
Abstract
Lysosome is a crucial organelle in charge of degrading proteins and damaged organelles to maintain cellular homeostasis. Transcription factor EB (TFEB) is the master transcription factor regulating lysosomal biogenesis and autophagy. Under external stimuli such as starvation, dephosphorylated TFEB transports into the nucleus to specifically recognize and bind to the coordinated lysosomal expression and regulation (CLEAR) elements at the promotors of autophagy and lysosomal biogenesis-related genes. The function of TFEB in the nucleus is fine regulated but the molecular mechanism is not fully elucidated. In this study, we discovered that miR-30b-5p, a small RNA which is known to regulate a series of genes through posttranscriptional regulation in the cytoplasm, was translocated into the nucleus, bound to the CLEAR elements, suppressed the transcription of TFEB-dependent downstream genes, and further inhibited the lysosomal biogenesis and the autophagic flux; meanwhile, knocking out the endogenous miR-30b-5p by CRISPR/Cas9 technique significantly increased the TFEB-mediated transactivation, resulting in the increased expression of autophagy and lysosomal biogenesis-related genes. Overexpressing miR-30b-5p in mice livers showed a decrease in lysosomal biogenesis and autophagy. These in vitro and in vivo data indicate that miR-30b-5p may inhibit the TFEB-dependent transactivation by binding to the CLEAR elements in the nucleus to regulate the lysosomal biogenesis and autophagy. This novel mechanism of nuclear miRNA regulating gene transcription is conducive to further elucidating the roles of miRNAs in the lysosomal physiological functions and helps to understand the pathogenesis of abnormal autophagy-related diseases.
Similar content being viewed by others


Introduction
The lysosome is a crucial organelle for macroautophagy (hereon autophagy), a basic physiological process for cells to maintain cellular homeostasis, which degrades autophagic cargoes such as incorrectly folded proteins, adipose grains, aging or damaged mitochondria, and endoplasmic reticulum [1, 2]. Transcription factor EB (TFEB), a basic helix-loop-helix–leucine-zipper transcription factor, is considered to be the master regulator of the lysosomal biogenesis and autophagy and mediates a variety of physiological processes that involve immune response, neurodegenerative diseases, metabolic diseases, and radiation protection [3,4,5,6]. Under normal conditions, TFEB is commonly phosphorylated at S142 and/or S211 by the kinase mTORC1 and retained in the cytoplasm by binding to the protein 14–3–3 [7, 8]. However, under external stimuli such as starvation, dephosphorylated TFEB is transported into the nucleus and binds to the coordinated lysosomal expression and regulation (CLEAR) element, a palindromic 10-base pair motif (GTCACGTGAC) at the promoters of autophagy and lysosomal biogenesis genes to activate their transcription [9, 10].
MiRNAs are a class of noncoding RNAs with a length of about 22 nucleotides, which play a momentous role in a range of biological processes and diseases [11,12,13,14]. In the nucleus of mammalian cells, the precursor miRNA (pri-miRNA) is transcribed by the polymerase II (pol II) and subsequently cleaved by Drosha and its essential cofactor DGCR8 to form the hairpin-like precursor RNA (pre-miRNA) with a length of about 65 nucleotides [15, 16]. The pre-miRNA is exported to the cytoplasm by Exportin-5 and further cleaved by Dicer to produce the double-stranded miRNA [17, 18]. Then, in the favor of Argonaute proteins combining with TRBP, one strand of the duplexes degrades and the other becomes a mature miRNA to regulate the expression of target genes [19,20,21,22]. The classic regulatory manner of miRNA is the posttranscriptional gene silencing (TGS) that miRNA assembles the miRNA-induced silencing complex to degrade the target mRNAs or suppress their translation by the seed sequence at the 5′ end [23, 24].
