
Abstract
Polymer field-effect transistors (PFETs) are among the most fascinating electronic devices because of their attractive properties, such as durability, environmental friendliness, and solution processability. To date, PFETs have been employed for the development of flexible displays, radio-frequency identification tags, flexible non-volatile memories, among others. Moreover, due to the above advantages, PFETs can be applied to disposable on-site analytical devices. In that regard, we have developed extended-gate type PFETs with molecular recognition biomaterials for protein sensing in aqueous media. The fabricated PFETs were used to successfully detect glycoproteins (immunoglobulin A, immunoglobulin G, and chromogranin A) without any complicated labeling processes. Since our proposed immunoassay, which is performed on the basis of extended-gate type PFETs, is rapid and easy-to-use, PFET will be an attractive platform for on-site monitoring devices in healthcare applications in the near future.
Similar content being viewed by others


Introduction
Biosensors consist of detection and transducer parts, and their capturing methods for target analytes rely on biomaterials, such as immunoproteins, enzymes, DNA, among others. The first biosensor device was reported by Clark and Lyons in 1962 [1], with Updike and Hicks also reporting “The Enzyme Electrode” at almost the same time [2]. These authors invented an amperometric oxygen electrode modified with glucose oxidase for the continuous monitoring of the glucose concentration in biological solutions (e.g., blood). To date, various types of biosensor devices have been developed and applied for preventative healthcare and rapid diagnoses of incipient diseases. In particular, bioassays for proteins are widely researched and developed because proteins are abundant in the human body and significantly correlate with mental, allergic, and lifestyle diseases, among others. In this regard, protein–protein interactions, such as immune-interactions, have been used to analyze biological fluids containing biomarkers because of their highly specific recognition ability for target analytes. For example, enzyme-linked immunosorbent assay (ELISA), which is a popular assay, has been widely used for protein determination in diagnostic applications. However, complicated processes, such as labeling and pre-treatment reactions, are necessary, while ELISA can detect target proteins specifically and sensitively. Thus, many research groups worldwide have also attempted to develop biosensor devices that utilize an electrochemical analyzer [3], quartz crystal microbalance (QCM) [4], or surface plasmon resonance (SPR) [5] due to easier detection systems for target proteins (Fig. 1a). However, conventional biosensors require relatively large and expensive systems for high throughput measurements, which means that development of on-site and easy-to-use assays based on these platforms can be challenging. By contrast, field-effect transistor (FET)-based biosensors [6] can enable one to easily miniaturize the sensing system. In particular, development of polymer FET (PFET)-based biosensors has attracted considerable attention because their features can be used to develop wearable and disposable healthcare monitoring systems.

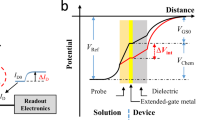
a Illustrated concept of immunosensors. b Device structure of the extended-gate type PFET. c Schematic illustration of the fabricated PFET utilizing a polymeric semiconductor (poly{2,5-bis(3-hexadecylthiophene-2-yl)thieno[3,2-b]thiophene}; PBTTT) and a self-assembled dielectric monolayer (tetradecylphosphonic acid; TDPA). d Transfer and output characteristics of the fabricated PFET device, which can be operated under low applied-voltage conditions (<|3| V)
PFETs are among the most fascinating devices in the field of electronic engineering owing to their mechanical durability and flexibility, solution-processability, and compact integration [7]. According to these advantages, light-weight, rollable (or stretchable), enlargeable, environmentally friendly, and low-cost electrical systems can be developed by utilizing PFETs. Therefore, PFET-based rollable active-matrix displays [8], radio-frequency identification (RFID) tags [9], and flexible non-volatile memories [10] have been successfully made. Among such applications, development of PFET-based bioassays for proteins is in its early stage because the design of devices and molecular recognition materials for PFET-based biosensors is not fully established. Hence, we propose a design for protein sensors based on PFET devices, which has potential to be a new attractive biosensing platform in various research fields. Our recent approaches for the development of protein sensors based on PFETs are summarized in this focus review, including the design strategy for developing PFET devices for protein sensing, as well as the problems that we have encountered.
