic diversity and inbreeding in all populations analyzed. We report highest rates of inbreeding for this species, which could reflect the anthropic impacts of selective cutting, fragmentation, and change in foraging behavior from pollinators and less availability and mobility of large dispersers. We detected by STRUCTURE, two genetic groups, Northern and Southern, which divide the Brazilian Atlantic Forest geographically. These groups have low genetic admixtures, but we found a region of lineage hybridization in the contact zone with low recent gene flow. Distribution pattern of this species corroborates results from previous studies reporting the Last Glacial Maximum (LGM) have shaped the structuring of the species through movements of forests’ expansion and contraction. The STRUCTURE analysis of each group revealed the presence of genetic subgroups with low rates of recurrent gene flow. Southern subgroups have higher rates of admixtures than the Northern subgroups, revealing greater historical connectivity of forests in this region.
Similar content being viewed by others

Introduction
The Atlantic Forest (Fig. 1) is the second-largest tropical forest in South America extending latitudinally from tropical to subtropical regions along the Brazilian coast, eastern Paraguay, and northeastern Argentina (Joly et al. 1999; Oliveira-Filho and Fontes 2000; Ribeiro et al. 2009). Longitudinal extension provides the difference in forest composition and rainfall regime, such that on the coast where the forests are wetter and inland, where forests are drier (Câmara 2003). In addition, this biome has strong seasonality and environmental gradients connected to complex topography, making it exceptionally diverse and encompassing various types of vegetation (Fundação Instituto Brasileiro de Geografia e Estatística 1993).

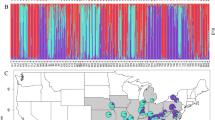
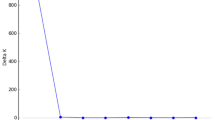
A Geographic distribution in the Brazilian Atlantic Forest (light greenish-gray) and lineage assignments of populations to Northern group (dark purple) and Southern group (dark green). The diagram represents the average of each proportion of assignment (Q) of the genetic groups for the population contemporary gene pool. In light gray the map of Brazil, highlighting the region sampled in dark gray. B ΔK statistic, based on the likelihood function, identified K = 2 as the most appropriate number of genetic groups. C Graph of cluster analysis in STRUCTURE. Each horizontal bar represents an individual. The colors represent the coefficient of association for each genetic group (see Table 1 for population codes). AL Alagoas, BA Bahia, DF Distrito Federal, ES Espírito Santo, MG Minas Gerais, SP São Paulo, PR Paraná, SC Santa Catarina, and RS Rio Grande do Sul.
Studies which analyzed Atlantic Forest species across geographic distribution reported latitudinal structure of genetic diversity and endemism in a north–south trend (Carnaval and Moritz 2008; Carnaval et al. 2009; Palma-Silva et al. 2009; d’Horta et al. 2011; Thomé et al. 2010; Aguiar-Melo et al. 2019; Turchetto-Zolet et al. 2013). The dynamics of the population’s expansion and contraction due to climatic oscillations of the Last Glacial Maximum (LGM) may imprint a signal in the genetic species over time (Hewitt 2000; Gugger et al. 2013). Bottlenecks, genetic drift, and consequent loss of genetic diversity was expected in species during forest contraction (Pauls et al. 2013). During forest expansion, it was expected an exchange of migrants between populations, promoting high genetic diversity which could increase genetic diversity per heterosis (Ortego et al. 2012). However, colonization of new habitats can result in loss of genetic diversity due to founder effects or due to the occurrence of outbreeding depression (Pauls et al. 2013; Collevatti et al. 2014).
The maintenance of biodiversity during the LGM’s climate change took place through the green areas which remained climate stable. Three areas were identified in the Atlantic Forest, the Pernambuco and Bahia refuge to the north and the São Paulo refuge to the south (Carnaval et al. 2008; Carnaval et al. 2009). Palaeoclimatic evidence shows that climatic variability impacted the Northern and the Southern regions of the Atlantic Forest differently. The northern part was influenced by the drier conditions of the Eastern Amazon during the LGM, becoming more humid in the Middle Holocene (6 kay BP). The opposite occurred in the south of the Atlantic Forest, in the western Amazon, and in the Andes, so that the LGM was more humid and the Holocene drier (Cheng et al. 2013; Carnaval et al. 2014). This alternation may have promoted the genetic isolation of lineages and divergence over time, through genetic drift.
The distribution of the Euterpe edulis Martius palm (Arecaceae) in the Atlantic Forest of Brazil, Argentina, and Paraguay and in gallery forests of the Brazilian Cerrado (Henderson et al. 1995), was influenced by climatic events that have occurred since the LGM, mainly by the reduction of habitat suitability which occurred in the Holocene, restricting the species to the coast but preserving genetic diversity (Carvalho et al. 2017). Data obtained from fragmented and exploited areas show that young individuals and adults respond differently to human pressure, but the consequences of the exploitation process as an increase in inbreeding, decrease in diversity and genetic structuring, was not identified in the populations as expected (Santos et al. 2015; Gaiotto et al. 2003; Carvalho et al. 2016; Konzen and Martins 2017; Lauterjung et al. 2019). Many of the works developed with E. edulis cover restricted sampling areas, and for this reason, it is interesting to recognize possible distribution patterns of E. edulis genetic diversity throughout the biome.
The aim of this study consisted in evaluating the diversity and structuring of E. edulis along the Brazilian Atlantic Forest, to provide new insights about the genetic variability of this key species, which can drive sustainable strategies for conservation and use. We expected to find a high genetic diversity of E. edulis and detect latitudinal genetic diversity patterns throughout its distribution in the Brazilian Atlantic Forest.