It is generally accepted that miRNAs mainly locate in the cytoplasm and function as the posttranscriptional regulators. However, in recent years, plenty of studies have shown that mature miRNAs also exist in the nucleus [11, 12, 25,26,27,28,29] and mediate the transcriptional gene activation or TGS in both animal and plant cells, which broadens the functional spectrum of miRNAs [19, 23]. miR-21 is the first miRNA found to be located in both the nucleus and the cytoplasm [24]. miR-373 was further found that it could target the gene promoters of E-cadherin (CDH1) and cold shock domain-containing protein C2, and induce gene expression [23]. In addition, Kim and his colleagues showed that miR-320, encoded by the promoter region of POLR3D, directed the association of RNA interference (RNAi) protein argonaute-1, polycomb group component EZH2, and tri-methyl histone H3 lysine 27 with the POLR3D promoter, leading to the transcriptional silencing of POLR3D expression [19]. Since then, many studies have shown that miRNA inhibited the transcription of a single gene or a group of genes, which involve a lot of cellular functions such as granulopoiesis and mitochondrial dysfunction [30, 31]. Nevertheless, there are few studies illuminating how miRNAs regulate the transcriptional activity of a transcription factor in the nucleus. In this study, we found that nuclear miR-30b-5p suppressed the transcriptional activity of TFEB by targeting its binding motif at the gene promoter regions containing the CLEAR element to inhibit lysosomal biogenesis and autophagy.
Materials and methods
Cell culture and drug treatment
HEK293 and HeLa cells were purchased from American Type Culture Collection (VA) and cultured in DMEM medium (Gibco, USA) containing 10% FBS and antibiotics (penicillin and streptomycin, 50 units/mL each). All cells were grown in a humidified incubator with 5% CO2 at 37 °C. Cells were starved with Earle’s balanced salt solution (Gibco) for 2 h or treated with DMSO (Sangon, China), or Torin1 (250 nM) (Tocris Bioscience, UK) for 2 h, or Bafilomycin A1 (100 nM) (Cell Signaling Technology, USA) for 18 h.
Isolation of nuclear and cytoplasmic protein and RNA
The nuclear and cytoplasmic isolation of protein in HEK293 cells was performed using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher, USA). Nuclear and cytoplasmic RNA was isolated as previously described [25]. Mature miR-30b-5p was detected using TaqMan MicroRNA assay kit (Invitrogen) by quantitative real-time PCR (qPCR). 47s pre-rRNA or tRNAlys was used as an internal control for the nucleus and cytoplasm, respectively.
Quantitative real-time PCR (qPCR)
Total RNA was extracted by Trizol reagent (Invitrogen, USA) from cells and reverse transcribed into cDNA using PrimeScript RT Master Mix (TaKaRa Biotechnology, Japan). qPCR was performed to detect mRNA expression of lysosomal genes by TB Green PCR Kit (TaKaRa Biotechnology) and GAPDH was used as an internal control. For mature miR-30b-5p detection, total RNA was reverse transcribed into cDNA using Taqman® MicroRNA Reverse Transcription Kit (Invitrogen). Taqman® MiRNA Assays (Invitrogen) were further used to analyze the expression of miR-30b-5p.
Constructs and transfections
The EGFP-mCherry-LC3B tandem reporter, EGFP-TFEB and EGFP-TFEBS211A plasmids were previously described [5]. EGFP–NES (nuclear export signal) plasmid was generated by cloning NES (CCCAAAAAGAAGAGGAAAGTG) into the EGFP-C3 vector. Plasmid DNA was transfected into cells with Lipofectamine 2000 (Invitrogen). Transfection of miRNA mimics (GenePharm, China), small interfering RNAs [si-TFEB: 5′-GAACAAGUUUGCUGCCCACAU-3′ [5]; siRNA targeting importin 8 gene (si-Importin8): 5′-UGAGCUCAAUCUAAGAAAUUT-3′, 5′-AUUUCUUAGAUUGAGCUCAUT-3′ [32]] (RiboBio, China) or their corresponding controls was performed using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer’s instructions with a concentration of 50 nM.
