
Abstract
In eukaryotic cells, lysosomes are digestive centers where biological macromolecules are degraded by phagocytosis and autophagy, thereby maintaining cellular self-renewal capacity and energy supply. Lysosomes also serve as signaling hubs to monitor the intracellular levels of nutrients and energy by acting as platforms for the assembly of multiple signaling pathways, such as mammalian target of rapamycin complex 1 (mTORC1) and adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK). The structural integrity and functional balance of lysosomes are essential for cell function and viability. In fact, lysosomal damage not only disrupts intracellular clearance but also results in the leakage of multiple contents, which pose great threats to the cell by triggering cell death pathways, including apoptosis, necroptosis, pyroptosis, and ferroptosis. The collapse of lysosomal homeostasis is reportedly critical for the pathogenesis and development of various diseases, such as tumors, neurodegenerative diseases, cardiovascular diseases, and inflammatory diseases. Lysosomal quality control (LQC), comprising lysosomal repair, lysophagy, and lysosomal regeneration, is rapidly initiated in response to lysosomal damage to maintain lysosomal structural integrity and functional homeostasis. LQC may be a novel but pivotal target for disease treatment because of its indispensable role in maintaining intracellular homeostasis and cell fate.
Similar content being viewed by others
Facts
-
The lysosome functions as the terminal degradation site and a cellular sensor and signaling hub.
-
Lysosomal damage is involved in different pathways of cell death.
-
Lysosomal dysfunction is related to various human diseases.
-
LQC may be a potential therapeutic target for human diseases.
Open questions
-
What are the effects of other nutrients, in addition to amino acids and growth factors, on lysosomal signaling?
-
Is there a specific anchored receptor involved in lysophagy?
-
Are there other mechanisms of cell death mediated by lysosomal damage, in addition to the release of cathepsins and hydrolases?
Introduction
The lysosome was first discovered by de Duve, who defined it as a monolayer vesicle rich in acidic hydrolases1. It was previously found to be an extremely important intracellular degradation site, serving as the terminal destination during phagocytosis and autophagy2. It has been demonstrated that lysosomes are capable of sensing and addressing cellular signals and playing essential roles in coping with stress stimuli3. For example, lysosomes are programmed to monitor the intracellular levels of nutrients and energy, thus engaging in nutrient sensing, metabolic regulation, and cellular homeostatic maintenance by providing an initial platform for the activation of mammalian target of rapamycin complex 1 (mTORC1) and adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK)4. Damage to lysosomes not only disrupts cellular clearance ability but also results in the leakage of multiple enzymes, triggering the activation of a series of cell death pathways5. Lysosomal damage is involved in various forms of cell death, such as apoptosis6,7, necroptosis8,9, pyroptosis10,11, and ferroptosis12,13, hinting that it is of great significance to the clarification of the potential mechanism of lysosomal damage and further exploration of efficient treatments based on lysosomal quality control (LQC). Indeed, the structural and functional abnormalities of lysosomes are critical for the pathogenesis and development of various diseases, including tumors14,15,16, neurodegenerative diseases17,18,19, inflammatory diseases20,21,22, and cardiovascular diseases23,24,25, making LQC a novel but pivotal target for treatments. Endolysosome damage consists of a series of specific cellular responses to lysosomal damage, including lysosomal repair, lysophagy, and lysosomal regeneration26, which have been identified as being crucial for maintaining intracellular homeostasis.
The structure and function of lysosomes
Lysosomal structure
Lysosomes are monolayer membrane organelles surrounded by a 7–10-nm-thick lipid membrane. The membrane vacuolar-type H+-ATPase (V-ATPase) is a distinct characteristic of lysosomes and can continuously pump H+ into lysosomes to maintain an acidic environment27. In addition to V-ATPase, other membrane proteins, such as lysosome-associated membrane proteins (LAMPs), ion channels, and multiple transporters, are the molecular bases for lysosomal function28. For example, LAMPs account for 80% of lysosomal membrane proteins, protecting the lysosomal membrane from acidic digestion through their highly glycosylated intracavitary parts29. Lysosomal transporters are needed for transporting the final products into the cytoplasm for further metabolic utilization30,31.
The pH value of the lysosomal lumen is maintained within a range from 3.5 to 5.5, which provides an optimal environment for the degradation of biological macromolecules3. To date, more than 60 kinds of acidic hydrolysis enzymes have been isolated from lysosomes. In addition to hydrolases, lysosomes can act as a store for various ions. Ca2+ is important for the function of lysosomes, affecting lysosome movement, membrane transport and repair, nutrient sensing, and organelle membrane contact32. Other ions, such as Na+, K+, Cl−, and Zn2+, are required for lysosomal responses, including transportation, the maintenance of membrane potential, and the activation of lysosomal enzymes33.
Lysosomal function
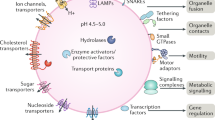
Lysosomes are the main digestive centers of eukaryotes and can effectively degrade biological macromolecules to maintain self-renewal capacity and meet energy needs2. Extracellular materials, such as pathogens and toxins, can be transported to lysosomes for degradation through phagocytosis34, while intracellular cytoplasmic macromolecules, unfolded or misfolded proteins, and even whole organelles are captured and transported to lysosomes, mainly through autophagy35,36. Recent studies have indicated that lysosomes are the primary centers of metabolic signaling pathways, have critical involvement in nutrient sensing, and are capable of monitoring the cellular metabolic state and further facilitate adaption to nutrient deprivation by activating mTORC1 and AMPK37,38,39 (Fig. 1).

Lysosomes function as terminal sites of phagocytosis and autophagy and as cellular sensors and signaling hubs. Cargos are transported to lysosomes for degradation by the phagosome formed by the closure of reshaped plasma membrane during phagocytosis or the autophagosome formed by the closure of the phagophore in macroautophagy, directly swallowing into lysosomes through membrane indentation, protuberance, or the separation of the lysosomal membrane in microautophagy, and chaperone binding mainly through LAMP2A in CMA. Lysosomes also function as mTORC1 and AMPK signaling hubs. Under conditions of nutrient enrichment, Rag GTPases, heterodimers formed by RagA/B and RagC/D, are activated and tethered to the lysosomal membrane, further recruiting mTORC1. Rag GTPases can be activated by Ragulator and inhibited by the GATOR1 complex. Ragulator is activated by SLC38A and lysosomal V-ATPase, which stimulated by arginine and leucine in the lysosomal lumen, respectively, while the GATOR1 complex is inhibited by GATOR2, which is stimulated by amino acids in the cytoplasm. Then, mTORC1 is activated by Rheb GTPases stimulated by GF through the inhibition of TSC. Upon glucose deficiency, Axin causes the dissociation and inactivation of mTORC1 through the inhibition of Ragulator. In addition, Axin recruits AMPK by interacting with LKB1 and induces AMPK activation by forming complexes with V-ATPase and Ragulator.
Phagocytosis and autophagy
Phagocytosis is programmed to eliminate invading pathogens and apoptotic cells40, making it crucial for maintaining intracellular homeostasis41,42. Cells undergo irreversible injury, and foreign stimuli, such as that from pathogens and associated toxins, are efficient in initiating phagocytosis43. Phagocytosis is completed by the following processes: the plasma membrane is reshaped by actin polymerization and local exocytosis, closing at the distal end to form early phagosomes43; then, to promote further digestion, phagosomes gradually fuse with lysosomes, which involves membrane remodeling, acidification of phagocytes, and the formation of oxidizing and degrading environments44.