Materials and methods
Study species
Euterpe edulis (Arecaceae) is a palm threatened with extinction, found in the Atlantic Forest of Brazil, Argentina, and Paraguay, and in gallery forests of the Brazilian Cerrado (Henderson et al. 1995). This species is widely distributed in the biome, occurring in different vegetation types within the Atlantic Forest, including Ombrophylous, Deciduous, and Restinga forests (Henderson et al. 1995; Gatti et al. 2008), mainly associated with humid environments or close to watercourses (Galetti and Fernandez 1998).
The fruits of E. edulis are small and round (1 to 1.5 cm diameter), weighing about 1 g. The seed constitutes 85% of the fruit and the mesocarp is fleshy and very thin, being an important food resource for birds and mammals (Bicudo et al. 2014; Galetti et al. 2013). During the ripening process, the color turns from green to black (Bicudo et al. 2014). Currently, the pulp extracted from ripe fruits is an income source for many smallholder communities due to its similarity to açai (Bourscheid 2011).
Euterpe edulis is a species of a single nonstoloniferous strain. It is monoic and pollinated by small Trigonas bees (Reis and Kageyama 2000) having a low rate of self-pollination (Gaiotto et al. 2003). For many years this palm was known by the flavor of the edible meristem (heart of palm), much appreciated in cooking (Reis and Kageyama 2000). Target of intense exploitation since the 1960s, many populations of the species have been extinct locally or drastically reduced due to indiscriminate cutting (Fantini et al. 2000).
Sampling
We checked the occurrence and distribution of the species through Species Link (www.splink.org.br), Reflora (floradobrasil.jbrj.gov.br), and ICMBio (www.icmbio.gov.br) to select the sampling areas. We sampled 26 locations with natural populations of Euterpe edulis, 25 from the Brazilian Atlantic Forest Biome and one from the Cerrado Biome (Table 1 and Supplementary Material, Fig. S1). Samples ranged from 10 to 29 adult individuals from conserved vegetation areas, maintaining a distance of at least 5 meters between individuals. Euterpe edulis forms agglomerates of adult and young plants. We sought to sample different agglomerates from the same area to compose the samples. The population codes, localities, geographic coordinates, number of specimen, and references are shown in Table 1. The pairwise geographic distance (Km) among populations is shown in Table S1.
DNA extraction, microsatellite genotyping
We obtained the samples from the cortex of 527 adult individuals of E. edulis. Total genomic DNA was extracted according to a previously published protocol by Carvalho et al. (2019). The genotyping was performed using ten microsatellite markers dye-labeled for E. edulis published by Gaiotto et al. (2001) (see Table S2). These markers are codominant, highly variable, and widely used in the scientific community for work with E. edulis and species of the genus, which favors the comparison and interpretation of results. The selected microsatellite markers, the conditions for amplification and analysis of the fragments are displayed in Table S2.
We used one sample as a standard for fragment size. This sample was genotyped in all plates. When we detected changes in the standard’s size, we corrected all other samples accordingly. We used this approach to avoid variation between different plates. Alleles were sized in an automated DNA sequencer (3500 Genetic Analyzer—Applied Biosystems) using GeneMarker software v.2.4 (Hulce et al. 2011) and LIZ 500 Size Standard (Applied Biosystems).
Data analysis—genetic diversity and population structure using molecular data
Initially, null allele frequencies for each locus were estimated using the Micro-Checker software version 2.2.3 (Van Oosterhout et al. 2004) with the Oosterhout algorithm. Subsequently, we used the FSTAT software, version 2.9.3.2 (Goudet 2002), to calculate the linkage disequilibrium between loci in all populations. The GDA software (Lewis and Zaykin 2002) was used to calculate the number of private alleles (Apriv), the number of alleles per locus (A/locus), and the observed heterozygosity (HO). We also estimated the allelic richness (AR) and expected heterozygosity (HE)—considering the differences in sample size among populations under analysis using the software FSTAT 2.9.3.2 (Goudet 2002). Finally, the software INEST 2.2 (Chybicki and Burczyk 2009) was used to estimate a corrected F accounting for the potential impacts of null alleles. This analysis was performed with the model based on a Bayesian approach (IIM), with 500000 Markov chain Monte Carlo interactions and burn-in of 50000.
The Mantel test was used to assess the correlation between geographical and genetic distances, as a predictor for isolation by distance (IBD). The pairwise Rousset’s genetic distances FST/(1–FST) and the pairwise geographic distances (log transformed) of populations were carried out using the SPAGeDi (Hardy and Vekemans 2002). The correlation was evaluated using the Mantel test with 100.000 permutations, using the software R, package Vegan. Also, a clustering analysis was performed by the UPGMA method in the R software, using the pairwise Nei’s standard distance among populations, which was estimated in the SPAGeDi.
To analyze E. edulis’ genetic structure, we used the Bayesian approach of STRUCTURE 2.3.4 with admixture model (Pritchard et al. 2000) and frequency parameters of correlated alleles (Falush et al. 2003), with no prior information on the structure of the population. We used the parallelization of STRUCTURE executed in the program StrAuto (Chhatre and Emerson 2017) under Linux environment. This analysis allowed the smallest computational time. The run parameters were a burn-in period of 25,0000 steps and 1,000,000 sampling steps with 20 independent repetitions of the Markov Monte Carlo Chain (MCMC) executions.