Pull-down assay
Single-stranded 4×CLEAR (5′-CCGGGTCACGTGACCCCAGGGTCACGTGACCCTGCGGGTCACGTGACCCTGCGGGTCACGTGACCCCC-3′) and Mut-4×CLEAR (5′-CCGGGAATCGTGACCCCAGGGAATCGTGACCCTGCGGGAATCGTGACCCTGCGGGAATCGTGACCCCC-3′) elements labeled with biotins at their 3′ ends were synthesized in Sangon. Single biotinylated miR-30b-5p and miRNA control were purchased from RiboBio. The biotinylated nucleotides were dissolved in wash/binding buffer (0.5 M NaCl, 20 mM Tris–HCl, 1 mM EDTA, pH 7.4) at a final concentration of 20 pmol/μl. After incubated with streptavidin-coated magnetic beads (New England BioLabs, USA) at room temperature for 1 h, they were incubated with nuclear RNA or protein at room temperature for 3 h. Then, the beads/RNA or beads/protein complexes were washed four times with the wash/binding buffer. The RNA was eluted from the streptavidin beads with Trizol reagent and analyzed by small RNA-seq using BGISEQ-500 Sequencing technology (Beijing Genomics Institution, China). Protein elution was performed by radio-immunoprecipitation assay (RIPA) lysis buffer (Beyotime, China) and detected by western blot.
Western blot
The cells or tissues were lysed by cell lysis buffer [25 mM Tris-HCl (pH 7.6), 1% NP-40, 150 mM NaCl and 1% sodium deoxycholate] adding a protease inhibitor cocktail (Roche). The samples were prepared and separated by SDS-PAGE. Then, they were transferred to PVDF membranes and incubated with primary antibodies. Mouse anti-SQSTM1/p62 (ab56416), rabbit anti-GLA (ab168341), rabbit anti-H2B (ab1790), rabbit anti-TPP1 (ab195234), and rabbit anti-LC3 (ab51520) antibodies were purchased from Abcam (USA). Mouse anti-GFP (SC-9996) was purchased from Santa Cruz (USA). Rabbit anti-LAMP1 (9091), rabbit anti-TFEB (37785), rabbit anti-mTOR (2983), rabbit anti-phospho-mTOR (5536), rabbit anti-p70S6K (2708), and anti-phospho-p70S6K (9234) antibodies were obtained from Cell Signaling Technology. α-Tubulin (ThermoFisher, PA5-22060) or GAPDH (Millipore, C87727) was used as a loading control. The Goat anti-mouse IgG (H + L) (115–035–003) and Goat anti-rabbit IgG (H + L) (111–035–003) secondary antibodies, coupled to horseradish peroxidase, were purchased from Jackson ImmunoResearch. After incubated with secondary antibodies, chemiluminescence was detected by Immobilon Western Chemiluminescent HRP Substrate (Millipore) and analyzed by ImageQuant software (GE Healthcare, USA).
Luciferase assays
4×CLEAR(5′-CCGGGTCACGTGACCCCAGGGTCACGTGACCCTGCGGGTCACGTGACCCTGCGGGTCACGTGACCCCC-3′), Mut-4×CLEAR (5′-CCGGGAATCGTGACCCCAGGGAATCGTGACCCTGCGGGAATCGTGACCCTGCGGGAATCGTGACCCCC-3′), and the promoter regions of six lysosomal biogenesis genes, including CTSA (−274 to +282 bp), CTSD (−500 to +50 bp), MCOLN1 (−500 to +50 bp), LAMP2 (−500 to +50 bp), ARSB (−200 to +50 bp), and NEU1 (−50 to +100 bp), were synthesized by Synbio Tech (China) [10]. Then, they were cloned into pGL4.10 luciferase reporter vectors. HEK293 cells were transfected with miR-30b-5p or miR-control (50 nM) and after 24 h, these cells were transfected with the reporter constructs (0.5 μg) in 24-well plates. Forty-eight hours after transfection, cell extracts were prepared and the luciferase activity was measured by the Dual-Luciferase Reporter Assay System (Promega).
Fluorescence resonance energy transfer (FRET) assay
Single-stranded DNA probe and miRNA mimics labeled with the Black Hole Quencher 1 (BHQ1) dye and FAM dye (BioSune) were dissolved in 1×annealing buffer (20 mM Tris–HCl pH 8.0, 100 mM NaCl, and 10 mM EDTA) at a final concentration of 20 μM. After a hybridization at 72 °C for 5 min and followed by slow cooling to room temperature, the FAM fluorescence intensities were detected with an excitation wavelength of 492 nm and an emission wavelength of 518 nm.