Autophagy is characterized by vesicles swallowing cytoplasmic proteins or organelles and further fusing with lysosomes for content degradation, which is essential for intracellular metabolic homeostasis and the renewal of certain organelles35. Generally, autophagy is divided into macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA)45. In contrast to macroautophagy, which is termed generalized autophagy, autophagosomes are not formed during microautophagy; instead, the lysosome directly swallows lysosomes through the indentation, protuberance or separation of the lysosomal membranes46. CMA is completed when a chaperone binds and transports a target protein to lysosomes for degradation47. Heat shock protein (Hsp) 70, the molecular chaperone that plays a critical role in CMA, is capable of recognizing and binding substrates for lysosomal degradation48. In addition, Hsp70 promotes the multimerization of LAMP type 2a, the lysosomal receptor involved in CMA, to form a translocation complex49,50, in which a translocation channel is further formed51 through which only unfolded substrates are permitted to pass52. Similarly, other molecular chaperones are required in CMA, including Hsp90, which regulates the stabilization of the translocation complex49, Hsp40, Hsp70-interacting protein (Hip), and Hsp70-Hsp90-organizing protein (Hop), which facilitates the translocation process53. Selective autophagy is programmed to degrade aggregates of proteins, invading pathogens, and damaged organelles, such as peroxisomes, mitochondria, lysosomes, nuclei, and endoplasmic reticula (ER)54,55. The ubiquitin-dependent sensor system is critical for targeting and degrading distinct substrates56. Autophagy-specific receptors further tether the ubiquitinated cargo to autophagosomes that carry microtubule-associated protein 1 light chain 3 (LC3)/GABAA receptor-associated protein (GABARAP) and integrate with lysosomes for degradation57,58.
Cellular sensor and signaling hub
Lysosomes are key hubs for integrating signals in response to nutrients and energy37,39. mTORC1 and AMPK, both of which are important metabolic regulators, assemble their signal centers on the surface of lysosomes to form a switch for anabolic and catabolic processes37,39,59.
mTORC1 is an important regulatory molecule of cell proliferation and metabolism because it senses intracellular signals, such as those from amino acids and growth factors, further triggering certain pathways to promote cell growth60. Intracellular signals can activate mTORC1 mainly through Rag GTPases and Ras homolog enriched in brain (Rheb) GTPases at the surface of lysosomes stimulated by amino acids and growth factors, respectively61. Rag GTPases are heterodimers formed by the combination of RagA/B and RagC/D, which can be tethered to the lysosomal membrane by Ragulator61. Under enrich nutrition conditions, the activation of Ragulator by amino acid signals enables Rag GTPases to bind and recruit mTORC161. The changes in amino acids in the lysosomal lumen and cytoplasm are sensed by different mechanisms. In the lysosomal lumen, arginine and leucine facilitate the activation of Ragulator by stimulating SLC38A, a sodium-coupled amino acid transporter and lysosomal V-ATPase on the lysosomal membrane, thereby promoting the binding of Rag GTPases and mTORC162,63,64. In the cytoplasm, amino acids, such as arginine and leucine, can lead to the activation of the GATOR2 complex, which further enhances the binding capacity of Rag GTPases and mTORC1 by inhibiting the hydrolysis of the GATOR1 complex by Rag GTPases65,66.
In the GTP-bound form, Rheb reportedly contributes to the activation of mTORC1 on the surface of lysosomes, and this process is disturbed by the activation of tuberous sclerosis complex (TSC)67. When the inhibitory status of TSC is induced by growth factors, the activation of mTORC1 augments cellular anabolism by stimulating the biosynthesis and inhibiting autophagy60,68. It was found that mTORC1 induces ribosomal biogenesis by phosphorylating and promoting the translation of S6 kinases69. The inhibitory effect of mTORC1 on autophagy is induced by phosphorylating and suppressing the nuclear translocation of transcription factor EB (TFEB)70.
AMPK is essential for the regulation of cellular metabolism. Upon glucose deficiency, Axin, a scaffolding protein that is capable of inhibiting the activity of Ragulator, triggers the dissociation and inactivation of mTORC1 on the lysosomal membrane71. In addition, Axin recruits AMPK by interacting with liver kinase B1 (LKB1) and induces AMPK activation by forming complexes with V-ATPase and Ragulator4. This activation of AMPK signaling reportedly enhances cellular energy storage processes by increasing glucose uptake, promoting autophagy, and inhibiting catabolism through the disruption to mTORC1 activation72.
Lysosomal quality control
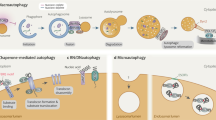
Lysosomal membrane permeabilization (LMP) or lysosomal complete rupture is the common form of lysosomal damage, and it is a potential hazard to cell fate and function73. Maintaining lysosomal structural integrity and functional homeostasis through LQC favors the physiological functions of cells. Under normal conditions, glycosylation of lysosomal membrane proteins can effectively maintain membrane stability by preventing the lysosomal membrane from being destroyed by lumen proteolytic enzymes28. However, LMP with the loss of membrane integrity and increased permeability occur after persistent exposure to oxygen radicals, optical damage, and other factors74,75, thereby resulting in the release of cathepsins and hydrolases into the cytoplasm76,77. When the LMP is not repaired, persistent lysosomal rupture may lead to the massive release of lysosomal contents, cascade hydrolysis of cytoplasmic contents, and extensive cytoplasmic acidification, thus contributing to irreversible cell damage76. In response to various stimuli, endolysosome damage leads to the formation of specific cellular elements via efficient LQC, which includes lysosomal repair, lysophagy, and lysosomal regeneration26 (Fig. 2).

Lysosomal quality control includes lysosomal repair, lysophagy, and lysosomal regeneration. The damaged lysosomal membrane can be repaired by Hsp70 and ESCRT. In lysophagy, damaged lysosomes are ultimately transported to normal lysosomes for degradation through phagosomes formed by phagophores. Two pathways for damaged lysosomes have been identified: Gals and SCFFBXO27. After binding with TRIM16, Gal3 accumulates at damaged lysosomes and recruits ULK1, Beclin 1, and ATG16L1. Then, Gal3 is modified by the K63 ubiquitin chain and binds to LC3 on the phagophore via p62. Gal8 accumulates at damaged lysosomes and binds to LC3 on the phagophore via NDP52. SCFFBXO27 is recruited to damaged lysosomes and ubiquitinates LAMP2, thereby binding to LC3 on the phagophore via p62. In addition, the inhibition of mTORC1 by the loss of lysosomes can enable TFEB to bind with CLEAR and promote lysosomal generation.