We allowed the model to analyze the probability of individuals belonging to different Bayesian groups (K; the number of sampling sites plus 3, here K = 29) by analyzing the set of allele frequencies in each locus (Evanno et al. 2005; Pritchard et al. 2010). The best K was identified in Structure Harvester (Earl and VonHoldt 2011) using the ΔK method (∆K = m(|L′′(K)|)/s[L(K)]; Evanno et al. 2005). The ΔK graph shows the distribution peak at the best value of K groups for the analyzed samples. These analyses allowed the detection of two distinct clusters with low admixture (see results). A separate STRUCTURE analysis was run with each of the genetic groups to identify subgroups. In these new analyses we considered K = 15 and K = 17. The CLUMPACK software (Kopelman et al. 2015) was used to align the 20 independent runs of STRUCTURE obtained for the best K value and to generate graphical outputs. For each population, we used the membership coefficients generated with CLUMPACK to obtain a diagram of the relative contribution of each lineage to the contemporary gene pool.
To assess whether stepwise mutations could have contributed significantly to genetic differentiation among populations or not, we applied an allele-size permutation test (Hardy et al. 2003) implemented in the SPAGeDi (Hardy and Vekemans 2002). This test compares the global RST values taking into account the allele sizes by stepwise mutations with a distribution of pRST values which consider allele identities by infinite allele model. If Global RST values >95% of the pRST indicates stepwise mutations contributed to population differentiation and can be interpreted as presence of phylogeographical pattern (Hardy et al. 2003). We used 1000 allele-size permutations to run the test.
Partitioning of genetic variability within and among groups was tested by analysis of molecular variance (AMOVA) between the groups and subgroups derived from STRUCTURE, using ARLEQUIN v.3 (Excoffier et al. 2005). In ARLEQUIN, we also computed pairwise FST and RST of populations and tested the significance using 1000 permutations with 95% confidence intervals.
Contemporary gene flow rates (m) were assessed using the BAYESASS edition 3 software (Wilson and Rannala 2003), which estimates the proportion of migrants per generation over the past three generations. E. edulis requires about 10 years to reach its reproductive maturity (Leitman et al. 2013), and exhibits an overlapping of generations. Therefore, all estimates of gene flow must consider the past 30 years, and genetic processes after intensive habitat fragmentation and specie’s exploitation in the study area. We used the genetic groups and subgroups detected by STRUCTURE to identify the clusters of populations. We carried out five independent simulations with random initial seeds following the recommendations of Chávez-Pesqueira and Núñez-Farfán (2016). Each run consisted of an MCMC of 108 iterations, sampled every 1000 iterations, with 107 iterations discarded as burn-in. The values of the MCMC admixture parameters for migration rates (gene flow), allele frequencies, and inbreeding coefficients were adjusted to achieve the recommended acceptance rates in the literature (Wilson and Rannala 2003). This procedure was repeated for each subcluster reported by STRUCTURE. The stationarity of the MCMC for each execution was confirmed using TRACER (Rambaut and Drummond 2007).
Results
Genetic diversity and genetic differentiation
One of the 10 loci (EE52) exhibited null allele frequency above 0.2 and was discarded from all subsequent analyses. A total of 178 alleles were identified for the remaining nine loci; the number of alleles per locus ranged from 14 (EE9) to 24 (EE54) (Supplementary Material, Table S2). Pairwise comparisons among the 9 loci revealed no evidence of linkage disequilibrium. Observed and expected heterozygosity values per population ranged from 0.36 (JIBO and VALE) to 0.68 (CAPE) and from 0.54 (TALH) to 0.81 (ITAT), respectively, rendering mean values of 0.54 and 0.75. The values of inbreeding coefficient (F) were high in almost all populations, ranging from 0.03 (CAPE and IBIC) to 0.28 (VALE) with an overall mean of 0.07. Only nine of 26 populations had private alleles; TALH, AZUL, ARAR, and PAL2 (a single private allele per population); ITAP (two private alleles); DESC and SAGU (three private alleles per population); and IBGE and VALE (five private alleles per populations) (Table 1).
Regarding global RST, the average among all sample populations was significantly higher than the permuted value (RST = 0.44, pRST = 0.19, P = 0.003; Supplementary Material, Table S3). This suggests that stepwise mutations contributed to population differentiation in E. edulis and thus RST should be the estimator preferred over FST (Hardy et al. 2003). Pairwise RST values ranged from 0.03 (between IBIC and IGUA populations) to 0.81 (between VERA and OURO populations); all pairwise RST values were significant (P < 0.05), except for 16 pairs that displayed low population differentiation (RST < 0.04; Supplementary Material, Table S4).
Genetic structure
The Bayesian clustering analysis completed with the software STRUCTURE suggested two geographic groups (best K = 2, Fig. 1A, B) for E. edulis. The first one denominated the Northern group (depicted in purple; Fig. 1A, C) consisted of populations sampled in the north of the Brazilian Atlantic Forest (from Alagoas to Espírito Santo states), and the second group denominated the Southern group (depicted in green; Fig. 1A, C) consisted of populations sampled in the south of the Brazilian Atlantic Forest (from Rio de Janeiro to the Rio Grande do Sul states); the only population of E. edulis sampled in the Cerrado (IBGE population) was grouped in the Northern group (Fig. 1A, C). Most populations showed a proportion of assignment over 90% to either cluster Northern or Southern (Table 1). In particular, the highest levels of admixture were detected in populations near the geographic contact between genetic groups, as the ARAR, ITAT populations from Rio de Janeiro state, and the ARCO and VALE populations from Espírito Santo state (Fig. 1A and Table 1).
The subsequent STRUCTURE analyses carried out separately for the Northern and the Southern groups are shown in Fig. 2. A value of K = 2 was found for both analyses (Fig. 2B, C, D). The populations within each group (Northern or Southern) were separated according to their geographic location, giving rise to the Northeast and the Central-Southeast subgroups as well as the Southeast and Sub-South subgroups (Fig. 2). The correlation between geographical and genetic distances was significant by the Mantel test (0.59; P = 0.0001), however the clustering pattern obtained in the STRUCTURE analysis was corroborated by UPGMA method (Fig. S2).

