
Abstract
Background and objectives
The EYS gene is an important cause of autosomal recessive retinitis pigmentosa (arRP). The objective of this study is to report on novel pathogenic variants in EYS and the range of associated phenotypes.
Subjects and methods
This retrospective case series at a tertiary referral centre for inherited retinal diseases describes patients with an IRD and at least two variants in the EYS gene. Phenotyping included multimodal retinal imaging; genotyping molecular genetic analysis using targeted next generation sequencing. Sanger sequencing verification and analysis of novel variants using in silico approaches to determine their predicted pathogenicity.
Results
Eight male and four female patients were included. Age at onset ranged from 11 to 62 years with variable symptom presentation; ten patients showed classical features of retinitis pigmentosa, albeit with great variation in disease severity and extent. Two patients had atypical phenotypes: one with localised inferior sector pigmentation and a mild RP phenotype with changes predominantly at the posterior pole. Eighteen variants in EYS were identified, located across the gene: six were novel. Eight variants were missense, two altered splicing, one was a whole exon duplication and the remainder were predicted to result in premature truncation of the protein.
Conclusion
The marked variability in severity and age of onset in most patients in this ethnically diverse cohort adds to growing evidence that that mild phenotypes are associated with EYS variants. Similarly, the two atypical cases add to the growing diversity of EYS disease as do the six novel pathogenic variants described.
Similar content being viewed by others

Introduction
Inherited retinal degenerations (IRDs) are a group of hereditary retinal disorders that are characterised by progressive loss of vision. IRDs can display marked phenotypic and genetic heterogeneity with variable age of onset of symptoms and rate of progression. Retinitis pigmentosa (RP) (OMIM #268000) collectively incorporates many genetic causes of IRD, with a worldwide prevalence of 1 in 4000 [1]. RP is characterised by progressive loss of rod photoreceptors with symptoms of nyctalopia and peripheral visual field loss, followed usually by secondary cone loss. Fundus appearances typically include intraretinal pigment deposition, vessel attenuation and optic disc pallor [1]. To date, over 80 different genes have been associated with RP [2, 3], however in ~40% of cases the causative gene has not yet been identified [1].
In 1998 an RP locus (RP25) was mapped to a 16 cM region on chromosome 6 (6p12-q15) in four Spanish families [4]. Subsequently the region was refined to a 2.67 cM region [5]. Two groups simultaneously identified the human orthologue of the Drosophila eyes shut/spacemaker (eys) gene EYS (OMIM #612424) at this locus [6, 7]. Subsequently mutations in EYS were reported to be one of the most frequent causes of autosomal recessive RP (arRP) in particular in the Japanese and Israeli populations, accounting for up to a third of simplex and arRP cases [8, 9], but with a frequency of approximately 5% in European and North American populations [10].
EYS is one of the largest genes expressed in the eye comprising 44 exons spanning over 2 Mb of genomic DNA [7, 11]. EYS encodes a 3165 amino acid protein (accession number ENST00000370621.3), which contains multiple epidermal growth factor (EGF)–like domains at its N-terminus, a probable coiled-coil domain, and five Laminin G domains interspersed by more EGF-like repeats (Fig. 1) [7, 11]. The EYS protein is now known to have four isoforms (3144aa, 619aa, 594aa and 3165aa), all of which are expressed in human retinal rod, cone and ganglion cells, where they localise to the cytoplasm, centrosome and ciliary axoneme [12]. Isoforms 2 and 3 are also expressed in human testicular cells [12], although their function there has not yet been investigated. The EYS protein is thought to be involved in stabilising the photoreceptor ciliary axoneme, and the organisation and function of other microtubule structures within retinal cells in humans [12]. The involvement of each EYS isoform in the pathogenesis of RP is unknown, but the high degree of variation in the domain structure between variants suggests they may perform multiple functions [12].

The protein consists of EGF-like and EGF-like calcium binding domains at the N terminal end, a coiled coil domain and 5 laminin G binding domains at the C-terminal end. The mutations identified in our cohort are marked, those novel to this study are shown above the structure and those reported previously are shown below (see Table 3 for details).
We characterise the genotype and phenotype a cohort of twelve patients from different ethnicities with EYS variants, describing milder and diverse phenotypes and report novel gene variants.