Fluorescence in situ hybridization (FISH)
HEK293 cells were fixed with 4% paraformaldehyde for 10 min and washed three times for 1 min in PBS at room temperature. Then, 0.1% Triton X-100 was used to increase the permeability of cells. Cells were preincubated in the hybridization solution (Exiqon) for 20 min at 55 °C. FAM-labeled microRNA Detection Probes (Exiqon) was added into cells and hybridized at 55 °C for 1 h followed by an extensive wash with SSC buffer. After incubated in block buffer for 30 min at room temperature, cells were treated with anti-FAM reagent (Roche) for a color reaction. All samples were incubated with DAPI (5 μg/ml) for nuclear staining and imaged using the AIR laser-scanning confocal microscope (TCS SP8; Leica, Germany).
CRISPR/Cas9-modified nucleotides knockdown
As previously described, two sgRNAs fragments: 5′-CACCGAACTCTTTACTAAGTTTGTA-3′ and 5′-CACCGGCTGTAATACATGGATTGGC-3′, were cloned into px330-mCherry and px330-GFP vectors, respectively [33]. Then, the plasmids were co-transfected into HEK293 cells or HeLa cells, and transfected cells were enriched by flow cytometry. For HEK293 cells, the lines from a single cell were obtained, and the one with the lowest expression of mature miR-30b-5p was used for further experiments. For HeLa cells, the genome deletion of miR-30b-5p is based on a purified population.
Chromatin immunoprecipitation (ChIP) assay
MiR-30b-5p and miR-control mimics were transfected into HEK293 cells at 50 nM final concentration. Forty-eight hours after transfection, cells were cross-linked by 1% formaldehyde for 10 min at room temperature. For ChIP assay in vivo, livers were harvested, cut into 1–3 mm pieces and cross-linked by 1% formaldehyde for 10 min at room temperature. ChIP assay was performed using the ChIP Assay Kit (ThermoFisher) according to the manufacturer’s handbook. Rabbit anti-RNA pol II (05–623) and rabbit anti-TFEB (37785) antibodies were purchased from Millipore and Cell Signaling Technology, respectively. Normal rabbit IgG (Santa Cruz) was used as a native control. qPCR was performed to assess the promoters of CLCN7, MCOLN1, CTSD, GLA and data were analyzed by normalizing to chromatin input.
Immunofluorescence staining and fluorescence imaging
The cells were washed with PBS and fixed with 4% paraformaldehyde in PBS (pH 7.4) for 10 min. Then, the cells were permeated with 0.1% Triton X-100 for 5 min and blocked with 0.5% FBS in PBST (PBS containing 0.1% tween-20). Immunocytochemical staining was performed using rabbit anti-LAMP1 antibody and then the cells were incubated with Alexa Fluor 488 donkey anti-rabbit secondary antibodies (ThermoFisher). For live cell staining, the cells were stained using Lysotracker red DND-99 (ThermoFisher) for 10 min. The tandem EGFP-mCherry-LC3 probe was transfected into the related cells for 48 h and then fixed with 4% paraformaldehyde in PBS (pH 7.4) for 10 min. All samples were observed using fluorescence microscopy (Nikon, Japan) or confocal microscopy (TCS SP8; Leica, Germany). For immunofluorescence staining on tissue sections, the slices were fixed with 4% paraformaldehyde in PBS (pH 7.4) overnight and permeated with 0.1% Triton X-100 for 10 min. After being blocked with 4% goat serum in PBS, the slices were incubated with mouse anti-SQSTM1/p62 (ab56416) and Rabbit anti-LAMP1 (9091) overnight and washed with PBS for three times. Alexa Fluor 488 goat anti-mouse or Alexa Fluor 647 goat anti-rabbit second antibodies were used. Then, the samples were observed using confocal microscopy.