Lysosomal repair
When LMP occurs, endogenous protective mechanisms are initiated to repair membrane damage, and prevent the release of lumenal hydrolases73. Previous studies documented the protective effects of Hsp70 for maintaining lysosomal integrity, showing that it can stabilize lysosomal membrane by binding to lipid bisphosphonates and enhancing the activity of acidic sphingolipase78,79. Recent studies showed that endosomal sorting complexes required for transport (ESCRT) was critical for the repair of damaged lysosomal membranes80,81,82. ESCRT is a highly conserved transport system commonly found in yeast and other eukaryotic cells, and it is programmed to transport ubiquitinated proteins into lysosomes80. ESCRTs can repair small perforations in the lysosomal membrane at an early stage of lysosomal damage, which are reportedly recruited in only a few minutes80. Further study revealed that the Ca2+ efflux from damaged lysosomes enhanced the lipid-binding activity of apoptosis-linked gene-2-interacting protein X (ALIX), a component of the ESCRT complex, for efficient binding to the lysosomal membrane. Moreover, Tsg101, another component of the ESCRT complex, was shown to be indispensable, as deficiency of ALIX or Tsg101 led to the inability to repair damaged lysosomes and irreversible cell death81. Currently, the precise mechanism by which ESCRT repairs lysosomal membranes is not clear, but it might be involved in inducing the formation of filamentous helices on the membrane surface and the contraction of pores in the lipid bilayer82.
Lysophagy
When lysosomal membrane damage cannot be reversed, selective autophagy of lysosomes is initiated to ensure the effective clearance of damaged lysosomes, a process named lysophagy73. Similar to selective autophagy, the ubiquitination of impaired lysosomes is the major factor driving and modulating lysophagy55. Two ubiquitination pathways regulate lysophagy, and they involve galectins (Gals) and SCFFBXO27 26,83,84,85. Gals are clearly crucial for lysophagy because they recognize glycoproteins in the lysosomal membrane after lysosomal rupture26,83,84. Gal3 is able to sense damaged lysosomes, where they accumulate to induce further lysophagy by recruiting autophagy regulatory factors, including uncoordinated-51-like kinase 1 (ULK1), Beclin 1, and autophagy-related protein 16L1 (ATG16L1) after binding with E3 ubiquitin ligase-tripartite domain containing protein 16 (TRIM16)86. Then, Gal3 is modified with a K63 ubiquitin chain and binds to LC3 on the phagophore through autophagy receptor p62, and the phagophore is encapsulated, forming an autophagosome for subsequent degradation86. Other galectins, such as Gal8 and Gal9, can also be recruited to damaged lysosomes and induce lysophagy via different pathways84,86. For example, Gal8 directly binds to LC3 on the phagophore through autophagy receptor NDP52, in turn driving damaged lysosomes toward an autophagy pathway84. In addition to galectins, other molecules can be ubiquitinated on the damaged lysosomes85. Skp1/CUL1/F-box (SCF) is a ligase complex, and F-box protein is the receptor of the substrate of the SCF ubiquitin-ligase complex. When lysosomes were disrupted by LLOME treatment, SCFFBXO27 was rapidly recruited to the broken lysosomes and ubiquitinated LAMP-2 to activate lysophagy85. Given that lysophagy is a normal lysosome-dependent pathway that eliminates damaged lysosomes, the timely reestablishment of lysosomal numbers is crucial for the maintenance of lysosomal function.
Lysosomal regeneration
Lysosomal biogenesis requires the integration of both cellular endocytosis and protein biosynthesis, which is realized through the formation of the membrane structure of lysosomes and the provision of lysosomal proteins, respectively3,87,88,89. The lysosomal membrane is constituted by both the primary lysosomes secreted by the Golgi and vesicles from the plasma membrane and the endocytic pathway3. Newly synthesized proteins targeted to lysosomes can be transported either directly into the lysosome through the trans-Golgi network depending on receptors, including mannose-6-phosphate receptor, sortilin, and lysosomal integral membrane protein type 287,88, or indirectly ingested into the lumen by endocytosis89.
Under physiological conditions, the number of lysosomes is continuously maintained by dynamic homeostasis balanced between formation and degradation83. Lysosomal regeneration is initiated when lysosomes are lost83. Bioinformatics and functional genome analysis have revealed that the promoter regions of many lysosomal genes contain one or more repeats of the 10-base pair motif (GTCACGTGAC), known for coordinating lysosomal expression and regulation (CLEAR) elements90,91. TFEB, a member of the MiT/TFE family, has been found to directly bind with the CLEAR element and further promote lysosomal renewal92. Further study confirmed that TFEB contributed to the expression levels of a large number of genes involved in lysosomal function, including exocytosis, phagocytosis, endocytosis, and autophagy90. For example, the inhibition of mTOR enabled TFEB dephosphorylation and translocation the later increased the expression of genes encoding lysosomal proteins, such as V-ATPases, lysosomal transmembrane proteins, and hydrolases93.
In general, endolysosome damage is initiated rapidly in response to lysosomal damage, and multiple processes are involved in the repair of damaged lysosomes and cell survival. Thus, it is of great significance to identify the molecular mechanisms of LQC, and its relationship with the occurrence and development of clinical diseases.
Lysosomal damage and cell fate
Persistent lysosomal damage poses a serious threat to cell fate. Lysosomal injury-associated cell death was first identified by de Duve, who defined the lysosome as a “suicide bag”94. In fact, the degree of lysosomal damage is the determinant of the type of cell death. Early studies suggested that moderate lysosomal damage can induce cell apoptosis, while extensive damage resulted in a substantial number of cells undergoing irreversible necrosis. Accumulating evidence has indicated that lysosomal damage is closely related to the development of apoptosis6,95, necroptosis8,10, pyroptosis11, and ferroptosis13,96 (Fig. 3).

Apoptosis appears to be triggered by lysosomal damage in a mitochondria-dependent manner. Cathepsins are released from damaged lysosomes and cleave BID into tBid, in turn promoting the oligomorphism of BAX, which can be further enhanced by cathepsin-induced degradation of Bcl-2. Then, BAX is transferred to the mitochondrial outer membrane, causing excessive formation of the MPTP. Through the MPTP, cyto C is released into the cytoplasm and promotes the formation of apoptosomes, further inducing apoptosis. Necroptosis is stimulated by the inhibition of lysosomal function, resulting in significant accumulation of necrosome components (RIPK1 and RIPK3) and hydrolyzed caspase-8 by the release of cathepsin D. Necroptosis executor (MLKL) is phosphorylated by necrosomes and translocated to the cell membrane or organelle membrane, thereby leading to necroptosis. Pyroptosis is induced by damaged lysosomes through the cleavage of GSDMD into GSDMD-N by the release of cathepsin G, and activating NLRP3 and caspase-1 by the release of cathepsin B. Subsequently, pyroptosis leads to cell perforation and the massive release of IL-1β and IL-18. Additionally, ferroptosis can be exacerbated by the release of ROS produced by Fe2+ and H2O2 from damaged lysosomes.