Materials and methods
This study was conducted in adherence to the tenets of the Declaration of Helsinki with local approval from the Essex 2 Research Ethics Committee (reference 08/H0302/96) with written informed, patient consent.
Literature search
An extensive literature search was performed to find studies reporting genotype or phenotype analysis of arRP patients identified to have EYS pathogenic sequence variants.
Study population
The clinical data of all patients identified by the clinical laboratory to have two or more pathogenic sequence variants in EYS were reviewed retrospectively.
Clinical data
Clinical notes, imaging and investigation results were reviewed for all patients and available family members. Data extracted included basic demographics (age, sex, ethnicity), pedigree and details of any family history, including details of consanguinity, ocular symptoms, ocular co-morbidity or systemic disorders. Examination and investigations included best-corrected visual acuity, and where available, Goldmann visual fields, fundus photography, autofluorescence imaging (AF), spectral domain optical coherence tomography (OCT) (OCT-Spectralis, Heidelberg Engineering, Heidelberg) and electrodiagnostic testing in accordance with ISCEV standards [13]. Patients in this study are represented by an alphabetical identifier A-L.
Molecular genetic analysis
Molecular genetic testing was performed by the Oxford Regional Genetics Laboratory. Enrichment for the EYS gene was achieved as part of a customised HaloPlex enrichment system kit (Agilent technologies, Stockport, UK) designed to capture the coding exons and at least 10 bp of the flanking introns of 111 retinal genes in the Oxford NGS IRD phenotype-based gene panel. HaloPlex reactions were prepared as per manufacturer’s instructions. Libraries were pooled into batches of 14 and sequenced on an Illumina MiSeq instrument (Illumina) using a MiSeq v3 kit as per manufacturer’s instructions. Reads were aligned using BWA [14] and variant pathogenicity was called using Platypus [15]. All reported variants identified by NGS were confirmed by Sanger sequencing. In silico analysis was performed on the variants identified using three different prediction methods to determine the deleteriousness of the variants: Polyphen [16, 17] and Mutation Taster [18].
Results
Literature search
A total of 66 studies report 449 distinct RP causing EYS sequence variants. These consist of 219 missense, 43 gross deletions, 92 small deletions, 39 splicing, 32 small insertions, 5 small indels, 12 gross insertions, 4 regulatory and 3 complex rearrangement. However, only 28 studies report phenotypic data (Supplementary Table S1).
Cohort characteristics
Twelve unrelated arRP patients with pathogenic sequence variants in EYS were identified. Of these nine were male and three female; five were Northern European, three South Asian, one Middle Eastern, one East Asian, two unrecorded ethnicities; age at most recent visit ranged from 42 to 77 years (see Table 1). Phenotypic information was available for 11 of the 12 patients. Summaries of these phenotypes are provided in Fig. 2 and Supplementary Table S3 with corresponding descriptions of genotype in Tables 2 and 3. A schematic showing the position of sequence variants in relation to the gene structure is shown in Fig. 1.

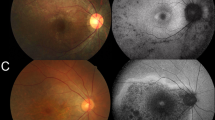
For each patient, fundus photographs (or an Optos® widefield image) is provided in the uppermost panel, a representative Optical Coherence Tomography slice in the centre and Fundus Autofluorescence images in the lowermost panels. For patients E&J, fundus photographs were not available. Please see Supplementary Fig. S1 for imaging of other patients.
Clinical presentation and phenotype
The age at onset of first symptom was variable, ranging from 11–62 years. The most common presenting symptom was nyctalopia, but peripheral and central field loss were also reported, and two patients were asymptomatic. Visual acuity ranged from 0.00 LogMAR to only being able to see hand movements. Where available, Goldman perimetry measured from 20 to 150o of horizontal field (see Table 1).
The phenotype was relatively consistent, with varying degrees of pigment, vessel attenuation and optic disc pallor with loss of the photoreceptor layer seen on imaging with central sparing in all but the most advanced cases (see Table 1 and Fig. 2). Two subjects had distinct phenotypes (patients E&F). Patient E, homozygous for the variant c.5834delA, had a phenotype consistent with inferior sector RP and was asymptomatic. Patient F, a compound heterozygote for c.7868G>A, and c.8196_8200delCTTC, had minimal visual loss with a mild limited RP phenotype characterised by an inferior band of mottled early atrophic appearing retinal pigment epithelium, with a corresponding band of decreased AF signal in this area, accompanied by a circumferential concentric ring of increased AF signal confined to the posterior pole. Electrophysiology testing confirmed mild disease with rod and cone ERGs showing reduced amplitudes and but normal implicit times, and visual fields were full.