Device design of the PFET-based protein sensor
Principally, PFET-based biosensors can detect target biomolecules captured on the surface of semiconductors or electrodes because the carrier concentration in the semiconducting layer (i.e., channel conductance) is affected by the surface charge of the target proteins [11]. In previous reports, capture of analytes using PFET biosensors depends on non-specific physical-adsorption [12], which means that selective detection of biomolecules using PFET biosensors is not fully understood. In addition, sensing of biomolecules with PFET biosensors is usually performed under dry conditions because organic semiconductor materials are sensitive to moisture; degradation of PFETs by water can readily occur [13]. Needless to say, biomolecule sensing should be carried out in aqueous media. We thus designed an extended-gate type PFET to use as a sensing system in water with high accuracy and reproducibility (vide infra).
A low-voltage operable extended-gate type PFET used as a protein sensing platform
The fundamental operation principle for PFETs follows that of metal-oxide-semiconductor field-effect transistors (MOSFETs). In typical MOSFETs, amorphous silicon (a-Si) or polycrystalline silicon (p-Si) is utilized as the semiconductor material. Although Si-based FETs are prevalent components in electrical circuit systems, fabrication of ultra-flexible electronic systems based on Si-FETs is difficult because of their rigidity. On the other hand, PFETs are widely studied in the field of flexible and conformable electronics (vide supra). In addition, the fabrication process for PFETs is much simpler compared to that for Si-FETs [7]. Hence, PFETs are suitable devices for disposable sensing platforms. More importantly, polymer semiconductors have high durability against mechanical strain [14] and high uniformity in thin-film deposition, meaning that polymeric-material-based devices are suitable for flexible sensors with high reproducibility and stability. The device structure includes three terminal electrodes (source, drain, and gate) and dielectric (capacitor) and semiconductor layers. Charge transport (i.e., hole and/or electron density) in the semiconductor layer is controlled by changing the applied voltage at the gate terminal. Thus, PFETs can function as switching devices in electrical circuits. To employ PFETs as protein sensor platforms, the channel conductance should be directly or indirectly affected by charged proteins captured on the devices. In addition, the sensor devices should be operated under low-voltage conditions to avoid the electrolysis of water and signal noise. However, operation of conventional PFETs requires a relatively high applied voltage (>10 V) due to their low carrier-mobility and/or high contact resistance [7].
Ultimately, we decided to employ an extended-gate type structure for the fabrication of PFET-based protein sensors [15] (Fig. 1b). PFET (=the transducer) is separated from the extended-gate (=the sensing portion), meaning that degradation of PFET by water can be prevented. Additionally, the extended-gate part can be easily modified with various molecular recognition materials, such as antibodies. To reduce the operation voltage of the device, we used a bilayered capacitor that consisted of aluminum oxide and a tetradecylphosphonic acid (TDPA) self-assembled monolayer (SAM) as the gate dielectric in PFET [16] (Fig. 1c). This ultra-thin capacitor enabled us to generate high carrier concentrations in the channel region, which means that the operation voltage of the PFET device could be reduced. Moreover, a π-conjugated polymer (i.e., poly{2,5-bis(3-hexadecylthiophene-2-yl)thieno[3,2-b]thiophene}, PBTTT [17, 18]) was employed as the semiconductor in PFET (Fig. 1c). PBTTT analogues have been widely applied to PFETs because of their high-carrier mobility, solution-processability and high uniformity in thin-film formation. Given these advantages, PBTTT is a suitable material for developing PFET-based protein sensors.
The basic electrical characteristics of the fabricated PFET are shown in Fig. 1d. The fabricated PFET exhibited excellent characteristics, with no hysteresis in the linear and saturation regions. The representative field-effect mobility and the on/off drain current ratio were estimated to be 10−2 cm2/Vs and ≥103, respectively. The observed performance for the low-voltage operable (<|3| V) PFET was reproducible under ambient conditions, indicating that the designed device could be applied to the detection of target proteins in water.