Mass spectrometry
Total protein lysates from HEK293 cells transfected with miR-30b-5p or miR-control mimics for 48 h were prepared by RIPA lysis buffer containing 1:1000 phenylmethanesulfonyl fluoride (Beyotime). Proteins were precipitated with pre-cold acetone and dried by SpeedVac. The pellet was dissolved in 8 M urea; 100 mM Tris-HCl, pH 8.5. 5 mM TCEP (ThermoFisher), and 10 mM iodoacetamide (Sigma) were subsequently added into the solution for reduction and alkylation, respectively. The solution was then incubated at room temperature for 30 min. The protein mixture was diluted four times and digested overnight at 37 °C with trypsin at 1:40 (w/w) (Promega). Mass spectrometry was performed in the National Center for Protein Science—Shanghai following the standard protocol. The tryptic digested peptides were analyzed using a modified MudPIT separation that has been described previously [34]. Data-dependent tandem mass spectrometry (MS/MS) analysis was performed with an Orbitrap Elite mass spectrometer (ThermoFisher). The acquired MS/MS data were analyzed against a Swiss-Prot Homo sapiens database (November 2018) using PEAKS Studio 8.5 (Bioinformatics Solutions, Canada). A protein was identified when at least one unique peptide was matched. Protein quantification was based on label-free quantitative analysis. Furthermore, spectrum counts were normalized with the total identified spectra per biological sample and the proteins with statistically higher or lower peptide counts in the participants were considered as different expressions. For cellular component analysis, 3771 proteins with a threshold of 0.5 ≤ Group Profile (miR-30b-5p/miR-control) ≤1.5 were selected and analyzed using FunRich version 3.1.3 (March 2017) database [35].
AAV injection and starvation in mice
All animal experiments were performed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The 6–8 weeks old male C57BL mice were purchased from Shanghai Jihui Experimental Animal Breeding Co., Ltd (Shanghai, China) and fed under specific pathogen-free conditions at National Center for Protein Science—Shanghai. AAV-mCherry and AAV-mCherry-miR-30b-5p plasmids were generated by cloning pri-miR-30b-5p into PGMAAV-MA2 vectors in Genomeditech (Shanghai, China). Twenty-eight mice, randomly divided into four groups (n = 7 for each group), were injected with AAV-mCherry or AAV-mCherry-miR-30b-5p (MOI = 1 × 1012) via tail vein for 9 weeks and starved for 48 h.
Statistical analysis
Data of at least three independent experiments analyzed by GraphPad Prism software (version 7, GraphPad Software) were presented as mean values with standard deviation (SD) and p < 0.05 was considered as significant. Student’s t test (two tailed) was employed to analyze data. For fluorescence quantification, 50 cells were analyzed.
Results
Screening of candidate miRNAs binding to the CLEAR element
To investigate whether nuclear miRNAs moderate the TFEB-dependent transcriptional regulation, the nuclear RNAs from HEK293 cells (Fig. S1A) were incubated with the DNA fragment containing the 4×CLEAR element (GTCACGTGAC), the binding motif of TFEB. DNA fragment with the mutant 4×CLEAR element (GAATCGTGAC) was used as the negative control (Mut-4×CLEAR). By pull-down assay, binding of TFEB to the 4×CLEAR element, but not the Mut-4 CLEAR element, was confirmed (Fig. S1B). Then, small RNA-sequencing was used to detect the nuclear miRNAs that were pulled down by the 4×CLEAR element and the mutant 4×CLEAR element, respectively (Fig. 1a). The threshold for selecting the miRNAs specific binding to the 4×CLEAR element was set as CLEAR_TPM > 100 and (CLEAR_TPM − Mut_CLEAR_TPM)/Mut_CLEAR_TPM > 0.4. Twenty-eight miRNAs were identified (Fig. 1b) and transfected into HEK293 cells with the 4×CLEAR luciferase reporter (Fig. 1c). We noticed that the normalized activity from the 4×CLEAR luciferase reporter decreased 46% with miR-30b-5p transfection, comparing to that with nontargeted miRNA control transfection (miR-control, p < 0.001; Fig. 1c). Motifs complementary to miR-30b-5p were observed on the promoters of lysosomal biogenesis genes, CTSD, ARSB, and NEU1, which were known containing the CLEAR element (Fig. 1d) [10]. The direct interaction between miR-30b-5p and the DNA fragment of CTSD gene promoter was validated by FRET assay. Fluorescent dye FAM labeled on miR-30b-5p or Mut-miR-30b-5p was used as the donor, and BHQ1 dye labeled on single-stranded DNA probe was used as the quencher. It showed that fluorescence from miR-30b-5p probe, instead of Mut-miR-30b-5p probe (the predicted binding motif of the CLEAR element was mutated; Fig. 1d), was decreased by 35% (p < 0.01; Fig. 1e), indicating the specific binding of miR-30b-5p to CTSD gene promoter. In summary, we identified that a novel miRNA, miR-30b-5p, could bind to the CLEAR element and suppress the transcriptional activation of CLEAR element-dependent luciferase reporter activity.