Fundoscopy
The fundoscopic appearance of the majority of patients demonstrated the peripheral intraretinal bone spicule pigmentation, vessel attenuation and disc pallor expected in RP. Patients E and F showed a different phenotype with limited location of disease.
Optical coherence tomography
In the majority of patients, OCT demonstrated generalised atrophy of photoreceptor layers, with macular sparing of the photoreceptor layers in all but the most advanced cases. There was only one patient with cystic macular oedema (G), which was very mild, but patient D had, in addition, an epiretinal membrane. Patient F had a partial thickness macular hole and patients D, H and J had hyperreflective dots in the interdigitation zone (Supplementary Table S3).
Fundus autofluorescence
In all patients, there was an increased signal within the arcades, except for patient E, where there was a band of increased AF external to the inferior atrophic sector RP. In addition, in patients B, C, G and K there was a ring of increased AF within the posterior pole.
Electroretinography
Electrophysiology results were available for three patients and were consistent with both rod and cone system involvement showing reduced a, b amplitudes and increased implicit times in response to both dark adapted and photopic flash stimuli (Supplementary Table S2).
Molecular genetic analysis
All twelve patients described herein have bi-allelic pathogenic sequence variants in EYS. Four patients were homozygous, and the remaining eight were compound heterozygote (Table 3). Eighteen variants in EYS were identified in total, six of which were novel (Table 3, Fig. 1). As the patients were screened using a panel of 111 IRD genes, variants in other genes were also described (Table 2). None of these variants described were considered to be pathogenic, either because they were seen in a heterozygous state in genes causing autosomal recessive disease or because the phenotype described for the gene did not fit. For example, patients E and F both had a single variant in ABCA4 but phenotypically did not have Stargardt disease. However patient D (from consanguineous parents) as well as being homozygous for an EYS variant (c.1308C>A, p.(Cys436*), also had a heterozygous CRX variant (c.292C>T, p.(Arg98*) and we cannot rule out involvement of this gene as samples for segregation analysis were not available.
Sixteen variants, six of which were small deletions, resulted in missense or nonsense mutations, one was a whole exon duplication (exon 14) and one was a splicing mutation. One novel missense mutation, c.6389G>C, p.(Cys2130Ser), was found in two unrelated patients, G and L. Patient G is a compound heterozygote and the other change, which is a small deletion is also novel, c.9159_9165delGAGCTAT, p.(Met3053fs16*). Patient L is homozygous for the change (Table 3). A previously reported small deletion, c.8196_8200delCTTTC, p.(Phe2733fs33*) [19] was also seen twice in our cohort, once as a homozygous change (Patient A) and once in a compound heterozygote (Patient F). All variants identified were analysed using three different in silico prediction methods: Polyphen2, SIFT and Mutation Taster, which predict whether or not the changes are pathogenic (Table 3).
Discussion
In this study, we characterise the genotype and phenotype of twelve patients with EYS variants from a variety of ethnic backgrounds, reporting an expanded phenotype and novel gene variants. To date 219 point variants (145 missense and 74 nonsense), 39 splicing, 4 regulatory, 92 small deletions, 32 small insertions, 5 indels, 43 gross deletions, 12 gross insertions and 3 complex rearrangements) have been reported [20]. We identified a similar distribution of variants in our cohort with ten point mutations, six small deletions, one splicing and one whole exon duplication.
Reports from some case series have raised the possibility that EYS sequence variants may cause more rapidly progressive arRP than ciliopathies associated with other RP genes [21, 22]. By contrast in our cohort there was significant heterogeneity in the type and age of onset of symptoms and extent of degenerative changes. There did not appear to be any obvious relation between the severity of disease and the type of mutation, nor its location in the gene. Indeed, several patients in our cohort maintained acuity and field beyond the sixth decade, suggesting marked variability of expression in EYS associated RP. This supports the observation made by Pierrache et al. that EYS associated IRD is variable [23]. However, as the EYS gene encodes four different isoforms, further work to determine the roles they play in the retina may uncover a genotype-phenotype correlation.