Apoptosis
Apoptosis is defined as programmed cell death. Currently, two main pathways are widely accepted as mediating apoptosis, namely exogenous apoptosis via cell surface death receptors, and intrinsic apoptosis dependent on mitochondria involvement97. Intrinsic apoptosis is initiated under exposure to internal stimuli, such as growth factor deprivation, hypoxia, DNA damage, oxidative stress, and calcium overload98. In this pathway, apoptosis is trigger by the release of cytochrome C (cyto C) from mitochondria, which further induces the activation of caspase cascades99. Lysosomal damage triggers apoptosis through the endogenous pathway. When LMP or lysosomal rupture occurs, cathepsins are released from lysosomes and cleave BH3-interacting domain death agonist (BID) into tBid fragments, which promote the oligomorphism of B-cell lymphoma-2 (Bcl-2)-associated X protein6. Then, the oligomorphic complex is transferred to the outer mitochondrial membrane, causing the excessive formation of the mitochondrial permeability transition pore (MPTP)6. Cyto C is released through the MPTP into the cytoplasm, where it augments the formation of apoptosomes6,7. In summary, the leakage of lysosomal enzymes from damaged lysosomes is critical for apoptosis in the mitochondrial-dependent pathway. Interestingly, the release of H2O2 from damaged mitochondria also accounts for the fragmentation of lysosomal membranes100. In addition, cathepsins are capable of degrading Bcl-2, thereby leading to exacerbated cellular apoptosis5. Timely recognition and prompt interference of lysosomal damage are efficient measures for preventing cellular apoptosis. For example, the apoptosis of HepG2 cells can be significantly inhibited by specific cathepsin B inhibitors95,101. The inhibition of cathepsin B and cathepsin L by interfering with the mitochondrial pathway markedly protected astrocytes from apoptosis102.
Necroptosis
The traditional view suggests that necrosis is a nonprogrammed or “accidental” cell death. However, it has been demonstrated that necrosis can be driven and regulated by certain molecular mechanisms, similar to apoptosis, in a process called necroptosis103. The morphological characteristics of necroptosis are similar to those of necrosis, but its initiation is noncaspase-dependent104,105. The currently accepted concept suggests that necroptosis is mediated by receptor-interacting protein kinase (RIPK) and mixed-lineage kinase domain-like protein (MLKL). Pasparakis et al.106 found that tumor necrosis factor (TNF)-α was capable of activating RIPK1, promoting the phosphorylation of RIPK3, and thus forming RIPK1–RIPK3 complex, named a necrosome. MLKL was then phosphorylated by the necrosome and translocated to the cell membrane or organelle membrane, thus leading to necroptosis107. It was recently revealed that the inhibition of lysosomal function after traumatic spinal cord injury led to the significant accumulation of RIPK1, RIPK3, and MLKL in neurons, which was alleviated by the increased activation of autophagy-lysosome pathways8. Caspase-8 is a major negative regulator of necroptosis by interfering with RIPK activation and preventing the formation of necrosomes9. The release of cathepsin D induced necroptosis through the proteolysis of caspase-8 and promotion of RIPK1 activation, implying that lysosomal dysfunction might be crucially involved in the onset of necroptosis9.
Pyroptosis
Pyroptosis is characterized by both induction of a proinflammatory response and programmed cell death, which is realized mainly through the activation of caspase-1 and the proteolysis of GSDMD, accompanied by cell perforation and excessive release of IL-1β and IL-18108. The cathepsins released from damaged lysosomes play important roles in the process of pyroptosis. It has been shown that cathepsin G effectively induces the development of pyroptosis by cleaving GSDMD into GSDMD-N10. The activation of caspase-1 primarily depends on NOD-like receptors that recruit caspase-1 and further induce caspase-1 activation by binding with NOD-like receptor protein (NLRP)3 inflammasomes109. In addition, released cathepsin B can activates NLRP3 and caspase-1 by binding to the leucine-rich repeat domain of NLRP3, hinting to the critical involvement of lysosomal damage in cellular pyroptosis11.
Ferroptosis
Ferroptosis is a form of regulated cell death characterized by excessive intracellular accumulation of iron-dependent lipid hydroperoxides110. Either the overproduction of the Fe2+ that is involved in the formation of lipid hydroperoxides through the Fenton reaction111 or the inactivation of glutathione peroxidase 4, which is programmed to degrade lipid hydroperoxides, leads to intracellular lipid hydroperoxide accumulation to a lethal level, ultimately triggering ferroptosis112. An imbalance between intracellular reactive oxygen species (ROS) production and clearance is among the major causes of cellular ferroptosis110. The lysosome is one of the main storage sites for iron and essential for maintaining the levels of intracellular ROS and iron. Upon exposure to H2O2, the free iron in lysosomes increases the production of ROS, which can then be released through an unstable lysosomal membrane to trigger the activation of cell death pathways12. Most of the iron is in the form suitable for binding with ferritin in the cytoplasm, which can be degraded by autophagy-lysosome pathway, causing the release of active iron113. The inhibition of the autophagy–lysosome pathway was reportedly capable of slowing drug-induced ferroptosis, indicating that the autophagy–lysosome pathway appeared to be associated with the occurrence of ferroptosis13. However, ferroptosis can also be restricted by inhibiting lysosomal function through the downregulation of cathepsin or V-ATPase activity96.
Lysosomes and various diseases
A growing body of evidence indicates that the structural and functional stability of lysosomes are essential for maintaining cellular homeostasis2,3. Lysosomal dysfunction might play an important role in the development of various diseases, including tumors14,15,16, neurodegenerative diseases17,18,19, inflammatory diseases20,21,22, and cardiovascular diseases23,24,25 (Table 1).
Tumors
Catabolic hyperactivity is one of the main characteristics of tumor cells, and it is the major reason for tumor progression and metastasis14. The rapid proliferation of tumor cells is guaranteed by high synthesis rates, which largely depend on the degradation and recycling of cellular components28. The autophagy–lysosome system is fully activated to meet the metabolic needs of tumor cells28. For example, TFEB was found to maintain high levels of autophagic activity by upregulating lysosomal function in pancreatic ductal adenocarcinoma (PDA)15. The knockout of autophagy-related gene (ATG) 5 or ATG7 and the inhibition of lysosomal function significantly diminished the invasion and progression of PDA, suggesting that downregulation of lysosomal function is associated with inhibited tumor progression114. A number of clinical trials have been conducted to treat tumors by targeting lysosomes, such as through the anti-breast-cancer effects of saponin D, which blocks autophagosome–lysosome fusion115. Therefore, disturbing the autophagy–lysosome function is efficient for delaying the occurrence and development of tumors, and it is gradually becoming a promising target for tumor treatment.
In addition, cathepsin B is considered to be involved in proteolytic cascade destruction of the extracellular matrix, which further remodels the tumor environment by enabling angiogenesis, tumor migration, invasion, and even metastasis16. The overexpression of cathepsin B promoted the invasiveness and progression of breast cancer cells116,117, while downregulation of cathepsin B function by shRNA significantly reduced bone metastasis in mice breast cancer models118. The abnormal distribution of lysosomes is critically involved in carcinogenesis. Under normal conditions, lysosomes are mainly located in the peripheral and perinuclear regions of eukaryocytes119. During the process of carcinogenesis, lysosomes significantly transfer from the perinuclear region to the peripheral cytoplasm, which led to enhanced lysosomal exocytosis28. Furthermore, lysosomal exocytosis of hydrolytic enzymes promoted the invasion of tumor cells and degradation of the extracellular matrix, thereby contributing to cancer progression120. Thus, the regulation of lysosomal distribution should be taken seriously in anticancer therapy.