Working principle of PFET-based sensor devices for protein detection
Electrical detection of various proteins was realized using the fabricated extended-gate type PFETs functionalized with biomaterials (e.g., streptavidin, antibodies, etc.). As shown in Fig. 1b, we immersed the extended-gate and reference (=Ag/AgCl) electrodes into the target solutions. The extended-gate electrode acted like a floating-gate electrode, which means that the charge of the proteins captured on the electrode affected the electrical characteristics of PFET. These characteristics (e.g., transfer characteristics) should follow Eq. (1) [19],
where IDS is the observed drain current; W and L are the channel width and length, respectively; μ is the field-effect mobility; C is the capacitance of the gate dielectric; VTH is the threshold voltage; and VGS is the gate voltage. Moreover, ΔVTH, which is derived by adding the charged proteins (=the charge density, Q), can be expressed in the following Eq. (2),
meaning that the transfer characteristics are influenced by the charge strength of the proteins captured on the extended-gate electrode [20]. The shift of the interfacial potential between the analyte solution and extended-gate electrode can be triggered by a partial charge in the target protein (=residues in the protein adjacent to the electrode). Therefore, the stacking property and density of the molecular recognition materials should be controlled to secure a reproducible response in the PFET sensor. In this regard, we employed a SAM as the anchor layer for the receptors because the spontaneous formation of SAMs is highly reproducible. Here, concrete examples of protein sensing using the fabricated PFET are shown in the following chapters.
Electrical protein detection using PFET
Detection of immunoglobulin G (IgG) with or without biotin-labeling
To evaluate the sensing ability of the fabricated PFET, we first attempted to detect a biotin-labeled IgG through a biotin-streptavidin interaction. Due to the extreme low-dissociation constant of the biotin-streptavidin interaction (Kd ~ 10−15 M) [21], the biotin-labeling process for the detection of target proteins is occasionally employed. IgG is a representative major player in the humoral immune system and is ubiquitous, which means that monitoring the IgG concentration in biological fluids can reveal human health conditions [22]. In that regard, IgG is widely utilized as a biomarker for several illnesses (e.g., connective tissue diseases [22], food allergy [23], etc.) in diagnosis tests. To recognize biotin-labeled IgG, streptavidin was immobilized through multi-step reactions at the sensing portion in the device. The surface of the gold extended-gate was first covered by 10-carboxy-1-decanethiol, followed by fixation of streptavidin through amide formation using N,N’-diisopropylcarbodiimide (DIC) and N-hydroxysulfosuccinimide (sulfo-NHS). To quench unreacted sulfo-NHS esters, 2-aminoethanol was subsequently added (Fig. 2a). Characterization of the sensing electrode is important in advance of the sensing application. Thus, the surface properties of the functionalized gold electrode were investigated step-by-step using contact angle goniometry (CAG) and photoemission yield spectroscopy (PYS) in air. The CAG measurement showed a slight increase in the hydrophobicity of the gold electrode treated with 10-carboxy-1-decanethiol (42.8°) in comparison with that of the untreated electrode (39.7°), indicating that the hydrophobic decyl chain was introduced onto the gold electrode (Fig. 2b). In addition, the PYS measurement exhibited a deeper shift for the work function of the electrode before (4.53 eV) and after (5.02 eV) the formation of SAM (Fig. 2c), meaning that the gold surface was decorated with the electronegative group (i.e., the carboxy group) [24]. After the amide coupling reaction, a dramatic decrease of the contact angle (16.3°) with no photoelectric effect was observed, probably because the gold extended-gate was fully covered by the charged macromolecule (i.e., streptavidin). According to the above-mentioned results, it is noted that fabrication of the sensing electrode for the biotin-labeled IgG was successfully carried out. We then performed a titration experiment for biotin-labeled IgG in Dulbecco’s phosphate-buffered saline (PBS) in the presence of a 0.1 wt% bovine serum-albumin (BSA) interferent. As expected, the PFET device with the sensing electrode responded to the addition of the biotin-labeled IgG (Fig. 2d). The observed negative shift of the transfer curve with the increasing IgG concentration indicated that the positively charged molecule (i.e., the biotin-labeled IgG) was captured on the surface of the extended-gate electrode [25]. The limit of detection [26] (LOD) estimated from the relationship between the IgG levels and threshold voltage was approximately 8 nM (Fig. 2e). Notably, the electrical response of the PFET device was observed in the presence of a large amount of BSA interferent, suggesting that the electrical changes were derived from the specific interaction of the biotin-labeled IgG and streptavidin on the extended-gate electrode.