a Schematic depicting the workflow of screening for miRNAs binding to the CLEAR element. Nuclear RNA extraction from HEK293 cells was co-incubated with the CLEAR elements labeled with biotins. Result of the pull-down assay was analyzed by small RNA-sequencing and the luciferase assay was performed to further evaluate the candidate miRNAs. b Result of small RNA-sequencing: the threshold of candidate miRNAs was set as CLEAR_TPM > 100 and (CLEAR_TPM − Control_TPM)/Control_TPM > 0.4. c HEK293 cells were transfected with the reporter constructs containing the 4×CLEAR elements for 24 h and then re-transfected with the related candidate miRNAs. Forty-eight hours later, the luciferase assay was operated. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. d Schematics of intact/mutated miR-30b-5p and the CLEAR element potential binding site on it. e FRET assays detecting the binding between miR-30b-5p and the CLEAR elements of CTSD promoters. Data were presented as mean ± SD of three independent experiments, **p < 0.01.
MiR-30b-5p inhibited the transactivation of CLEAR element-containing genes in the nucleus
Given that the usual subcellular location of the CLEAR element is the nucleus, the distribution pattern of miR-30b-5p was examined. FAM-labeled miR-30b-5p detection probes were used to observe the distribution of endogenous miR-30b-5p, and FAM-labeled U6 snRNA detection probes were used as a nuclear location control. The FISH assay showed miR-30b-5p was located in both cytoplasm and nucleus; whereas U6 snRNA exhibited the dominant nuclear location (Fig. 2a). To avoid the effect of FAM dye on miR-30b-5p distribution, the small RNA-sequencing for miR-30b-5p confirmed its expression in both cytoplasmic and nuclear fractions from HEK293 cells (Fig. S2A). The role of miR-30b-5p on the TFEB-dependent transactivation was further investigated by ChIP assay. In the miR-30b-5p-transfected HEK293 cells, the enrichment of TFEB on promoter fragments of the CLEAR element-containing genes, including CLCN7, MCOLN1, CTSD, and GLA, compared with the scramble miRNA-transfected cells (miRNA control) (p < 0.05; Fig. 2b). In the miR-30b-5p knockout HEK293 cells by CRISPR/CAS9 tool (miR-30b-5p+/−; Fig. S2B), the enrichment of TFEB on the promoter fragments of above four genes was consistently increased, compared with the wild-type (WT) cells (p < 0.05; Fig. 2c). The enrichment of pol II on the same promoter fragments of above four genes showed the similar changes as TFEB when the expression level of miR-30b-5p was altered (p < 0.01), except no significant changes on the CLCN7 and GLA promoter fragments in the miR-30b-5p-transfected cells (Fig. S2D, E). Moreover, the luciferase reporter activities of the CLEAR element-containing genes, such as CTSA, CTSD, MCOLN1, and LAMP2, were significantly reduced in the miR-30b-5p-transfected cells (p < 0.05; Fig. 2d) and increased in the miR-30b-5p+/− cells (p < 0.01; Fig. 2e), using the dual-luciferase reporter assay. Consistently, mRNA levels of the CLEAR element-containing genes, including CLCN7, MCOLN1, CTSD, GLA, and TPP1, were significantly reduced in the miR-30b-5p-transfected cells (p < 0.001; Fig. 2f) and increased in the miR-30b-5p+/− cells (p < 0.01; Fig. 2g), using qPCR assay. Similar results were also observed in HeLa cells (Fig. S2C, F, G). To address whether the inhibition role of miR-30b-5p on transactivation of the CLEAR element-containing genes requires nuclear location, si-Importin8, a member of the karyopherin β which is commonly thought to mediate the cytoplasm-to-nucleus transport of mature miRNAs [36] was used, leading to 88% decrease of miR-30b-5p level in the nucleus (Fig. 2h) and the disappearance of inhibitory effect of miR-30b-5p on mRNA expression levels of above CLEAR element-containing genes (Fig. 2i). Collectively, these results confirmed that miR-30b-5p inhibited the transcriptional activation of CLEAR element-containing genes in the nucleus.