Neurodegenerative diseases
Neurons cannot dilute damaged organelles and cellular waste through cell division, implying the great need for an efficient intracellular clearance system121,122. Strikingly, lysosomes are essential for the functional maintenance and survival of neurons. An increasing number of studies have documented that failed lysosomal function is closely related to the occurrence and development of neurodegenerative diseases, including Gaucher disease, hereditary spastic paraplegias, and Alzheimer’s disease (AD)18,123,124,125. Gaucher disease is a rare autosomal recessive genetic disease that is caused by the excessive accumulation of glucose neuraminidase in macrophages because of the dysfunction and abnormal lysosomal transport of glucosaminidase123. The clinical manifestations of neurological Gaucher disease vary by subtype and manifest different symptoms123. Enzyme replacement therapy has achieved remarkable curative effects by enhancing the decomposition of glycosphingolipids in lysosomes, relieving the pressure on lysosomes, and improving lysosomal function123. Hereditary spastic paraplegias is a highly heterogeneous autosomal recessive hereditary disease most commonly caused by mutations in SPG11 and SPG15124. Lack of these two proteins can prevent lysosomal biogenesis and autophagosome maturity, leading to the ganglioside accumulation and neuronal death124. A recent study showed that alleviating ganglioside accumulation can significantly reduce the number of neuron deaths and thus improve the disease phenotype in zebrafish models with the SPG11 mutation124. Thus, alleviating the degradation pressure on lysosomes and improving lysosomal function are effective strategies for the management of neurodegenerative diseases. Additionally, lysosomal acidification is a major factor in the activity of most lysosomal hydrolytic enzymes and the regulation of lysosomal signals19. For example, the mutant presenilin 1, one of the main causes of familial AD, was found to cause a dysfunction in lysosomal acidification, leading to elevated lysosomal pH values125, lysosomal proteolysis injury, and AD-like pathologic alterations126. Similarly, two subtypes of early-onset Parkinson’s disease (PD), XPDS and Kufor-Rakeb syndrome, are both related to disorders of lysosomal acidification127,128, indicating that lysosomal deacidification accelerates the development of PD-like neuropathology. Some nerve agents that aggravate PD and PD-like symptoms have been shown to disturb pH values in lysosomes129,130.
Inflammatory diseases
Inflammation, deemed a protective response to pathogens, irritation, and injury131, can effectively remove invading pathogens and maintain tissue integrity by releasing inflammatory mediators, inducing inflammatory cell migration and aggregation132. However, excessive or sustained inflammation results in the destruction of tissues, followed by the development of inflammatory diseases. It has been generally accepted that lysosomes are critical to the initiation and recession of inflammatory responses37. In hyperuricemia, uric acid is supersaturated in urine and forms uric acid crystals, which are transported to lysosomes by the endocytosis of renal tubular epithelial cells, destroying the lysosomal membrane and causing lysosomal rupture, in turn, resulting in renal inflammation20. It was found that renal tubular epithelial cells with the Atg5 gene knocked out failed to isolate the damaged lysosomes, which caused irreversible cell damage in mouse models of acute hyperuricemic nephritis83. Similarly, the coexistence of lysosomes and digestive enzymes in pancreatic acinar cells is considered an early event in acute pancreatitis21. Activation of trypsin induces the leakage of lysosomes and the release of cathepsin B into the cytoplasm, which activates the cell death pathway and finally causes pancreatic injury mainly in the forms of cellular apoptosis and necrosis21. Taken together, these studies show that maintaining the integrity of the lysosomal structure and function or inhibiting the activity of cathepsin B, may be promising therapeutic targets for acute pancreatitis.
In the context of sepsis, stimulation of autophagy can lead not only to the removal of damaged organelles and redundant proteins, but also to the removal of invading pathogenic microorganisms, a process that has become a potential target for improving the survival and prognosis of septic complications133. The activity of autophagy increases significantly at the early stage of sepsis, which shows obvious advantages by promoting pathogen clearance, attenuating cell apoptosis, and modulating inflammation134. However, autophagy is remarkably inhibited during persistent exposure to septic challenge, and enhancing autophagic activity can significantly reduce the immunosuppression and organ dysfunction secondary to severe sepsis135,136. It was recently reported that a variety of drugs used to reverse sepsis-induced immunosuppression were related to induced autophagy activation136,137. In addition, mRNA expression of key genes in lysosomes, including acid hydrolases (such as cathepsin A/D), LAMP-1 and LAMP-2, was significantly upregulated following sepsis, hinting that overloaded lysosomal function might be an early marker for poor outcomes for patients with sepsis22.
Cardiovascular diseases
Given the critical involvement of autophagy in cardiovascular diseases, such as myocardial ischemic and reperfusion injury, cardiomyopathy, myocardial infarction, and heart failure138,139,140,141,142, the roles of lysosomes as the terminal sites of the autophagic process are indispensable. In myocardial ischemia and reperfusion injury, cardiomyocyte death can be caused by lysosomal consumption, which manifests as a decrease in the number of lysosomes and the downregulated expression of LAMP-1139. Both the enhancement of lysosomal function and promotion of autophagy were reportedly beneficial by alleviating cardiomyocyte death after myocardial ischemia and reperfusion injury139,143. In proteotoxic cardiomyopathy, cardiomyocytes showed impaired autophagic flux and reduced lysosome abundance; however, adeno-associated virus-TFEB transduction was sufficient to reverse cardiomyopathy pathology in a preclinical study142. Both dysfunctional lysosomes and impaired autophagic processes might contribute to the development of myocardial infarction and heart failure, which may be improved by enhancing autophagic activity140,141. Lysosomes are critically involved in the development and progression of cardiovascular diseases in autophagy-independent ways.
Atherosclerosis refers to lipid deposition (mainly cholesterol and cholesteryl ester) in the intima and subintima of arteries. Lipids induce the dysfunction of lysosomes in mice macrophages, as shown by an increase in lysosomal pH, decrease in proteolytic ability, and destruction of membrane integrity24. Lysosomal acid lipase is the main enzyme that hydrolyzes low-density lipoproteins and the modified forms of low-density lipoproteins, playing a pivotal role in lipid decomposition and prevention of excessive lipid accumulation144. Upon the development of atherosclerosis, excessive accumulation of free cholesterol leads to the suppression of lysosomal acid lipase activity and the accumulation of cholesterol esters in lysosomes145. In addition, deficiency of primary LAMP-2 is associated with the occurrence of Danon disease25, as evidenced by the pathological characteristics of massive autophagosome deposition in cardiomyocytes and skeletal muscle cells146,147. Similarly, mice with LAMP-2 knocked out show impaired autophagy and autophagosomes aggregation in cardiomyocytes, resulting in cardiac defects148. Taken together, studies show the critical involvement of lysosomes in the occurrence and development of cardiovascular diseases, which has been confirmed by their decreased number and significant dysfunction, as well as an impaired autophagic process, leading to aberrant cellular responses and even cell death. While upregulation of the autophagy–lysosome pathway or reduction in lysosomal stress may alleviate the progression of cardiomyocyte damage and prevent cell death, providing novel insights into disease therapy.