a Reaction scheme for the immobilization of streptavidin on the surface of the extended-gate electrode. b Water contact angle goniometry measurements on Au electrodes. c Photo-emission yield spectra for Au electrodes in air. Untreated Au (blue circle), 10-carboxy-1-decanethiol-treated Au (red diamond) and streptavidin-immobilized Au (white circle). d Transfer characteristics for PFET upon titration with biotin-labeled IgG in a PBS solution with 0.1 wt% bovine serum albumin. [IgG] = 0, 1, 5, 10, 20, and 50 μg·mL−1. e Changes in the threshold voltage (VTH) of the PFET device due to biotin-labeled IgG at various concentrations in a PBS solution with 0.1 wt% bovine serum albumin
The above results encouraged us to develop a label-free electronic assay for IgG [27]. The labelling process is not only one of the rate-limiting factors for protein determination assays but also alters the surface properties and natural activities of the target proteins. Hence, the development of a label-free assay for an analyte is desirable in a high-throughput sensing platform for various applications. The fabricated device can be used to directly detect the charge property of the protein captured on the electrode (Eq. 2), meaning that PFET is a suitable platform for an easy-to-handle and rapid immunoassay for proteins. We used a polyclonal antibody (pAb) against unlabeled IgG as the molecular recognition biomaterial, which was immobilized on the gold electrode in a similar way as the above protocols. Figure 3a shows a schematic structure for the electronic label-free immunosensor based on PFET. A shorter alkyl-chain (n = 5) based SAM was employed for label-free detection. The transfer characteristics of the PFET device were shifted upon addition of unlabeled IgG (Fig. 3b). This was the first example of electronic label-free immune sensing using PFETs.

a Schematic illustration of the extended-gate type PFET functionalized with pAb for unlabeled IgG detection. b Transfer characteristics of PFET upon titration with unlabeled IgG in a PBS solution with 0.1 wt% bovine serum albumin. [IgG] = 0–100 μg mL−1
Demonstration of label-free immunoglobulin A (IgA) sensing in simulated saliva
Blood and mucous secretions (i.e., saliva and tears) contain IgA [28]. In particular, secretory IgA in mucous secretions constitutes a part of mucosa-associated lymphoid tissue. Since a significant relationship between the IgA levels in saliva and psychological stress [29, 30] has been widely investigated, the detection of IgA can be not only useful as a marker for infection risk but also as a marker for mental stress. Although IgA is potentially an important marker for healthcare in daily life, conventional methods for IgA detection, such as ELISA, are complicated. Hence, we decided to develop an easy-to-use sensing system utilizing the PFET-based immunosensor for the IgA detection [31].
Practically, the non-invasive detection of IgA using PFET should be achieved in saliva. We thus carried out IgA titration in a simulated saliva solution containing interferential proteins, such as myeloperoxidase, lactoferrin, lysozyme, and amylase. The concentration of the interferential proteins simulated the actual conditions of human saliva [32,33,34,35] ([myeloperoxidase] = 3.6 μg/mL; [lactoferrin] = 1.0 μg/mL; [lysozyme] = 0.4 μg/mL; and [amylase] = 0.4 μg/mL). To achieve selective IgA detection under such competitive conditions, a biotinylated monoclonal antibody (mAb) against IgA (clone code: IgA5-3B) [36, 37] was immobilized on the extended-gate sensing electrode. Monoclonal antibodies possessing highly specific affinity for target proteins are produced by identical immune cells, meaning that the mAb-modified electrode was able to recognize the target IgA in the presence of an interferent. Moreover, in consideration of the large molecular size of IgA (320 kDa), we employed a SAM with a shorter alkyl chain (n = 3) spacer (Fig. 4a) because the distance from the electrode could affect the sensitivity and reproducibility of the assay. The influence of the charge of the captured molecules at the extended-gate/analyte solution interface on the FET characteristics is limited by the Debye screening effect [38]. Therefore, the interface design at the sensing electrode/analyte solution crucially affects the sensitivity of the device. As a result, the threshold voltage of the fabricated PFET was changed with the increasing IgA concentration in simulated saliva (Fig. 4b), suggesting that selective IgA detection without any labeling processes was successfully achieved. The linear-response range of the PFET-based immunosensor for IgA was estimated to be 0–10 μg/mL. Although the IgA levels in saliva are generally higher than 40 μg/mL, this sensing ability could be useful for healthcare monitoring because the low concentration of IgA in saliva is related to psychological stress or infectious diseases (e.g., dental caries [39], upper respiratory infection [40], etc.).

