
Abstract
Germline changes in the CDH1 tumor suppressor gene predispose to diffuse gastric cancer and lobular breast cancer. In carriers of deleterious germline CDH1 variants, prophylactic gastrectomy is recommended. In case of germline missense variants, it is mandatory to assess the functional impact on E-cadherin, the protein encoded by CDH1, and to predict their clinical significance. Herein, we have identified a recurrent germline missense variant, c.1679C>G, segregating with gastric cancer in three unrelated Spanish families. Through genetic, transcriptional, in silico and in vitro studies, we demonstrate the deleterious effect of the c.1679C>G variant and its association with hereditary diffuse gastric cancer, providing relevant data to relatives and allowing an accurate genetic counseling.
Similar content being viewed by others

Introduction
Hereditary diffuse gastric cancer (HDGC; OMIM #137215) is an autosomal dominant condition associated with heterozygous germline changes in the tumor suppressor gene CDH1, which encodes the key cellular adhesion protein E-cadherin. The cumulative risk of developing diffuse gastric cancer (DGC) for CDH1 alteration carriers by age 80 is reported to be 70% for men and 56% for women; women carriers also have a cumulative risk of 42% for developing lobular breast cancer (LBC) by the age of 80 [1]. There is currently no evidence of increased risks for other cancer types in CDH1 variant carriers [2].
In areas with a low incidence of gastric cancer, like Spain [3], germline variants in CDH1 are found in about 30–50% of families fulfilling the clinical criteria for HDGC [1, 2, 4] and it was reported recently that 6% of the cases might be explained by alterations in other genes [1].
Around 126 different deleterious variants and 29 unclassified variants of CDH1 have been reported to date [1, 2]. In variants of uncertain significance (VUS), it is crucial to study their functional outcome, in order to offer suitable clinical management to carriers. In the present study, we described three unrelated Spanish families with HDGC that carry the same missense variant in the CDH1 gene. We performed in silico, segregation, population, and functional studies to assess the pathogenicity of this variant.
Subjects and Methods
This material can be found in the Supplementary material.
Results
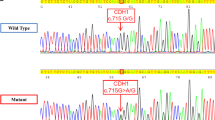
CDH1 genetic testing revealed the presence of the germline variant c.1679C>G in exon 11 in all individuals with gastric cancer from three unrelated Spanish families (Fig. 1). The putative effects of the variant were first explored by using nine in silico predictors, five of which suggested a damaging effect of the p.(T560R) change: SIFT: 0.04, PPHEN: 0.997, FATHMM: 0.71, MutationTaster: 0.997 and Align-GVGD: C65 (GV: 0.00-GD: 70.97). The variant frequency was then evaluated in a cohort of 730 healthy individuals from all over Spain, but it was not found in any of them. Furthermore, we assessed a possible founder effect, however, no common haplotype segregating with the variant was observed through microsatellite marker analysis.

Family pedigrees. The three probands were referred to our institution for genetic evaluation because of their family history. In each family, several individuals were diagnosed with gastric cancer at early ages (ranging from 27 to 45 years old). Two cases of breast cancer were found in the families, one confirmed as a LBC case. AMI acute myocardial infarction, CRC colorectal cancer, DGC diffuse gastric cancer, Gast gastrectomy (unknown if partial or total), HNC head and neck cancer, LBC lobular breast cancer, nc non-carrier of c.1679C>G, SRC signet ring cells, SubGast subtotal gastrectomy
cDNA analysis revealed that carriers of the c.1679C>G variant produced three transcripts of different sizes corresponding to the full-length transcript, a transcript with exon 11 deleted, and a transcript with a deletion of 32 nucleotides. In the control samples only the full-length transcript and the one in which exon 11 was skipped were detected (Fig. 2).

CDH1 transcript analysis. a cDNA fragments generated through RT-PCR in patients (named according to the pedigrees) and control samples. b Representative cDNA sequences of each cDNA fragment: one with a deletion of the last 32 nucleotides of exon 11 (GTACACAGCCCTAATCATAGCTACAGACAATG), one with a deletion of the entire exon 11, and one corresponding to the full-length fragment (arrowhead points the WT nucleotide c.1679C)
Since the c.1679C>G variant occurs in a pivotal site for post-translational processing of E-cadherin, and aberrant transcripts may be difficult to detect in vitro by colony screening, we decided to study the functional impact of the variant at the protein level, namely its effects on expression, localization, cell-cell adhesion, and invasion.
CHO cells transfected with a vector encoding the p.(T560R) E-cadherin produced a slight decrease in protein levels, together with a band mobility shift in the gel, despite similar transfection efficiencies (Fig. 3a–c).