Conclusions and perspectives
In-depth understanding of lysosomes has shown that they are not simple “digestive centers” but are main metabolic centers and signal hubs. The degradation function of lysosomes has been adequately studied for decades, but studies on the roles of lysosomes as cellular sensors and signal hubs are in an initial state. Although the mechanism of amino acid sensing has been clarified, the impacts of other nutrients on lysosomal signaling are largely unclear. As LQC is critically involved in maintaining cell viability and function, the general aspects of LQC have been gradually emphasized and revealed; nevertheless, some details remain unclear. For example, the interaction between components of the ESCRT complex and their precise mechanisms in lysosomal repair are largely unknown. Based on the current evidence, the selective degradation process of lysosomes via lysophagy is thought to be facilitated by the recognition of lysosomal proteins exposed to cytoplasm after rupture. Therefore, similar to its significance in organelle-specific autophagy, the importance of discovering specific lysosome-anchored receptors is self-evident. The critical involvement of lysosomal damage in different pathways of cell death has been widely studied, but the current focus is placed on the release of cathepsin-based contents caused by structural damage. Few studies have addressed cell death induced by the release of ions, other proteases, and/or the dysfunction of the degradation or signal sensing roles of lysosomes.
Lysosomes play key roles in the development and progression of various human diseases, mainly through the alteration of their degradation capacity and the release of their contents, resulting in catabolic hyperactivity in tumors, accumulation of abnormal substances that causes neurodegenerative and cardiovascular diseases, and excessive inflammatory responses in inflammatory diseases. Therapies targeting lysosomes might exert beneficial effects for disease remission. For example, the inhibition of lysosomal function in cancer cells can restrain tumor metabolism, thereby alleviating tumor progression. In addition, the enhancement of lysosomal function and lysosomal enzyme replacement can accelerate the clearance of accumulated pathogenesis-related proteins in neurodegenerative and cardiovascular diseases. Similarly, the protection of lysosomes from structural damage and functional impairment can remarkably alleviate the progression of inflammatory diseases. In view of the indispensable role of LQC in lysosomal structure and function maintenance, LQC might also serve as a potential therapeutic target for treating human diseases.
References
de Duve, C. The lysosome turns fifty. Nat. Cell Biol. 7, 847–849 (2005).
Appelqvist, H., Wäster, P., Kågedal, K. & Öllinger, K. The lysosome: from waste bag to potential therapeutic target. J. Mol. Cell Biol. 5, 214–226 (2013).
Perera, R. M. & Zoncu, R. The lysosome as a regulatory hub. Annu. Rev. Cell Dev. Biol. 32, 223–253 (2016).
Carroll, B. & Dunlop, E. A. The lysosome: a crucial hub for AMPK and mTORC1 signalling. Biochem. J. 474, 1453–1466 (2017).
Česen, M. H., Pegan, K., Spes, A. & Turk, B. Lysosomal pathways to cell death and their therapeutic applications. Exp. Cell Res. 318, 1245–1251 (2012).
Zhu, X. et al. Mastocarcinoma therapy synergistically promoted by lysosome dependent apoptosis specifically evoked by 5-Fu@nanogel system with passive targeting and pH activatable dual function. J. Control Release 254, 107–118 (2017).
Taylor, R. C., Cullen, S. P. & Martin, S. J. Apoptosis: controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 9, 231–241 (2008).
Liu, S. et al. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 9, 476 (2018).
Zou, J. et al. Poly IC triggers a cathepsin D- and IPS-1-dependent pathway to enhance cytokine production and mediate dendritic cell necroptosis. Immunity 38, 717–728 (2013).
Burgener, S. S. et al. Cathepsin G inhibition by serpinb1 and serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep. 27, 3646–3656 (2019).
Bruchard, M. et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the NLRP3 inflammasome and promotes tumor growth. Nat. Med. 19, 57–64 (2013).
Ostenfeld, M. S. et al. Effective tumor cell death by sigma-2 receptor ligand siramesine involves lysosomal leakage and oxidative stress. Cancer Res. 65, 8975–8983 (2005).
Torii, S. et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 473, 769–777 (2016).
Piao, S. & Amaravadi, R. K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 1371, 45–54 (2016).
Perera, R. M. et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365 (2015).
Schmitz, J. et al. Cathepsin B: active site mapping with peptidic substrates and inhibitors. Bioorg. Med. Chem. 27, 1–15 (2019).
Lie, P. P. Y. & Nixon, R. A. Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol. Dis. 122, 94–105 (2019).
Thelen, A. M. & Zoncu, R. Emerging roles for the lysosome in lipid metabolism. Trends Cell Biol. 27, 833–850 (2017).
Colacurcio, D. J. & Nixon, R. A. Disorders of lysosomal acidification—the emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res. Rev. 32, 75–88 (2016).
Kimura, T., Isaka, Y. & Yoshimori, T. Autophagy and kidney inflammation. Autophagy 13, 997–1003 (2017).
Talukdar, R. et al. Release of cathepsin B in cytosol causes cell death in acute pancreatitis. Gastroenterology 151, 747–758 (2016).
Ma, J. et al. Lysosome and cytoskeleton pathways are robustly enriched in the blood of septic patients: a meta-analysis of transcriptomic data. Mediat. Inflamm. 2015, 984825 (2015).
Liu, C. L. et al. Cysteine protease cathepsins in cardiovascular disease: from basic research to clinical trials. Nat. Rev. Cardiol. 15, 351–370 (2018).
Emanuel, R. et al. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arterioscler. Thromb. Vasc. Biol. 34, 1942–1952 (2014).
Nishino, I. et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 406, 906–910 (2000).
Papadopoulos, C. & Meyer, H. Detection and clearance of damaged lysosomes by the endo-lysosomal damage response and lysophagy. Curr. Biol. 27, R1330–r1341 (2017).
Mindell, J. A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 74, 69–86 (2012).
Pu, J., Guardia, C. M., Keren-Kaplan, T. & Bonifacino, J. S. Mechanisms and functions of lysosome positioning. J. Cell Sci. 129, 4329–4339 (2016).
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Saxton, R. A. et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 351, 53–58 (2016).
Wyant, G. A. et al. mTORC1 activator SLC38A9 is required to efflux essential amino acids from lysosomes and use protein as a nutrient. Cell 171, 642–654 e612 (2017).
Li, P., Gu, M. & Xu, H. Lysosomal ion channels as decoders of cellular signals. Trends Biochem. Sci. 44, 110–124 (2019).
Xiong, J. & Zhu, M. X. Regulation of lysosomal ion homeostasis by channels and transporters. Sci. China Life Sci. 59, 777–791 (2016).
Luzio, J. P., Parkinson, M. D., Gray, S. R. & Bright, N. A. The delivery of endocytosed cargo to lysosomes. Biochem. Soc. Trans. 37, 1019–1021 (2009).
Kaur, J. & Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 16, 461–472 (2015).
Singh, R. & Cuervo, A. M. Autophagy in the cellular energetic balance. Cell Metab. 13, 495–504 (2011).
Inpanathan, S. & Botelho, R. J. The lysosome signaling platform: adapting with the times. Front. Cell Dev. Biol. 7, 113 (2019).
Savini, M., Zhao, Q. & Wang, M. C. Lysosomes: signaling hubs for metabolic sensing and longevity. Trends Cell Biol. 29, 876–887 (2019).
Lamming, D. W. & Bar-Peled, L. Lysosome: the metabolic signaling hub. Traffic 20, 27–38 (2019).
Uribe-Querol, E. & Rosales, C. Control of phagocytosis by microbial pathogens. Front. Immunol. 8, 1368 (2017).
Lim, J. J., Grinstein, S. & Roth, Z. Diversity and versatility of phagocytosis: roles in innate immunity, tissue remodeling, and homeostasis. Front. Cell Infect. Microbiol. 7, 191 (2017).