The EYS variants detected in our cohort, including the six novel ones, are distributed across the whole gene (Fig. 1) which corroborates previous reported findings (see [24] for summary).
The three patients with the most severe disease (visual acuity <1.00logMAR in either eye (A, H, and I) all had EYS sequence variants located in different parts of the gene. Previous reports have suggested mutations in downstream regions of the gene can be more deleterious [25], however patient I’s variants are located relatively upstream, which is not in keeping with this. Apart from one mutation (c.8196_8200delCTTC), seen in two unrelated patients of South Asian heritage (A and F), there was no other mutation in this series that was particularly found in one ethnic group. The phenotype of A is quite different from F suggesting that the more severe phenotype seen in A is because there is no functional protein and F may have some residual function because the second variant is a milder mutation.
Although EYS mutations lead to recessive disease, the large coding sequences required for the retinal isoforms (cDNA 9.4–9.5 kb) exclude adeno-associated viral vectors for gene replacement therapy [26]. Alternative technologies such as CRISPR gene editing may however offer therapeutic options for long coding sequences [27] and for this reason is it critically important to document all known mutations in EYS, so that appropriate CRISPR targeting strategies can be developed. Significantly, six of the mutations we identified were single nucleotide transitions that could potentially be amenable to potential CRISPR therapies via adenosine or cytosine base editing applied to the plus or minus strand (c.76C>T, c.2137 + 1G>A, c.7868G>A, c.8054G>A, c.8842T>C, c.8897G>A) [28].
Seko et al. [29] recently, by using differentiated photoreceptor-like cells, showed that the manner of decay of the retina specific transcripts varied depending on the EYS gene mutation ranging from complete nonsense mediated decay (NMD) to minimal NMD. Further cell modelling work may help elucidate the relevance of these different isoforms in disease severity. However, given the heterogeneity of mutations suggested by this series, a larger cohort will be required to explore whether it is possible to demonstrate any phenotype–genotype correlation.
In the majority of previous studies, the EYS-associated IRD phenotype is consistent with typical RP with pigment. More recently however, Pierrache et al described a case of cone rod dystrophy with a ring of AF signal surrounding the arcades (in their study labelled patient XXV [23]); our patient F has very similar features on AF imaging with cone rod dysfunction, however these patients have different genotypes. Patient F is a compound heterozygote with a missense variant c.7868G>A (p.(Gly2623Glu)) and a frameshift variant c.8196_8200delCTTC (p.(Phe2733fs33*)) causing disease, whereas patient XXV has the homozygous nonsense variant c.9405T>A (p.Tyr3135*). The c.7868G>A variant has been reported before in a compound heterozygous patient, but with typical arRP and not sector RP [25]. The other EYS variant identified in patient F (c.8196_8200delCTTC) was also identified in patient A, who was homozygous for this variant, but had a typical ARRP phenotype; this same variant has been described in one other study, in one patient in a Japanese arRP cohort but phenotypic data were not presented [19].
The other mild phenotype we report is sector RP in patient E. EYS related sector RP has been reported once before by Bandah-Rozenfeld et al. [30]. Their patient had a homozygous null mutation in exon 43, and they suggest that this particular mutation may have escaped NMD, however their patient developed severe widespread RP over time. It is possible, as our patient E was 40 years old at presentation, that progression may occur with development of a more severe phenotype in time. Indeed, a clinical phenotype of sectoral RP may be seen to have more widespread manifestations on imaging modalities such as autofluorescence.
In summary, this study describes an ethnically diverse cohort of twelve patients with 18 pathogenic sequence variants in EYS, six of which were novel, located throughout the gene. We also report two atypical phenotypes of sector RP and cone rod dystrophy with limited mild disease. Of the twelve patients only three had the more typical severe RP as previously described associated with EYS. No clear correlation between genotype, phenotype or age at onset was identified. The EYS gene is an important cause of arRP and with the practice of whole genome sequencing becoming more widespread more copy number variations and intronic variants will be identified as reported by Zampaglione et al. [31]. Identification of EYS variants and a better understanding of the gene and its function will help enable progress towards a therapy.
Summary
What was known before
-
Pathological variants in the EYS gene can cause autosomal recessive retinitis pigmentosa.