A Geographic distribution in the Brazilian Atlantic Forest (light greenish-gray) and lineage assignments of populations to Northeast subgroup (light purple), Central-North subgroup (purple), Southeast subgroup (green) and Sub-South subgroup (light green). The diagram represents the average of each proportion of assignment (Q) of the genetic groups for the population contemporary gene pool. In light gray the map of Brazil, highlighting the region sampled in dark gray. ΔK statistic, based on likelihood function, identified K = 2 for the Northern (B) and Southern (C) genetic group as the most appropriate number of genetic groups. The colors in purple tones represent the North group, and the colors in green tones represent the South group. D Graph of the cluster analysis in STRUCTURE. Each horizontal bar represents an individual. The colors indicate an association coefficient for each genetic subgroup (see Table 1 for population codes). The rivers Paraná and Paranapanema favor the maintenance of remnants of the Brazilian Atlantic Forest along its course and are possible limits for the distribution of contemporary plant lineages (Martins 2011). AL Alagoas, BA Bahia, DF Distrito Federal, ES Espírito Santo, MG Minas Gerais, SP São Paulo, PR Paraná, SC Santa Catarina, and RS Rio Grande do Sul.
Contemporary migration rates were estimated as extremely low between groups (Northern and Southern) and between subgroups pairs (Northeast and the Central-Southeast; Southeast and Sub-South), with values of m ≤ 0.006 in all comparisons (Fig. 3). Consequently, extremely high migration rates (m > 0.99) were observed within each group or subgroup (Fig. 3).

A Rate of gene flow between the Northern (dark purple) and Southern (dark green) genetic groups. B Rate of gene flow between the Northeast (lilac) and Central-North (purple) genetic subgroups. C Rate of gene flow between the Southeast (green) and Sub-South (light green) genetic subgroups. The groups and subgroups analyzed were detected by population structure analysis using the STRUCTURE software. The circles correspond to each genetic group and subgroup and the size of the circles indicates the sample size (North groups with 230 individuals and south with 297 individuals; Northeast subgroups with 110 individuals, Central-North with 120 individuals, Southeast with 118 individuals and Sub-South with 179 individuals). The arrows inside the circle indicate the gene flow within the population and the horizontal arrows indicate the direction of gene flow between genetic groups and subgroups and their thickness varying with intensity.
Hierarchical AMOVA showed that 26.74% of the genetic variation was explained by differences between Northern and Southern groups, with an RST value of 0.53; on the remaining variation, 26.60% was observed among populations within groups, and 46.66% was within populations (Supplementary Material, Table S5). When AMOVA was evaluated within each group, Northern group had 33.03% of the genetic variation attributed to differences between Northeast and the Central-Southeast subgroups, while the Southern group had only 4.60% of the genetic variation attributed to differences between Southeast and Sub-South subgroups (Supplementary Material, Table S5). The genetic differentiation based on RST for the Northern subgroups and the Southern subgroups were of 0.58 and 0.15, respectively.
Overall, when populations of E. edulis were evaluated based on genetic groups, the mean genetic diversity was similar in both Northern and Southern groups (Table 1) and 37.64% of alleles are shared by both groups (Supplementary Material, Table S6). However, the Northern group had higher amounts of private alleles (Apriv = 18) compared to the Southern group (Apriv = 4). Analyzing the subgroups, the Center-Southeast has more private alleles (Apriv = 14) than the other subgroups and the Northeast has higher average alleles per locus (A/locus = 11.70; Central-Southeast, A/locus = 7.60; Southeast, A/locus = 6.70 and Sub-South, A/locus = 7.16) and a larger number of alleles considering all loci (Supplementary Material, Table S6).
Discussion
In this study, we used microsatellite markers to evaluate the genetic diversity, and structure of the palm E. edulis along the Brazilian Atlantic Forest. Our main results indicated: (1) high within-population genetic diversity and inbreeding coefficients (F range from 0.03 to 0.28); (2) the existence of phylogeographic pattern with two geographical genetic groups within the range of E. edulis, the Northern and Southern groups; (3) further subdivision of the Northern and Southern groups, the Northeast and Central-Southeast subgroups and the Southeast and Sub-South subgroups; and (4) limited genetic admixture between groups or subgroups.
High levels of genetic diversity and inbreeding of Euterpe edulis
The high levels of genetic diversity revealed in the E. edulis populations (mean HE = 0.75; Table 1) is in accordance with previous studies on this species (Carvalho et al. 2015; Santos et al. 2015; Carvalho et al. 2017; Novello et al. 2017) and reinforces further the influence of the large historical effective size of E. edulis populations to the conservation of their current genetic diversity (Carvalho et al. 2015; Santos et al. 2015; Soares et al. 2019).
We detected inbreeding in all populations (Table 1), including relevant levels in Brazilian conservation areas as BOCA (F = 0.14) and ITAP (F = 0.12). The highest inbreeding value was found in the VALE population (F = 0.28), followed by JIBO (F = 0.24), both with a history of logging and further preservation. In populations with low density, the Trigona spinipes bee can keep inbreeding low by moving pollen between geographically more distant palm trees (800 m maximum flight distance) (Zurbuchen et al. 2010; Côrtes et al. 2013; Carvalho et al. 2017). However, reestablishing the high density of population, some intercrossing among kinship could contribute to the increase of inbreeding generation after generation (Ghazoul 2005; Carvalho et al. 2017). Furthermore, there is a positive relationship between the density of mother plants and the number of dispersed seeds, promoting greater dispersion of related genotypes (Carvalho et al. 2021).