Rosales, C. & Uribe-Querol, E. Phagocytosis: a fundamental process in immunity. Biomed. Res. Int. 2017, 9042851 (2017).
Gordon, S. Phagocytosis: an immunobiologic process. Immunity 44, 463–475 (2016).
Fairn, G. D. & Grinstein, S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol. 33, 397–405 (2012).
Feng, Y., He, D., Yao, Z. & Klionsky, D. J. The machinery of macroautophagy. Cell Res. 24, 24–41 (2014).
Li, W. W., Li, J. & Bao, J. K. Microautophagy: lesser-known self-eating. Cell Mol. Life Sci. 69, 1125–1136 (2012).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Chiang, H. L., Terlecky, S. R., Plant, C. P. & Dice, J. F. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385 (1989).
Bandyopadhyay, U., Kaushik, S., Varticovski, L. & Cuervo, A. M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 28, 5747–5763 (2008).
Rout, A. K., Strub, M. P., Piszczek, G. & Tjandra, N. Structure of transmembrane domain of lysosome-associated membrane protein type 2a (LAMP-2A) reveals key features for substrate specificity in chaperone-mediated autophagy. J. Biol. Chem. 289, 35111–35123 (2014).
Yim, W. W. & Mizushima, N. Lysosome biology in autophagy. Cell Discov. 6, 6 (2020).
Salvador, N., Aguado, C., Horst, M. & Knecht, E. Import of a cytosolic protein into lysosomes by chaperone-mediated autophagy depends on its folding state. J. Biol. Chem. 275, 27447–27456 (2000).
Agarraberes, F. A. & Dice, J. F. A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci. 114, 2491–2499 (2001).
Mizushima, N. & Komatsu, M. Autophagy: renovation of cells and tissues. Cell 147, 728–741 (2011).
Yao, R. Q., Ren, C., Xia, Z. F. & Yao, Y. M. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy. https://doi.org/10.1080/15548627.2020.1725377 (2020).
Shaid, S., Brandts, C. H., Serve, H. & Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 20, 21–30 (2013).
Weidberg, H., Shvets, E. & Elazar, Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 80, 125–156 (2011).
Gatica, D., Lahiri, V. & Klionsky, D. J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242 (2018).
Hesketh, G. G., Wartosch, L., Davis, L. J., Bright, N. A. & Luzio, J. P. The lysosome and intracellular signalling. Prog. Mol. Subcell. Biol. 57, 151–180 (2018).
Saxton, R. A. & Sabatini, D. M. mTOR signaling in growth, metabolism, and disease. Cell 169, 361–371 (2017).
Sancak, Y. et al. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141, 290–303 (2010).
Wang, S. et al. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 347, 188–194 (2015).
Bar-Peled, L., Schweitzer, L. D., Zoncu, R. & Sabatini, D. M. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208 (2012).
Zoncu, R. et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683 (2011).
Peng, M., Yin, N. & Li, M. O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 543, 433–437 (2017).
Parmigiani, A. et al. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 9, 1281–1291 (2014).
Long, X., Lin, Y., Ortiz-Vega, S., Yonezawa, K. & Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 15, 702–713 (2005).
Ben-Sahra, I. & Manning, B. D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 45, 72–82 (2017).
Iadevaia, V., Liu, R. & Proud, C. G. mTORC1 signaling controls multiple steps in ribosome biogenesis. Semin. Cell Dev. Biol. 36, 113–120 (2014).
Martina, J. A., Chen, Y., Gucek, M. & Puertollano, R. mTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012).
Zhang, T. et al. Structural basis for Ragulator functioning as a scaffold in membrane-anchoring of Rag GTPases and mTORC1. Nat. Commun. 8, 1394 (2017).
Hardie, D. G., Ross, F. A. & Hawley, S. A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 (2012).
Papadopoulos, C., Kravic, B. & Meyer, H. Repair or lysophagy: dealing with damaged lysosomes. J. Mol. Biol. 432, 231–239 (2020).
Bove, J. et al. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy 10, 889–900 (2014).
Repnik, U., Hafner Česen, M. & Turk, B. Lysosomal membrane permeabilization in cell death: concepts and challenges. Mitochondrion 19 Pt A, 49–57 (2014).
Serrano-Puebla, A. & Boya, P. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann. N. Y. Acad. Sci. 1371, 30–44 (2016).
Gomez-Sintes, R., Ledesma, M. D. & Boya, P. Lysosomal cell death mechanisms in aging. Ageing Res. Rev. 32, 150–168 (2016).
Kirkegaard, T. et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 463, 549–553 (2010).
Petersen, N. H., Kirkegaard, T., Olsen, O. D. & Jaattela, M. Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle 9, 2305–2309 (2010).
Skowyra, M. L., Schlesinger, P. H., Naismith, T. V. & Hanson, P. I. Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360 (2018).
Radulovic, M. et al. ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. Embo J. https://doi.org/10.15252/embj.201899753 (2018).
Christ, L., Raiborg, C., Wenzel, E. M., Campsteijn, C. & Stenmark, H. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 42, 42–56 (2017).
Maejima, I. et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. Embo J. 32, 2336–2347 (2013).
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A. & Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418 (2012).
Yoshida, Y. et al. Ubiquitination of exposed glycoproteins by SCF(FBXO27) directs damaged lysosomes for autophagy. Proc. Natl Acad. Sci. USA 114, 8574–8579 (2017).
Chauhan, S. et al. TRIMs and Galectins globally cooperate and TRIM16 and Galectin-3 co-direct autophagy in endomembrane damage homeostasis. Dev. Cell 39, 13–27 (2016).
Braulke, T. & Bonifacino, J. S. Sorting of lysosomal proteins. Biochim Biophys. Acta 1793, 605–614 (2009).
Reczek, D. et al. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 131, 770–783 (2007).
Saftig, P. & Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10, 623–635 (2009).
Palmieri, M. et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 (2011).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Napolitano, G. & Ballabio, A. TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016).
Roczniak-Ferguson, A. et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5, ra42 (2012).
de Duve, C. Lysosomes revisited. Eur. J. Biochem. 137, 391–397 (1983).
Gao, C. et al. Tacrine induces apoptosis through lysosome- and mitochondria-dependent pathway in HepG2 cells. Toxicol. in Vitro 28, 667–674 (2014).
Gao, H. et al. Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 503, 1550–1556 (2018).
Hengartner, M. O. The biochemistry of apoptosis. Nature 407, 770–776 (2000).
Tait, S. W. & Green, D. R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 11, 621–632 (2010).
Riedl, S. J. & Salvesen, G. S. The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 8, 405–413 (2007).
Li, Z., Berk, M., McIntyre, T. M., Gores, G. J. & Feldstein, A. E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 47, 1495–1503 (2008).
Gao, C. et al. Phellinus linteus mushroom protects against tacrine-induced mitochondrial impairment and oxidative stress in HepG2 cells. Phytomedicine 20, 705–709 (2013).
Xu, M. et al. Inhibition of cysteine cathepsin B and L activation in astrocytes contributes to neuroprotection against cerebral ischemia via blocking the tBid-mitochondrial apoptotic signaling pathway. Glia 62, 855–880 (2014).
Galluzzi, L. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on cell death 2012. Cell Death Differ. 19, 107–120 (2012).