What this study adds
-
The Retinitis Pigmentosa caused by pathological variants in EYS is heterogeneous at the genetic level and in terms of severity and phenotypic pattern.
-
Atypical phenotypes (including central and sectoral RP) can be seen with EYS mutations.
References
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809.
Verbakel SK, van Huet RAC, Boon CJF, den Hollander AI, Collin RWJ, Klaver CCW, et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018;66:157–86.
Daiger SP Ret Net. 2020. https://sph.uth.edu/retnet/home.htm. Accessed 7 April 2020.
Ruiz A, Borrego S, Marcos I, Antinolo G. A major locus for autosomal recessive retinitis pigmentosa on 6q, determined by homozygosity mapping of chromosomal regions that contain gamma-aminobutyric acid-receptor clusters. Am J Hum Genet. 1998;62:1452–9.
Barragan I, Abd El-Aziz MM, Borrego S, El-Ashry MF, O’Driscoll C, Bhattacharya SS, et al. Linkage validation of RP25 Using the 10K genechip array and further refinement of the locus by new linked families. Ann Hum Genet. 2008;72(Pt 4):454–62.
Abd El-Aziz MM, Barragan I, O’Driscoll CA, Goodstadt L, Prigmore E, Borrego S, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet. 2008;40:1285–7.
Collin RW, Littink KW, Klevering BJ, van den Born LI, Koenekoop RK, Zonneveld MN, et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet. 2008;83:594–603.
Iwanami M, Oishi A, Ogino K, Seko Y, Nishida-Shimizu T, Yoshimura N, et al. Five major sequence variants and copy number variants in the EYS gene account for one-third of Japanese patients with autosomal recessive and simplex retinitis pigmentosa. Mol Vis. 2019;25:766.
Sharon D, Ben-Yosef T, Goldenberg-Cohen N, Pras E, Gradstein L, Soudry S, et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum Mutat. 2020;41:140–9.
Littink KW, van den Born LI, Koenekoop RK, Collin RW, Zonneveld MN, Blokland EA, et al. Mutations in the EYS gene account for approximately 5% of autosomal recessive retinitis pigmentosa and cause a fairly homogeneous phenotype. Ophthalmology. 2010;117:2026–33. 2033 e2021-2027.
Abd El-Aziz MM, O’Driscoll CA, Kaye RS, Barragan I, El-Ashry MF, Borrego S, et al. Identification of novel mutations in the ortholog of Drosophila eyes shut gene (EYS) causing autosomal recessive retinitis pigmentosa. Investig Ophthalmol Vis Sci. 2010;51:4266–72.
Alfano G, Kruczek PM, Shah AZ, Kramarz B, Jeffery G, Zelhof AC, et al. EYS is a protein associated with the ciliary axoneme in rods and cones. PLoS ONE. 2016;11:e0166397.
McCulloch DL, Marmor MF, Brigell MG, Hamilton R, Holder GE, Tzekov R, et al. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc Ophthalmol. 2015;130:1–12.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95.
Rimmer A, Phan H, Mathieson I, Iqbal Z, Twigg SRF, Consortium WGS, et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–8.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Arai Y, Maeda A, Hirami Y, Ishigami C, Kosugi S, Mandai M, et al. Retinitis pigmentosa with EYS mutations is the most prevalent inherited retinal dystrophy in Japanese populations. J Ophthalmol. 2015;2015:819760.
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–77.
McGuigan DB, Heon E, Cideciyan AV, Ratnapriya R, Lu M, Sumaroka A, et al. EYS mutations causing autosomal recessive retinitis pigmentosa: changes of retinal structure and function with disease progression. Genes (Basel). 2017;8:178. https://doi.org/10.3390/genes8070178.
Zelhof AC, Hardy RW, Becker A, Zuker CS. Transforming the architecture of compound eyes. Nature. 2006;443:696–9.
Pierrache LHM, Messchaert M, Thiadens A, Haer-Wigman L, de Jong-Hesse Y, van Zelst-Stams WAG, et al. Extending the spectrum of EYS-associated retinal disease to macular dystrophy. Investig Ophthalmol Vis Sci. 2019;60:2049–63.
Messchaert M, Haer-Wigman L, Khan MI, Cremers FPM, Collin RWJ. EYS mutation update: In silico assessment of 271 reported and 26 novel variants in patients with retinitis pigmentosa. Hum Mutat. 2018;39:177–86.