a Schematic illustration of the extended-gate type PFET functionalized with mAb for unlabeled secretory IgA detection. b Changes in the threshold voltage (VTH) of the PFET device due to unlabeled secretory IgA at various concentrations in a PBS solution with amylase (0.4 μg mL−1), lysozyme (0.4 μg mL−1), lactoferrin (1.0 μg mL−1), and myeloperoxidase (3.6 μg mL−1)
Sensitive label-free detection of chromogranin A (CgA)
CgA, which is expressed in neuroendocrine and neuronal tissues, is involved in the production of secretory granules and is cleaved to form small peptides by endogenous proteases [41]. The CgA-derived peptide contributes to the secretion of various hormones that belong to the neuroendocrine system (e.g., inhibition of insulin release [42]). CgA in serum is known to be a biomarker for various diseases, such as endocrine tumors [43], heart failure [44], hypertension [45], neuro diseases [46], among others. In addition, CgA in saliva can be utilized as a metal stress marker for anxiety [47] and depression [48]. To date, although label-free detection of CgA using a carbon nanotube (CNT)-based FET has been previously reported [49], the development of electrical device based biosensors for CgA detection is in its early stage. Toward that end, we demonstrated label-free CgA sensing based on the PFET-based immunosensor [50].
CgA detection was demonstrated in a commercially available artificial saliva (Saliveht® Aerosol, purchased from Teijin Pharma Co. Ltd., Chiyoda-ku, Tokyo, Japan) containing many types of ionic species, such as NaCl, KCl, CaCl2, MgCl2, and K2HPO4. We utilized a biotinylated mAb (clone code: LK2H10) against CgA [51], with the surface of the extended-gate electrode functionalized using the mAb-streptavidin complex. An increase in the CgA concentration caused electrical changes in the PFET characteristics (Fig. 5), suggesting that the fabricated PFET could be applied for selective detection of CgA in saliva. Notably, the LOD for the PFET-based immunosensor was estimated to be approximately 2 nM, implying that this value was comparable to that for a reported CNT-FET sensor for CgA [49]. The observed sensitivity was a few fold lower than the actual biological level of CgA (~0.3 nM) [52]. However, the main advantage of using PFETs in comparison with other types of electrochemical sensors is that its sensitivity is potentially improved by chemical approaches (the molecular design of the sensing portion) as well as device physical approaches (the design of the device structure). Thus, the detection signal can be enhanced by employing FET-based electrical circuits, such as a differential amplifier [53]. More importantly, the assay time for the fabricated immunosensor for CgA (~0.5 h) is less than the time required for ELISAs (~2.5 h) [54].

Changes in the threshold voltage (VTH) of PFET due to unlabeled CgA at various concentrations in artificial saliva containing NaCl, KCl, CaCl2, MgCl2, and K2HPO4
Conclusion and outlook
In this focus review, we summarized the development of extended-gate type PFETs for immunoassays. The fabricated PFET was able to be operated under low-voltage (<|3| V) conditions. Molecular recognition biomaterials (i.e., pAb and mAb) were introduced onto the electrode of the PFET, which offered label-free, rapid detection of target proteins, such as immunoglobulins in pseudo-biological fluids. The substrate of the sensing portion is made of a plastic film (PEN film), which means that our PFET biosensors can be used in a disposable and hygienic fashion.
In some cases, it was found that the sensitivity of the PFET immunosensors should be improved. To solve the above problem, device approaches, such as differential amplifiers, can be used [53]. In addition, molecular interactions between SAM and target proteins could be enhanced by conformational changes of SAM by external-stimuli [55]. Moreover, we preliminary succeeded in improving the sensitivity using chemical approaches, i.e., employment of artificial receptors instead of immune-proteins [56]. For example, sub-femtomolar detection of serum albumin was achieved by an extended-gate PFET with a coordination-bonding-based artificial receptor [57]. Moreover, the assay time in artificial receptor-attached PFETs was sped up compared to that of conventional bioassays that included the above PFET-based immunosensors. Since the development of PFET-based protein sensors is undergoing rapid progress, we believe that PFETs will be an attractive tool for on-site monitoring devices in healthcare applications in the near future.