Functional evaluation of p.(T560R) E-cadherin. a CHO cells transfected with vectors encoding the wild-type E-cadherin or the p.(T560R) variant were analyzed for protein levels by western blot. The empty vector was used as transfection control (Mock). Untransfected cells (parental) were also examined. α-Tubulin was used as a loading control. A diagram indicating putative N-glycosylation sites of the wild type and the p.(T560R) proteins are shown at the right. b Band intensity was determined and normalized against wild-type cells. The average intensity + SE of three independent experiments is shown. c Transfection efficiency was controlled by flow cytometry using GFP expression. d Cells were fixed and immunostained for E-cadherin (green). Nuclei were counterstained with DAPI (blue). In the enlarged image, arrows point to areas of irregular E-cadherin staining. e Cell invasive abilities through a Matrigel matrix. The relative number of invasive cells + SE is represented. f Slow aggregation assay showing distinct cell-cell adhesion phenotypes for wild-type and p.(T560R) E-cadherin-expressing cells. g Average area + SE of aggregates. * represents p ≤ 0.05 and ****p ≤ 0.0001
E-cadherin immunofluorescence showed that p.(T560R)-expressing cells presented a less intense and more irregular staining at the plasma membrane, when compared with the strong and well-defined membrane expression of cells expressing the wild-type protein. A diffuse pattern of E-cadherin expression could also be observed throughout the cytoplasm of mutant protein-expressing cells, but not in the wild-type counterparts (Fig. 3d).
p.(T560R) cells formed smaller and more diffuse cell aggregates than the wild-type cells (Fig. 3f, g). Consistent with this loss of cell-cell adhesion behavior, the p.(T560R) cells showed an increased ability to invade a Matrigel matrix (Fig. 3e).
Our observations regarding this variant have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and can be found under accession number SCV000693437.
Discussion
In the present work, we described three unrelated families with HDGC, in which we identified the same CDH1 variant: c.1679C>G. The clinical management of a germline missense variant conveys significant limitations for an efficient genetic counseling, especially in the case of HDGC, where early detection of the disease is very limited, leaving prophylactic gastrectomy as the only effective preventive measure. Thus, we performed a comprehensive in-depth analysis of the mentioned CDH1 variant that allowed us to classify it as a variant that affects E-cadherin functions and, therefore, to offer a precise counseling.
In fact, the initial approaches to assess the pathogenicity of the c.1679C>G variant (through in silico predictions, familial segregation, and population frequencies) suggested the involvement of the variant in HDGC. Furthermore, this variant had been reported twice before in the literature—in a young patient with DGC [5] and in a family of Indian origin also with DGC [6]—together with two unpublished cases reported to the Institute of Molecular Pathology and Immunology of the University of Porto (IPATIMUP; personal communication from R Seruca, 2016). Benusiglio et al. did not perform any type of functional analysis [5], while Yelskaya et al. described that the c.1679C>G variant disrupts normal splicing, which presumably leads to truncation of the protein [6]. In support of these findings, we also observed that the c.1679C>G change generates an alternative 5′ splice site, leading to the loss of 32 nucleotides in exon 11 or to a premature stop codon at position 576 of the protein. Remarkably, our study goes beyond the pathogenic consequences of aberrant splicing by also assessing the behavior of the variant p.(T560R) E-cadherin protein in vitro.
To verify the impact of the variant in vitro, we addressed protein expression and localization, and the two main functions of E-cadherin: the ability to establish cell-cell adhesion and its role as an invasion suppressor. These are well-recognized approaches to ascertain the pathogenicity of E-cadherin missense changes [7,8,9,10,11]. With these analyses, we demonstrated that the p.(T560R) change caused a decrease in protein levels and an abnormal pattern of expression in the cytoplasm (distinct from the correct plasma membrane localization), possibly due to an impairment of N-glycosylation of E-cadherin. Indeed, N-glycosylation of E-cadherin seems to be essential for its correct folding, trafficking, and stability at the plasma membrane [12,13,14]. We verified that the variant here described alters the consensus sequence NST in E-cadherin’s fourth extracellular domain (EC4), which is required for protein N-glycosylation. The N-558 residue has already been described as one of the most important sites of E-cadherin N-glycosylation and its dependent cell-cell adhesion [12]. Accordingly, we demonstrated that the p.(T560R) variant disrupts cell-cell aggregation and causes an increase in cellular invasion.
Given the common geographic origin of the families (province of Madrid, Spain), we explored a possible founder effect. The haplotype study indicated that a common ancestry of this variant event for the three families is unlikely. This result was expected for two reasons: (1) a large region of CDH1 (which includes our variant site) shows no linkage disequilibrium (GTEX), which suggests it might be very exposed to recombination; and (2) there are other individuals reported to carry this variant who have very distant geographic origins [5, 6].