Two genetic lineages in the Brazilian Atlantic Forest revealed by Microsatellite markers
We show that E. edulis has two genetic lines, to the North and South of the Brazilian Atlantic Forest (RST = 0.53). A large number of private alleles in the Northern group (Apriv = 18, Table 1) and the low levels of admixture observed between the genetic groups are indicative of long-time isolation of population, low historical gene flow, and differentiation by genetic drift. Similar values of genetic diversity between the Northern and Southern groups (Table 1) show the maintenance of genetic heterogeneity along the Atlantic Forest.
The analysis carried out within each genetic group showed the structuration of subgroups, with isolation after the differentiation of the Northern and Southern groups. The differentiation between the subgroups has similar intensity to the differentiation observed in the groups, due to the low genetic mixture between them (Fig. 2) and high RST = 0.58 (Table S5). We did not identify an increase in values of genetic diversity or private alleles in populations in the Northeast subgroup, as expected for areas next to refugees of Bahia and Pernambuco (Carnaval et al. 2009). However, we found a larger number of different alleles, suggesting the persistence of the species in this area (Petit et al. 2002). The high rate of private alleles in the IBGE and VALE populations (five each, Table 1) suggests small E. edulis refuges in the states of Espírito Santo state and Distrito Federal (Central-North subgroup). These regions showed habitat suitability throughout the LGM climate change as detected by Carvalho et al. (2017).
We found less differentiation between Southern subgroups (percentage variation = 4.6), higher mixing ratios between subgroup lineages, and a low number of private alleles (Apriv = 4, Table 1) as expected in areas with shorter isolation time (Szpiecha and Rosenberga 2011). This suggests that Southern subgroups come from a more recent structuring process than the subgroups of the Northern Group.
The current low gene flow between the groups and the subgroups (Fig. 3) arises possibly due to Atlantic Forest fragmentation, loss of large frugivores, and greater difficulties in seed movement (Carvalho et al. 2015; Carvalho et al. 2016; Carvalho et al. 2021) between populations. Thus, the genetic exchanges of groups and subgroups reflect conditions that are much older than those predicted by the gene flow analysis (Fig. 3).
The Southeast and Sub-South subgroups of E. edulis showed a limit of division between the states of São Paulo and Paraná. This delimitation is in accordance with the distribution limits of habitat suitability in different climatic cycles (Carvalho et al. 2017). Some features of this region are identified as isolating factors for some species, such as the plateau in the state of Paraná (Cortez et al. 2019), the Paraná and Paranapanema rivers (Martins 2011) and the transition between tropical and subtropical forests due to difference in annual precipitation and soil types (Cabanne et al. 2007; Cabanne et al. 2008; Thomé et al. 2010; d’Horta et al. 2011).
The diversity and endemism, Northern and Southern oriented, is a phylogeographic pattern already reported for other species in the Brazilian Atlantic Forest (Martins et al. 2007; Carnaval et al. 2014; Costa et al. 2017). Carnaval et al (2014) relate this to the influence of different climatic regimes during the Last Glacial Maximum (LGM = 21 kyr PB). The climatic fluctuation in the Quaternary helps to understand the suitability of habitat for E. edulis during the LGM. Our hypothesis is that these cycles of retraction and expansion may have led to the differentiation of the Northern and Southern groups of E. edulis, due to the loss of genetic variability and differentiation of populations during retractions and hybridization in contact zones of different groups during expansions.
The habitat suitability analysis for E. edulis showed that the Brazilian Atlantic Forest coast has remained stable throughout climate change and may represent refuge for the species (Carvalho et al. 2017). Phylogeography studies can help to understand the distribution of E. edulis genetic diversity through the dating of the divergence of the Northern and Southern groups and testing our hypothesis. The evolutionary history of the southern part of the Brazilian Atlantic Forest is complex and involves several processes (Martins 2011). Therefore, our study with E. edulis clearly provided useful information to help the understanding of the evolution of the southern part of the Brazilian Atlantic Forest.
The identification of different genetic lineages, in the Northern and Southern regions of the Brazilian Atlantic Forest, shows that it is necessary to increase the sampling effort in restoration projects to obtain seeds that contribute to the remaining gene pool of different geographic regions. Our work showed the importance of conservation areas for the preservation of E. edulis. Therefore, we support the creation of new units like the proposal for the Recôncavo Sul Baiano (Blengini et al. 2015) that includes the JIBO population. Struggling with illegal palm heart cutting is a challenge, mainly due to the difficulties of monitoring large conserved areas. We reinforce the importance of support and investment in inspection, as well as the use of the species in forest restoration projects.
Data availability
Microsatellite genotypes and geographic location of samples are available from the Agri-Environmental Research Data Repository at the University of Guelph (https://doi.org/10.5683/SP2/KRHVEH).