References
Clark LC Jr, Lyons C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann NY Acad Sci. 1962;102:29–45.
Updike SJ, Hicks GP. The enzyme electrode. Nature. 1967;214:986–8.
Grieshaber D, MacKenzie R, Vörös J, Reimhult E. Electrochemical Biosensors - Sensor Principles and Architectures. Sensors. 2008;8:1400–58.
Crosson C, Rossi C. Quartz crystal microbalance immunosensor for the quantification of immunoglobulin G in bovine milk. Biosens Bioelectron. 2013;42:453–9.
Islam N, Shen F, Gurgel PV, Rojas OJ, Carbonell RG. Dynamic and equilibrium performance of sensors based on short peptide ligands for affinity adsorption of human IgG using surface plasmon resonance. Biosens Bioelectron. 2014;58:380–7.
Cheng S, Hotani K, Hideshima S, Kuroiwa S, Nakanishi T, Hashimoto M, et al. Field effect transistor biosensor using antigen binding fragment for detecting tumor marker in human serum. Materials. 2014;7:2490–500.
Sirringhaus H. 25th Anniversary article: organic field-effect transistors: the path beyond amorphous silicon. Adv Mater. 2014;26:1319–35.
Gelinck G, Heremans P, Nomoto K, Anthopoulos TD. Organic transistors in optical displays and microelectronic applications. Adv Mater. 2010;22:3778–98.
Subramanian V, Fréchet JMJ, Chang PC, Huang DC, Lee JB, Molesa SE, et al. Progress toward development of all-printed RFID tags: materials, processes, and devices. Proc IEEE. 2005;93:1330–8.
Heremans P, Gelinck GH, Müller R, Baeg K-J, Kim D-Y, Noh Y-Y. Polymer and organic nonvolatile memory devices. Chem Mater. 2011;23:341–58.
Bergveld P. Thirty years of ISFETOLOGY: what happened in the past 30 years and what may happen in the next 30 years. Sens Actuators B. 2003;88:1–20.
Stoliar P, Bystrenova E, Quiroga SD, Annibale P, Facchini M, Spijkman M, et al. DNA adsorption measured with ultra-thin film organic field effect transistors. Bionsens Bioelectron. 2009;24:2935–8.
Kumaki D, Umeda T, Tokito S. Influence of H2O and O2 on threshold voltage shift in organic thin-film transistors: deprotonation of SiOH on SiO2 gate-insulator surface. Appl Phys Lett. 2008;92:093309.
Sekine T, Fukuda K, Kumaki D, Tokito S. The effect of mechanical strain on contact resistance in flexible printed organic thin-film transistors. Flex Print Electron. 2016;1:035005.
Minamiki T, Minami T, Kurita R, Niwa O, Wakida S, Fukuda K, et al. Accurate and reproducible detection of proteins in water using an extended-gate type organic transistor biosensor. Appl Phys Lett. 2014;104:243703.
Klauk H, Zschieschang U, Pflaum J, Halik M. Ultralow-power organic complementary circuits. Nature. 2007;445:745–8.
McCulloch I, Heeney M, Bailey C, Genevicius K, MacDonald I, Shkunov M, et al. Liquid-crystalline semiconducting polymers with high charge-carrier mobility. Nat Mater. 2006;5:328–33.
Umeda T, Kumaki D, Tokito S. Surface-energy-dependent field-effect mobilities up to 1 cm2/V s for polymer thin-film transistor. J Appl Phys. 2009;105:024516.
Horowitz G. Organic field-effect transistors. Adv Mater. 1998;10:365–77.
Sakata T, Fukuda R. Simultaneous biosensing with quartz crystal microbalance with a dissipation coupled-gate semiconductor device. Anal Chem. 2013;85:5796–800.
Stayton PS, Freitag S, Klumb LA, Chilkoti A, Chu V, Penzotti JE, et al. Streptavidin-biotin binding energetics. Biomol Eng. 1999;16:39–44.