a FISH assays showed the distribution of miR-30b-5p in the nucleus and the cytoplasm of HEK293 cells. U6 snRNA was used as a positive control and scramble as a negative control. Scale bar, 10 μm. The fluorescence quantification was performed. ChIP analysis of TFEB enrichment on the promoters of lysosomal genes in (b) wild-type or (c) miR-30b-5p+/− HEK293 cells using anti-TFEB antibody, with IgG antibody as a negative control. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. The luciferase activity of lysosomal promoters was measured in (d) wild-type or (e) miR-30b-5p+/− HEK293 cells. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. The mRNA levels of lysosomal genes in (f) wild-type or (g) miR-30b-5p+/− HEK293 cells were quantified by qPCR analysis, normalized relative to GAPDH. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. h Total RNA from HEK293 cells transfected with si-Importin8 for 48 h was separated into cytoplasmic and nuclear fractions and then subjected to qPCR. Data were presented as mean ± SD of three independent experiments, *p < 0.05 and **p < 0.01. i si-Importin8 and miR-30b-5p or miR-control were co-transfected into HEK293 cells and then qPCR was used to analyze the mRNA expression level of lysosomal genes. Data were presented as mean ± SD of three independent experiments, *p < 0.05.
Since TFEB could be translocated into the nucleus to transactivate the CLEAR element-containing genes, under starvation by mTORC1-mediated dephosphorylation of Ser142 or Ser211 [8], the direct interaction between miR-30b-5p and TFEB was analyzed by pull-down assay, and minimal TFEB could be pulled down by miR-30b-5p, compared to that by the CLEAR element (Fig. S3A), indicating no strong direct binding miR-30b-5p to TFEB. In addition, transfected miR-30b-5p did not change mTORC1 expression nor its downstream signaling (Fig. S3B). Notably, under EBSS-induced starvation, transfected miR-30b-5p could not change the TFEB protein expression level (Fig. S3C), as well as the nuclear translocation of TFEB protein induced by EBSS starvation, using confocal microscopy (Fig. S3D) and western blotting (Fig. S3E). To address whether miR-30b-5p would interfere with the process of nuclear transport, we constructed a plasmid (EGFP–NES) that expressed the fusion protein of the classic nuclear export sequence (NES) and EGFP, which mainly located in the cytoplasm; while the cells were treated with a nuclear export inhibitor-leptomycin B, EGFP–NES showed obvious nuclear localization. However, miR-30b-5p did not affect the nuclear transport of EGFP–NES at all (Fig. S3F). These data imply that the nuclear location of miR-30b-5p is independent of TFEB and the major effect of miR-30b-5p may be through TFEB-dependent transactivation of downstream target genes.