Degterev, A. et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 (2005).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Gong, Y. et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 18, 100 (2019).
Shi, J., Gao, W. & Shao, F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42, 245–254 (2017).
He, Y., Hara, H. & Nunez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41, 1012–1021 (2016).
Xie, Y. et al. Ferroptosis: process and function. Cell Death Differ. 23, 369–379 (2016).
Yu, F. et al. The role of lysosome in cell death regulation. Tumour Biol. 37, 1427–1436 (2016).
Stockwell, B. R. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017).
Hou, W. et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016).
Yang, A. et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 4, 905–913 (2014).
Wang, K., Tu, Y., Wan, J. B., Chen, M. & He, C. Synergistic anti-breast cancer effect of pulsatilla saponin D and camptothecin through interrupting autophagic-lysosomal function and promoting p62-mediated ubiquitinated protein aggregation. Carcinogenesis 41, 804–816 (2019).
Bengsch, F. et al. Cell type-dependent pathogenic functions of overexpressed human cathepsin B in murine breast cancer progression. Oncogene 33, 4474–4484 (2014).
Sevenich, L. et al. Transgenic expression of human cathepsin B promotes progression and metastasis of polyoma-middle-T-induced breast cancer in mice. Oncogene 30, 54–64 (2011).
Withana, N. P. et al. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res. 72, 1199–1209 (2012).
Zhao, Q., Gao, S. M. & Wang, M. C. Molecular mechanisms of lysosome and nucleus communication. Trends Biochem. Sci. https://doi.org/10.1016/j.tibs.2020.06.004 (2020).
Machado, E. et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 1, e1500603 (2015).
Lawrence, R. E. & Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 21, 133–142 (2019).
Nixon, R. A. The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 (2013).
Stirnemann, J. et al. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int. J. Mol. Sci. 18, 441 (2017).
Boutry, M., Morais, S. & Stevanin, G. Update on the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 19, 18 (2019).
Coffey, E. E., Beckel, J. M., Laties, A. M. & Mitchell, C. H. Lysosomal alkalization and dysfunction in human fibroblasts with the Alzheimer’s disease-linked presenilin 1 A246E mutation can be reversed with cAMP. Neuroscience 263, 111–124 (2014).
Lee, J. H. et al. Presenilin 1 maintains lysosomal Ca(2+) homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 12, 1430–1444 (2015).
Kinouchi, K., Ichihara, A. & Itoh, H. Functional characterization of (pro)renin receptor in association with V-ATPase. Front. Biosci. 16, 3216–3223 (2011).
Park, J. S., Blair, N. F. & Sue, C. M. The role of ATP13A2 in Parkinson’s disease: clinical phenotypes and molecular mechanisms. Mov. Disord. 30, 770–779 (2015).
Funakoshi-Hirose, I. et al. Distinct effects of methamphetamine on autophagy-lysosome and ubiquitin-proteasome systems in HL-1 cultured mouse atrial cardiomyocytes. Toxicology 312, 74–82 (2013).
Pal, R. et al. NADPH oxidase promotes Parkinsonian phenotypes by impairing autophagic flux in an mTORC1-independent fashion in a cellular model of Parkinson’s disease. Sci. Rep. 6, 22866 (2016).
Weiss, U. Inflammation. Nature 454, 427 (2008).
Neurath, M. F. Resolution of inflammation: from basic concepts to clinical application. Semin. Immunopathol. 41, 627–631 (2019).
Oami, T. et al. Blocking liver autophagy accelerates apoptosis and mitochondrial injury in hepatocytes and reduces time to mortality in a murine sepsis model. Shock 50, 427–434 (2018).
Ho, J. et al. Autophagy in sepsis: degradation into exhaustion? Autophagy 12, 1073–1082 (2016).
Ren, C., Zhang, H., Wu, T. T. & Yao, Y. M. Autophagy: a potential therapeutic target for reversing sepsis-induced immunosuppression. Front. Immunol. 8, 1832 (2017).
Zhang, H., Feng, Y. W. & Yao, Y. M. Potential therapy strategy: targeting mitochondrial dysfunction in sepsis. Mil. Med. Res. 5, 41 (2018).
Yang, X. et al. Hydroxytyrosol attenuates LPS-induced acute lung injury in mice by regulating autophagy and sirtuin expression. Curr. Mol. Med. 17, 149–159 (2017).
De Meyer, G. R. et al. Autophagy in vascular disease. Circ. Res. 116, 468–479 (2015).
Ma, X., Godar, R. J., Liu, H. & Diwan, A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 8, 297–309 (2012).
Nagoor Meeran, M. F. et al. α-Bisabolol protects against β-adrenergic agonist-induced myocardial infarction in rats by attenuating inflammation, lysosomal dysfunction, NLRP3 inflammasome activation and modulating autophagic flux. Food Funct. 11, 965–976 (2020).
Zhang, S. et al. Knockout of Eva1a leads to rapid development of heart failure by impairing autophagy. Cell Death Dis. 8, e2586 (2017).
Ma, X. et al. Transcription factor EB activation rescues advanced αB-crystallin mutation-induced cardiomyopathy by normalizing desmin localization. J. Am. Heart Assoc. 8, e010866 (2019).
Li, J., Xiang, X. & Xu, Z. Cilostazol protects against myocardial ischemia and reperfusion injury by activating transcription factor EB (TFEB). Biotechnol. Appl Biochem. 66, 555–563 (2019).
Dubland, J. A. & Francis, G. A. Lysosomal acid lipase: at the crossroads of normal and atherogenic cholesterol metabolism. Front. Cell Dev. Biol. 3, 3 (2015).
Zhang, H. Lysosomal acid lipase and lipid metabolism: new mechanisms, new questions, and new therapies. Curr. Opin. Lipidol. 29, 218–223 (2018).
Endo, Y., Furuta, A. & Nishino, I. Danon disease: a phenotypic expression of LAMP-2 deficiency. Acta Neuropathol. 129, 391–398 (2015).
Beertsen, W. et al. Impaired phagosomal maturation in neutrophils leads to periodontitis in lysosomal-associated membrane protein-2 knockout mice. J. Immunol. 180, 475–482 (2008).
Tanaka, Y. et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 406, 902–906 (2000).
Acknowledgements
This work was supported by grants from the National Natural Science Foundation (Nos. 81730057, 81801935) and the National Key Research and Development Program of China (No. 2017YFC1103302).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by B. Zhivotovsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, Sy., Yao, Rq., Li, Yx. et al. Lysosomal quality control of cell fate: a novel therapeutic target for human diseases. Cell Death Dis 11, 817 (2020). https://doi.org/10.1038/s41419-020-03032-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-03032-5
This article is cited by
-
The discovery of regional neurotoxicity-associated metabolic alterations induced by carbon quantum dots in brain of mice using a spatial metabolomics analysis
Particle and Fibre Toxicology (2024)
-
The race to uncover fission factors for lysosomal organelles heats up
Nature (2024)
-
Mechanical overload-induced release of extracellular mitochondrial particles from tendon cells leads to inflammation in tendinopathy
Experimental & Molecular Medicine (2024)
-
Erythronecroptosis: an overview of necroptosis or programmed necrosis in red blood cells
Molecular and Cellular Biochemistry (2024)
-
Diversity and complexity of cell death: a historical review
Experimental & Molecular Medicine (2023)