In conclusion, we report a CDH1 missense variant associated with gastric cancer in three unrelated families. The pathogenicity of this variant was explored in two scenarios: a novel splicing event and a possible p.(T560R) E-cadherin mutant protein. Overall, our results indicate a damaging effect in both situations. On the one hand, the c.1679C>G variant generates an alternative transcript (as a result of the novel splicing site) with a 32-nucleotide deletion, which is expected to be degraded by nonsense-mediated decay (NMD) or, in case it produces a protein, this protein would be truncated and non-functional. On the other hand, if the full-length transcript with the variant nucleotide is produced in vivo, we have shown that the p.(T560R) form of E-cadherin protein has a deleterious effect by decreasing its expression, altering its subcellular localization, interfering with cell-cell interactions, and increasing cell invasiveness. Thus, our study provides useful information to offer a better management of the patients, and suggests that individuals harboring this variant should undergo intensive screening and prophylactic gastrectomy.
References
Hansford S, Kaurah P, Li-Chang H, et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015;1:23–32.
van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet. 2015;52:361–74.
Corso G, Marrelli D, Pascale V, Vindigni C, Roviello F. Frequency of CDH1 germline mutations in gastric carcinoma coming from high- and low-risk areas: metanalysis and systematic review of the literature. BMC Cancer. 2012;12:8.
Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015;16:e60–e70.
Benusiglio PR, Malka D, Rouleau E, et al. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J Med Genet. 2013;50:486–9.
Yelskaya Z, Bacares R, Salo-Mullen E, et al. CDH1 missense variant c.1679C G (p.T560R) completely disrupts normal splicing through creation of a novel 5’ splice site. PLoS ONE. 2016;11:e0165654.
Brito LA, Yamamoto GL, Melo S, et al. Rare variants in the epithelial cadherin gene underlying the genetic etiology of nonsyndromic cleft lip with or without cleft palate. Hum Mutat. 2015;36:1029–33.
Zhang L, Xiao A, Ruggeri J, et al. The germline CDH1 c.48 GC substitution contributes to cancer predisposition through generation of a pro-invasive mutation. Mutat Res. 2014;770:106–11.
Vogelaar IP, Figueiredo J, Van rooij IALM, et al. Identification of germline mutations in the cancer predisposing gene CDH1 in patients with orofacial clefts. Hum Mol Genet. 2013;22:919–26.
Figueiredo J, Seruca R. Germline missense mutants in hereditary diffuse gastric cancer. In: Corso G, Roviello F, editors. Spotlight on familial and hereditary gastric cancer. The Netherlands: Dordrecht: Springer; 2013. p. 77–86.
Corso G, Figueiredo J, Biffi R, et al. E-cadherin germline mutation carriers: clinical management and genetic implications. Cancer Metastas Rev. 2014;33:1081–94.
Carvalho S, Catarino TA, Dias AM, et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene. 2016;35:1619–31.
Zhou F, Su J, Fu L, et al. Unglycosylation at Asn-633 made extracellular domain of E-cadherin folded incorrectly and arrested in endoplasmic reticulum, then sequentially degraded by ERAD. Glycoconj J. 2008;25:727–40.
Pinho SS, Carvalho S, Marcos-Pinto R, et al. Gastric cancer: adding glycosylation to the equation. Trends Mol Med. 2013;19:664–76.
Acknowledgements
We thank the families and their caregivers for their participation and for providing consent to use the information obtained in this study. This work was supported by the Spanish Ministerio de Economía y Competitividad (MINECO) and the Fondo Europeo de Desarrollo Regional (FEDER) in the framework of project PI14/00459. FPI fellowship BES‐2015‐071383 to LP-C, funded by MINECO and the European Social Fund (ESF). Functional studies were supported by FEDER through the COMPETE 2020 – Operational Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through FCT – Fundação para a Ciência e a Tecnologia/ Ministério da Ciência, Tecnologia e Inovação in the framework of the projects “Institute for Research and Innovation in Health Sciences” – POCI-01-0145-FEDER-007274, PTDC/BIM-ONC/0171/2012, PTDC/BIM-ONC/0281/2014, PTDC/BBB-IMG/0283/2014, doctoral grant SFRH/BD/108009/2015-SM, and post-doctoral grant SFRH/BPD/87705/2012-JF. We acknowledge the funding provided by the American Association of Patients with Hereditary Gastric Cancer “No Stomach for Cancer” through the project “Today's present, tomorrow's future on the study of germline E-cadherin missense mutations.” We thank Dr. Rui M Ferreira for his assistance with FoldX structural modeling.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Pena-Couso, L., Perea, J., Melo, S. et al. Clinical and functional characterization of the CDH1 germline variant c.1679C>G in three unrelated families with hereditary diffuse gastric cancer. Eur J Hum Genet 26, 1348–1353 (2018). https://doi.org/10.1038/s41431-018-0173-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0173-8
This article is cited by
-
CDH1 mutations recurrence and global clustering in genetically tested families with hereditary diffuse gastric cancer syndrome: results from a systematic study
Familial Cancer (2023)
-
Germline CDH1 variants in hereditary diffuse gastric cancer syndrome with focus on younger women
Journal of Cancer Research and Clinical Oncology (2023)
-
Case report: mutation analysis of primary pulmonary lymphoepithelioma-like carcinoma via whole-exome sequencing
Diagnostic Pathology (2019)