Kay RA, Wood KJ, Bernstein RM, Holt PJ, Pumphrey RS. An IgG subclass imbalance in connective tissue disease. Ann Rheum Dis. 1988;47:536–41.
Gocki J, Bartuzi Z. Role of immunoglobulin G antibodies in diagnosis of food allergy. Post Dermatol Alergol. 2016;33:253–6.
De Boer B, Hadipour A, Mandoc MM, van Woudenbergh T, Blom PWM. Tuning of metal work functions with self-assembled monolayers. Adv Mater. 2005;17:621–5.
Chen CP, Ganguly A, Lu CY, Chen TY, Kuo CC, Chen RS, et al. Ultrasensitive in situ label-free DNA detection using a GaN nanowire-based extended-gate field-effect-transistor sensor. Anal Chem. 2011;83:1938–43.
Miller JN, Miller JC. Statistics and Chemometrics for Analytical Chemistry. 6th edn. Harlow, UK: Pearson; 2010.
Minamiki T, Minami T, Kurita R, Niwa O, Wakida S, Fukuda K, et al. A label-free immunosensor for IgG based on an extended-gate type organic field effect transistor. Materials. 2014;7:6843–52.
Fagarasan S, Honjo T. Intestinal IgA synthesis: regulation of front-line body defences. Nat Rev Immunol. 2003;3:63–72.
Nagai H, Narita Y, Ohtaki M, Saito K, Wakida S. A single-bead analysis on a disk-shaped microfluidic device using an antigen-immobilized bead. Anal Sci. 2007;23:975–9.
Shirtcliff EA, Granger DA, Schwartz E, Curran MJ. Use of salivary biomarkers in biobehavioral research: cotton-based sample collection methods can interfere with salivary immunoassay results. Psychoneuroendocrinology. 2001;26:165–73.
Minamiki T, Minami T, Sasaki Y, Kurita R, Niwa O, Wakida S, et al. An organic field-effect transistor with an extended-gate electrode capable of detecting human immunoglobulin A. Anal Sci. 2015;31:725–8.
Zakowski JJ, Bruns DE. Biochemistry of human alpha amylase isoenzymes. Crit Rev Clin Lab Sci. 1985;21:283–322.
Nater UM, Rohleder N, Gaab J, Berger S, Jud A, Kirschbaum C, et al. Human salivary alpha-amylase reactivity in a psychosocial stress paradigm. Int J Psychophysiol. 2005;55:333–42.
Glimvall P, Wickström C, Jansson H. Elevated levels of salivary lactoferrin, a marker for chronic periodontitis? J Periodontal Res. 2012;47:655–60.
Thoma EL, Jefferson MM, Joyner RE, Cook GS, King CC. Leukocyte myeloperoxidase and salivary lactoperoxidase: identification and quantitation in human mixed saliva. J Dent Res. 1994;73:544–55.
Fox JG, Perkins S, Yan L, Shen Z, Attardo L, Pappo J. Local immune response in Helicobacter pylori-infected cats and identification of H. pylori in saliva, gastric fluid and faeces. Immunology. 1996;88:400–6.
Flies AS, Grant CK, Mansfield LS, Smith EJ, Weldele ML, Holekamp KE. Development of a hyena immunology toolbox. Vet Immunol Immunopathol. 2012;145:110–9.
Sakata T, Matsuse Y. In situ electrical monitoring of cancer cells invading vascular endothelial cells with semiconductor-based biosensor. Genes Cells. 2017;22:203–9.
Hegde M, Devadiga D, Shetty C, Shetty A. Correlation between dental caries and salivary immunoglobulin in adult Indian population: an in vivo study. J Res Dent. 2013;1:22–5.
Smith DJ, Taubman MA, Ebersole JL. Ontogeny and senescence of salivary immunity. J Dent Res. 1987;66:451–6.
D’amico MA, Ghinassi B, Izzicupo P, Manzoli L, Di Baldassarre A. Biological function and clinical relevance of chromogranin A and derived peptides. Endocr Connect. 2014;3:R45–R54.
Portela-Gomes GM, Gayen JR, Grimelius L, Stridsberg M, Mahata SK. The importance of chromogranin A in the development and function of endocrine pancreas. Regul Pept. 2008;151:19–25.