Di Y, Huang L, Sundaresan P, Li S, Kim R, Ballav Saikia B, et al. Whole-exome sequencing analysis identifies mutations in the EYS gene in retinitis pigmentosa in the indian population. Sci Rep. 2016;6:19432.
Lipinski DM, Thake M, MacLaren RE. Clinical applications of retinal gene therapy. Prog Retin Eye Res. 2013;32:22–47.
Fry LE, Peddle CF, Barnard AR, McClements ME, MacLaren RE. RNA editing as a therapeutic approach for retinal gene therapy requiring long coding sequences. Int J Mol Sci. 2020;21:777. https://doi.org/10.3390/ijms21030777.
Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19:770–88.
Seko Y, Iwanami M, Miyamoto-Matsui K, Takita S, Aoi N, Umezawa A, et al. The manner of decay of genetically defective EYS gene transcripts in photoreceptor-directed fibroblasts derived from retinitis pigmentosa patients depends on the type of mutation. Stem Cell Res Ther. 2018;9:279.
Bandah-Rozenfeld D, Littink KW, Ben-Yosef T, Strom TM, Chowers I, Collin RW, et al. Novel null mutations in the EYS gene are a frequent cause of autosomal recessive retinitis pigmentosa in the Israeli population. Investig Ophthalmol Vis Sci. 2010;51:4387–94.
Zampaglione E, Kinde B, Place EM, Navarro-Gomez D, Maher M, Jamshidi F, et al. Copy-number variation contributes 9% of pathogenicity in the inherited retinal degenerations. Genet Med. 2020;22:1079–87. https://doi.org/10.1038/s41436-020-0759-8.
Pierrottet CO, Zuntini M, Digiuni M, Bazzanella I, Ferri P, Paderni R, et al. Syndromic and non-syndromic forms of retinitis pigmentosa: a comprehensive Italian clinical and molecular study reveals new mutations. Genet Mol Res. 2014;13:8815–33.
Barragán I, Borrego S, Pieras JI, Pozo MG-d, Santoyo J, Ayuso C, et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum Mutat. 2010;31:E1772–800.
Audo I, Sahel JA, Mohand-Said S, Lancelot ME, Antonio A, Moskova-Doumanova V, et al. EYS is a major gene for rod-cone dystrophies in France. Hum Mutat. 2010;31:E1406–35.
Mucciolo DP, Sodi A, Passerini I, Murro V, Cipollini F, Borg I, et al. Fundus phenotype in retinitis pigmentosa associated with EYS mutations. Ophthalmic Genet. 2018;39:589–602.
Hosono K, Ishigami C, Takahashi M, Park DH, Hirami Y, Nakanishi H, et al. Two novel mutations in the EYS gene are possible major causes of autosomal recessive retinitis pigmentosa in the Japanese population. PLoS ONE. 2012;7:e31036.
Jespersgaard C, Fang M, Bertelsen M, Dang X, Jensen H, Chen Y, et al. Molecular genetic analysis using targeted NGS analysis of 677 individuals with retinal dystrophy. Sci Rep. 2019;9:1219.
Acknowledgements
The authors would like to thank the Eye Research Group Oxford (ERGO) for clinical evaluation and management of the patients, as well as the patients who gave their consent to participate in this study.
Funding
SD is supported by the NIHR (Thames Valley CRN), as are the research nursing staff who aided in this study. REM and the Eye Research Group Oxford (ERGO) are supported in part by the NIHR Oxford Biomedical Research Centre. MJG is supported by a Clinical Research Training Fellowship from the Wellcome trust (Grant Number 205151/Z/16/Z).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
None of the authors declares a competing interest relating to the described work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Cundy, O., Broadgate, S., Halford, S. et al. “Genetic and clinical findings in an ethnically diverse retinitis pigmentosa cohort associated with pathogenic variants in EYS”. Eye 35, 1440–1449 (2021). https://doi.org/10.1038/s41433-020-1105-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-020-1105-8
This article is cited by
-
Genotypic and phenotypic profiles of EYS gene-related retinitis pigmentosa: a retrospective study
Scientific Reports (2022)
-
EYS-Associated Sector Retinitis Pigmentosa
Graefe's Archive for Clinical and Experimental Ophthalmology (2022)