Change history
10 August 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41437-022-00556-8
References
Aguiar-Melo C, Zanella CM, Goetze M, Palma-Silva C, Hirsch LD, Neves B et al. (2019) Ecological niche modeling and a lack of phylogeographic structure in Vriesea incurvata suggest historically stable areas in the southern Atlantic Forest. Am J Bot https://doi.org/10.1002/ajb2.1317
Bicudo MOP, Ribani RH, Beta T (2014) Anthocyanins, phenolic acids and antioxidant properties of juçara fruits (Euterpe edulis M.) along the on-tree ripening process. Plant Foods Hum Nutr https://doi.org/10.1007/s11130-014-0406-0
Blengini IAD, Cintra MAMU, Caiafa AN (2015) Proposta de Unidade de Conservação da Serra da Jiboia. Gambá, Salvador, BA, https://www.gamba.org.br/wp-content/uploads/2016/03/Proposta-Final.pdf Accessed 05 May 2022
Bourscheid K (2011) Euterpe edulis—Palmito juçara. In: Coradin L, Siminski A, Câmara, Reis A (Eds) Espécies nativas da flora brasileira de valor econômico atual ou potencial: plantas para o futuro - Região Sul. Ministério do Meio Ambiente, Brasília, D, p 179–183
Cabanne GS, d’Horta FM, Sari EHR, Santos FR, Miyaki CY (2008) Nuclear and mitochondrial phylogeography of the Atlantic forest endemic Xiphorhynchus fuscus (Aves: Dendrocolaptidae): Biogeography and systematics implications Molecular. Mol Phylogenet Evol https://doi.org/10.1016/j.ympev.2008.09.013
Cabanne GS, Santos FR, Miyaki CY (2007) Phylogeography of Xiphorhynchus fuscus (Passeriformes, Dendrocolaptidae): vicariance and recent demographic expansion in southern Atlantic forest. Biol J Linn Soc https://doi.org/10.1111/j.1095-8312.2007.00775.x
Câmara IG (2003) Brief history of conservation in the Atlantic Forest. In: Galindo Leal C, Câmara IG (Eds.) The Atlantic Forest of South America: Biodiversity Status, Threats, and Outlook. CABS and Island Press, Washington, p 31–42
Carnaval AC, Moritz C (2008) Historical climate modelling predicts patterns of current biodiversity in the Brazilian Atlantic Forest J Biogeogr https://doi.org/10.1111/j.1365-2699.2007.01870.x
Carnaval AC, Moritz C, Hickerson M, Haddad C, Rodrigues M (2009) Stability predicts diversity in the Brazilian Atlantic Forest hotspot. Science https://doi.org/10.1126/science.1166955
Carnaval AC, Waltari E, Rodrigues MT, Rosauer D, VanDerWa J, Damasceno R et al. (2014) Prediction of phylogeographic endemism in an environmentally complex biome. Proc R Soc Lond https://doi.org/10.1098/rspb.2014.1461
Carvalho CDS, Garcia C, Lucas MS, Jordano P, Cortes MC (2021) Extant fruit‐eating birds promote genetically diverse seed rain, but disperse to fewer sites in defaunated tropical forests. J Ecol https://doi.org/10.1111/1365-2745.13534
Carvalho CS, Ballesteros-Mejia L, Ribeiro MC, Côrtes MC, Santos AS, Collevatti RG (2017) Climatic stability and contemporary human impacts affect the genetic diversity and conservation status of a tropical palm in the Atlantic Forest of Brazil Conserv Genet https://doi.org/10.1007/s10592-016-0921-7
Carvalho CS, Galetti M, Colevatti RG, Jordano P (2016) Defaunation leads to microevolutionary changes in a tropical palm. Sci Rep https://doi.org/10.1038/srep31957
Carvalho CS, Ribeiro MC, Côrtes MC, Galetti M, Collevatti RG (2015) Contemporary and historic factors influence differently genetic differentiation and diversity in a tropical palm. Heredity https://doi.org/10.1038/hdy.2015.30
Carvalho MS, Noia LR, Ferreira MFS, Ferreira A (2019) DNA de alta qualidade isolado a partir do córtex de Euterpe edulis Mart. (Arecaceae). Cienc Florest https://doi.org/10.5902/1980509824130
Chávez-Pesqueira M, Núñez-Farfán J (2016) Genetic diversity and structure of wild populations of Carica papaya in Northern Mesoamerica inferred by nuclear microsatellites and chloroplast markers. Ann Bot https://doi.org/10.1093/aob/mcw183
Cheng H, Sinha A, Cruz FW, Wang X, Edwards RL, d’Horta FM et al. (2013) Climate change patterns in Amazonia and biodiversity. Nat Commun https://doi.org/10.1038/ncomms2415
Chhatre VE, Emerson KJ (2017) StrAuto: automation and parallelization of STRUCTURE analysis. BMC Bioinform https://doi.org/10.1186/s12859-017-1593-0
Chybicki IJ, Burczyk J (2009) Simultaneous Estimation of Null Alleles and Inbreeding Coefficients. J Hered https://doi.org/10.1093/jhered/esn088
Collevatti RG, Lima-Ribeiro MS, Terribile LC et al. (2014) Recovering species demographic history from multi-model inference: the case of a Neotropical savanna tree species. BMC Evol Biol https://doi.org/10.1186/s12862-014-0213-0
Côrtes MC, Uriarte M, Lemes MR, Gribel R, Kress WJ, Smouse PE et al. (2013) Low plant density enhances gene dispersal in the Amazonian understory herb Heliconia acuminata. Mol Ecol https://doi.org/10.1111/mec.12495
Cortez MBS, Sforça DA, Alves FM, Vidal JD, Alves-Pereira A, Mori GM, Andreotti IA et al. (2019) Elucidating the Clusia criuva species ‘complex’: cryptic taxa can exhibit great genetic and geographical variation. Biol J Linn Soc https://doi.org/10.1093/botlinnean/boz004
Costa PC, Lorenz-Lemke AP, Furini PR, Honorio Coronado EN, Kjellberg F, Pereira RA (2017) The phylogeography of two disjunct Neotropical Ficus (Moraceae) species reveals contrasted histories between the Amazon and the Atlantic Forests. Biol J Linn Soc https://doi.org/10.1093/botlinnean/box056