Conlon JM. Granin-derived peptides as diagnostic and prognostic markers for endocrine tumors. Regul Pept. 2010;165:5–11.
Biomarkers Definition Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95.
O’Toole D, Grossman A, Gross D, Delle-Fave G, Barkmanova J, O’Connor J, et al. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Biochemical Markers. Neuroendocrinology. 2009;90:194–202.
Bartolomucci A, Pasinetti GM, Salton SRJ. Granins as disease-biomarkers: translational potential for psychiatric and neurological disorders. Neuroscience. 2010;170:289–97.
Rai B, Kaur J. Salivary stress markers and psychological stress in simulated microgravity: 21 days in 6o head-down tilt. J Oral Sci. 2011;53:103–7.
Katsuura S, Kamezaki Y, Yamagishi N, Kuwano Y, Nishida K, Masuda K, et al. Circulating vascular endothelial growth factor is independently and negatively associated with trait anxiety and depressive mood in healthy Japanese university students. Int J Psychophysiol. 2011;81:38–43.
Wang CW, Pan CY, Wu HC, Shih PY, Tsai CC, Liao KT, et al. In situ detection of chromogranin A released from living neurons with a single-walled carbon-nanotube field-effect transistor. Small. 2007;3:1350–5.
Minamiki T, Minami T, Sasaki Y, Wakida S, Kurita R, Niwa O, et al. Label-free detection of human glycoprotein (CgA) using an extended-gated organic transistor-based immunosensor. Sensors. 2016;16:2033.
Wilson BS, Lloyd RV. Detection of chromogranin in neuroendocrine cells with a monoclonal antibody. Am J Pathol. 1984;115:458–68.
Nishikawa Y, Jun L, Futai Y, Yanaihara N, Iguchi K, Mochizuki T, et al. Region-specific radioimmunoassay for human chromogranin A. Biomed Res. 1998;19:245–51.
Fukuda K, Minamiki T, Minami T, Watanabe M, Fukuda T, Kumaki D, et al. Printed organic transistors with uniform electrical performance and their application to amplifiers in biosensors. Adv Electron Mater. 2015;1:1400052.
Stridsberg M, Eriksson B, Öberg K, Janson ET. A comparison between three commercial kits for chromogranin A measurements. J Endocrinol. 2003;177:337–41.
Yeung CL, Iqbal P, Allan M, Lashkor M, Preece JA, Mendes PM. Tuning specific biomolecular interactions using electro-switchable oligopeptide surfaces. Adv Funct Mater. 2010;20:2657–63.
Minamiki T, Minami T, Koutnik P, Anzenbacher P Jr, Tokito S. Antibody- and label-free phosphoprotein sensor device based on an organic transistor. Anal Chem. 2016;88:1092–5.
Minamiki T, Sasaki Y, Tokito S, Minami T. Label-free direct electrical detection of a histidine-rich protein with sub-femtomolar sensitivity using an organic field-effect transistor. ChemistryOpen. 2017;6:472–5.
Acknowledgements
We gratefully acknowledge financial support from the Towa Foundation for Food Science and Research, the Kanamori Foundation, the Descente and Ishimoto Memorial Foundation for the Promotion of Sports Science, Tateishi Science and Technology Foundation, Society for Research on Umami Taste, and Japan Society for the Promotion of Science (JSPS, Grant-in-Aid for Scientific Research, Nos. 16J08092, 17H04882, 17K14489, and 18J21190). We also thank Prof. S. Tokito (Yamagata University), Prof. O. Niwa (Saitama Institute of Technology), Dr. R. Kurita and Dr. S. Wakida (National Institute of Advanced Industrial Science and Technology) for their technical support and valuable feedback. YS is also grateful to the JSPS Research Fellowships for Young Scientists.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Minamiki, T., Sasaki, Y., Su, S. et al. Development of polymer field-effect transistor-based immunoassays. Polym J 51, 1–9 (2019). https://doi.org/10.1038/s41428-018-0112-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-018-0112-0
This article is cited by
-
Chemical sensing based on water-gated polythiophene thin-film transistors
Polymer Journal (2021)
-
Structural effect of fluorophore on phenylboronic acid fluorophore/cyclodextrin complex for selective glucose recognition
Frontiers of Chemical Science and Engineering (2020)