d’Horta FM, Cabanne GS, Meyer D, Miyaki CY (2011) The genetic effects of Late Quaternary climatic changes over a tropical latitudinal gradient: diversification of an Atlantic Forest passerine. Mol Ecol https://doi.org/10.1111/j.1365-294X.2011.05063.x
Earl DA, Von Holdt BM (2011) STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet Resources https://doi.org/10.1007/s12686-011-9548-7
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Laval G, Schneider S (2005) Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform PMCID: PMC2658868
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics PMCID: PMC1462648
Fantini AC, Guries R, Ribeiro RJ (2000) Produção de palmito (Euterpe edulis Martius Arecaceae) na Floresta Ombrófila Densa: potenciais, problema e possíveis soluções. In: Reis MS, Reis A (Eds.) Euterpe edulis Martius (Palmiteiro) Biologia, Conservação e Manejo. Herbário Barbosa Rodrigues, Itajaí, p 256–280
Fundação Instituto Brasileiro de Geografia e Estatística (1993) Recursos naturais e meio ambiente: Uma visão do Brasil. Fundação Instituto Brasileiro de Geografia e Estatística, Rio de Janeiro
Gaiotto FA, Brondani RPV, Grattapaglia D (2001) Microsatellite markers for heart of palm–Euterpe edulis and E, oleracea Mart, (Arecaceae). Mol Ecol Notes https://doi.org/10.1046/j.1471-8278.2001.00036.x
Gaiotto FA, Grattapaglia D, Vencovsky V (2003) Genetic structure, mating system, and long-distance gene flow in heart of palm (Euterpe edulis Mart.). J Hered 94(5):399–406. https://doi.org/10.1093/jhered/esg087
Galetti M, Fernandez JC (1998) Palm heart harvesting in the Brazilian Atlantic forest: changes in industry structure and the illegal trade. J Appl Ecol https://doi.org/10.1046/j.1365-2664.1998.00295.x
Galetti M, Guevara R, Côrtes MC, Fadini R, Von Mattes S, Leite AB et al. (2013) Functional extinction of birds drives rapid evolutionary changes in seed size. Science https://doi.org/10.1126/science.1233774
Gatti MG, Campanello PI, Montti LF, Goldstein G (2008) Frost resistance in the tropical palm Euterpe edulis and its pattern of distribution in the Atlantic Forest of Argentina. For Ecol Manag https://doi.org/10.1016/j.foreco.2008.05.012
Ghazoul J (2005) Pollen and seed dispersal among dispersed plants. Biological Reviews Cambridge Philosophical Society. https://doi.org/10.1017/S1464793105006731
Goudet J (2002) FSTAT: a program to estimate and test gene diversities and fixation indices (version 2.9.3.2). http://www2.unil.ch/popgen/softwares/fstat.htm
Gugger PF, Ikegami M, Sork VL (2013) Influence of late Quaternary climate change on present patterns of genetic variation in valley oak, Quercus lobata. Mol Ecol https://doi.org/10.1111/mec.12317
Hardy OJ, Charbonnel N, Fréville H, Heuertz M (2003) Microsatellite allele sizes: a simple test to assess their significance on genetic differentiation. Genetics https://doi.org/10.1093/genetics/163.4.1467
Hardy OJ, Vekemans X (2002) SPAGEDI: a versatile computer program to analyses spatial genetic structure at the individual or population levels. Mol Ecol Notes https://doi.org/10.1046/j.1471-8286.2002.00305.x
Henderson A, Galeano G, Bernal R (1995) Field guide to the palms of the Americas. Princeton University Press, Princeton, NJ, p 352
Hewitt G (2000) The genetic legacy of the Quaternary ice ages. Nature https://doi.org/10.1038/35016000
Hulce D, Li X, Snyder-Leiby T, Johathan Liu CS (2011) GeneMarker® Genotyping Software: tools to increase the statistical power of DNA fragment analysis. J Biomol Screen PMCID: PMC3186482
Joly C, Aidar M, Klink CA, McGrath DG, Moreira AG, Moutinho P et al. (1999) Evolution of the Brazilian phytogeography classification systems: implications for biodiversity conservation. Ciên e Cul 51:331–348
Konzen ER, Martins, MP (2017) Contrasting levels of genetic diversity among populations of the endangered tropical palm Euterpe edulis Martius, Cerne https://doi.org/10.1590/01047760201723012237.
Kopelman NM, Mayzel J, Jakobsson M, Rosenberg NA, Mayrose I (2015) Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol Ecol Resour https://doi.org/10.1111/1755-0998.12387
Lauterjung MB, Montagna T, Bernardi AP, Silva JZ, Freitas NCC, Steiner, F et al. (2019) Temporal changes in population genetics of six threatened Brazilian plant species in a fragmented landscape. For Ecol Manag https://doi.org/10.1016/j.foreco.2018.12.058
Leitman P, Judice DM, Barros FSM, Prieto PV (2013) Arecaceae, In: Martinelli G, Moraes MA (org) Livro Vermelho da Flora do Brasil. CNCFlora, Rio de Janeiro, pp 187–195
Lewis PO, Zaykin D (2002) Genetic data analysis: Computer program for the analysis of allelic data. http://phylogeny.uconn.edu/software/
Martins FM (2011) Historical biogeography of the Brazilian Atlantic forest and the Carnaval—Moritz model of Pleistocene refugia: what do phylogeographical studies tell us? Biol. J Linn Soc https://doi.org/10.1111/j.1095-8312.2011.01745.x
Martins FM, Ditchfield AD, Meyer D, Morgante JS (2007) Mitochondrial DNA phylogeography reveals marked population structure in the common vampire bat, Desmodus rotundus (Phyllostomidae). J Zoolog Syst Evol https://doi.org/10.1111/j.1439-0469.2007.00419.x
Novello M, Viana JPG, Alves-Pereira A, Silvestre EA, Nunes HF, Pinheiro JB et al. (2017) Genetic conservation of a threatened Neotropical palm through community-management of fruits in agroforests and second-growth forests. For Ecol Manag https://doi.org/10.1016/j.foreco.2017.06.059
Oliveira-Filho A, Fontes M (2000) Patterns of floristic differentiation among Atlantic forests in southeastern Brazil and the influence of climate. Biotropica https://doi.org/10.1111/j.1744-7429.2000.tb00619.x
Ortego J, Riordan EC, Gugger PF, Sork VL (2012) Influence of environmental heterogeneity on genetic diversity and structure in an endemic southern Californian oak. Mol Ecol https://doi.org/10.1111/j.1365-294X.2012.05591.x
Palma-Silva C, Lexer C, Paggi GM, Barbará T, Bered F, BodaneseZanettini MH (2009) Range-wide patterns of nuclear and chloroplast DNA diversity in Vriesea gigantea (Bromeliaceae), a neotropical forest species. Heredity 103:503–512
Pauls SU, Nowak C, Bálint M, Pfenninger M (2013) The impact of global climate change on genetic diversity within populations and species. Mol Ecol https://doi.org/10.1111/mec.12152
Petit RJ, Csaiklb UM, Bordácsbc S, Burgb K, Coartd E, Cottrelle J et al. (2002) Chloroplast DNA variation in European white oaks. Phylogeography and patterns of diversity based on data from over 2600 populations. For Ecol Manag https://doi.org/10.1016/S0378-1127(01)00645-4
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics https://doi.org/10.1093/genetics/155.2.945
Pritchard JK, Wen X, Falush D (2010) Documentation for structure software: Version o2.3. http://web.stanford.edu/group/pritchardlab/structure.html
Rambaut A, Drummond AJ (2007) TRACER version 1.4. http://beast.bio.ed.ac.uk/Tracer. Accessed 05 May 2022
Reis A, Kageyama PY (2000) Dispersão de sementes de Euterpe edulis Martius Palmae. In: Reis MS, Reis A (Eds.) Euterpe edulis Martius (Palmiteiro): biologia, conservação e manejo. Herbário Barbosa Rodrigues, Itajaí, p 60–92
Ribeiro MC, Metzger JP, Martensen AC, FPonzoni FJ, Hirota MM (2009) The Brazilian Atlantic Forest: How much is left, and how is the remaining forest distributed? Implications for conservation. Biol Conserv https://doi.org/10.1016/j.biocon.2009.02.021
Santos AS, Cazetta E, Morante Filho JC, Baumgarten J, Faria D, Gaiotto FA (2015) Lessons from a palm: genetic diversity and structure in anthropogenic landscapes from Atlantic Forest, Brazil. Conserv Genet https://doi.org/10.1007/s10592-015-0740-2
Soares LASS, Cazetta E, Santos LR, França DS, Gaiotto FA (2019). Anthropogenic disturbances eroding the genetic diversity of a threatened palm tree: a multiscale approach. Front Genet https://doi.org/10.3389/fgene.2019.01090
Szpiecha ZA, Rosenberga NA (2011) On the size distribution of private microsatellite alleles. Theor Popul Biol https://doi.org/10.1016/j.tpb.2011.03.006
Thomé MTC, Zamudio KR, Giovanelli JGR, Haddad CFB, Baldissera Jr FA, Alexandrino J (2010) Phylogeography of endemic toads and post-Pliocene persistence of the Brazilian Atlantic Forest Mol Phylogenet Evol https://doi.org/10.1016/j.ympev.2010.02.003
Turchetto-Zolet AC, Pinheiro F, Salgueiro F, Palma-Silva C (2013) Phylogeographical patterns shed light on evolutionary process in South America. Mol Ecol https://doi.org/10.1111/mec.12323
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) Micro-checker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes https://doi.org/10.1111/j.1471-8286.2004.00684.x
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics https://doi.org/10.1093/genetics/163.3.1177
Zurbuchen A, Landert L, Klaiber J, Müller A, Hein S, Dorn S (2010) Maximum foraging ranges in solitary bees: only few individuals have the capability to cover long foraging distances. Conserv Biol https://doi.org/10.1016/j.biocon.2009.12.003
Acknowledgements
We thank all the providers of plant material as listed in Table S7 in the Supplementary Information, and Ramon A. Braz, Vanderlei F. de Araujo, and Amanda F. Ulbinski for helping in collecting plant material. We would like to thank all colleagues at the Biometrics and Genetics and Plant Improvement laboratories from the Universidade Federal do Espírito Santo, for helping to process and organize the samples. We are grateful to Daniele S. França, Horlei V. Ribeiro, and Vanessa P. Leite from the Centro de Biotecnologia e Genética at the Universidade Estadual de Santa Cruz for their support with genotyping of microsatellite markers. Financial support to this study was obtained from the Conselho Nacional de Pesquisa (CNPq, Brazil) (Researcher productivity fellowship FAG #312065/2021-3; AF; MFSF #311840/2019-1), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, Brazil)—Finance Code 001, and from the Fundação de Amparo à Pesquisa do Espírito Santo (FAPES, Vitória—ES, Brazil) in partnership with VALE.
Author information
Authors and Affiliations
Contributions
AGP designed research, data analysis and wrote the paper. TCS data analysis and wrote the paper. JHS-G and GBC coordinated sampling. LBA and FN helped with performed molecular biology. FAG assistance with microsatellite genotyping and wrote the paper. MFSF conception and design of the project, secured funding and wrote the paper. AF conception and design of the project, data analysis, secured funding and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor Thomas Meagher.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pereira, A.G., da Silva Ferreira, M.F., da Silveira, T.C. et al. Patterns of genetic diversity and structure of a threatened palm species (Euterpe edulis Arecaceae) from the Brazilian Atlantic Forest. Heredity 129, 161–168 (2022). https://doi.org/10.1038/s41437-022-00549-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00549-7