Overexpressing miR-30b-5p suppressed lysosomal biogenesis and autophagy
As TFEB is the master transcription factor in the regulation of lysosomal biogenesis and autophagy and miR-30b-5p inhibited its activity on the lysosomal genes, there remains to be investigated that miR-30b-5p would affect lysosomal biogenesis and autophagy through regulating the transcriptional activity of TFEB. In order to investigate the downstream targets of miR-30b-5p, mass spectrometry was performed in HEK293 cells transfected with miR-30b-5p and miR-control. The results showed that the protein expression levels of most lysosomal genesis-related genes were inhibited with transfected miR-30b-5p (Fig. 3a, b), confirmed by Western blotting for LAMP1, TPP1, and GLA (Fig. 3c). Furthermore, miR-30b-5p was capable to reduce the increased protein levels of above lysosomal genes under nutrition deprivation or Torin1 (a specific mTORC1 inhibitor) treatment (p < 0.01; Fig. 3c, d). Subsequently, we analyzed the levels of the lysosome in the presence of miR-30b-5p via immunofluorescence staining for LAMP1, a classic lysosomal marker. Meanwhile, Lysotracker, a lysosomal fluorescent probe was also used as a lysosomal marker to measure the biogenesis of lysosomes. miR-30b-5p decreased the fluorescent intensities of LAMP1 (Fig. 3e) and Lysotracker (Fig. 3f) under both normal and nutrition deprivation conditions. Similar results were also observed in HeLa cells (Fig. S4A–C). These results indicated that overexpressed miR-30b-5p inhibited the lysosomal biogenesis.


a Mass spectrometry showed that the expression of most lysosomal genes decreased in miR-30b-5p overexpression cells. b 3771 proteins with a threshold of 0.5 ≤ Group Profile (miR-30b-5p/miR-control) ≤1.5 in MS analysis were selected and cellular component analysis were performed using FunRich version 3.1.3 (March 2017) database. HEK293 cells were transfected with miR-30b-5p or miR-control for 48 h. After being treated with EBSS (c) or Torin1 (d) for 2 h, cell lysate was obtained and subjected to immunoblot analysis using the indicated antibodies. The quantification was presented as mean ± SD of three independent experiments, *p < 0.05 and **p < 0.01. e Similar transfection as in (c, d) was performed. Forty-six hours after transfection, the cells were incubated with EBSS for 2 h. Then, the cells were stained using LAMP1 antibody (green). Scale bar, 10 μm. The relative fluorescence densities of LAMP1 were quantified and presented as mean ± SD of three independent experiments, *p < 0.05 and **p < 0.01. f Similar treatment as in (e) was performed and the cells were stained with Lysotracker and visualized. Scale bar, 10 μm. The relative fluorescence densities of Lysotracker were shown. Data were presented as mean ± SD of three independent experiments, *p < 0.05. HEK293 cells transfected with miR-30b-5p or miR-control were incubated with (g) EBSS for 2 h or (h) bafilomycin A1 (100 nM) for 6 h. Forty-eight hours after transfection, the cells were processed for western blotting analysis. The relative amounts of LC3-II and SQSTM1/p62 versus GAPDH were determined by Western blotting and the relative densities were shown beneath. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, and ****p < 0.0001. i HEK293 cells overexpressing miR-30b-5p or miR-control were transfected with mCherry-EGFP-LC3 for 24 h. After being treated as in (g, h), the fluorescence images were captured by confocal microscopy. Scale bar, 10 μm. The quantification data of yellow (autophagosomes) or red (autolysosomes) were shown. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, and ****p < 0.0001.
Lysosomal biogenesis is an indispensable process of the autophagy–lysosome pathway and TFEB also regulates the expression of autophagy-related genes. Thus, we wondered that miR-30b-5p was able to influence autophagy. Decreased level of LC3-II (a marker of autophagosome) and increased level of SQSTM1/p62 (an autophagic substrate) were detected with miR-30b-5p transfection under both normal and EBSS starvation conditions (Fig. 3g). Considering that LC3-II reflects both the biogenesis and degradation of autophagosomes, we employed Bafilomycin A1, a V-ATPase inhibitor which prevented the autophagosome–lysosome fusion, to explore the change of autophagosomes and lysosomes, respectively. With Bafilomycin A1 treatment, the level of LC3-II was still decreased in miR-30b-5p-transfected cells compared with control cells, indicating that miR-30b-5p inhibited the biogenesis of autophagosome (Fig. 3h). To further investigate the effect of miR-30b-5p on autophagic flux, the tandem EGFP-mCherry-LC3 probe was employed to monitor the autophagy–lysosome process. Yellow dots, fusing mCherry and GFP signals, reflect autophagosomes; while red dots, with mCherry signals alone, represent autolysosomes, because GFP signals are quenched in acidic lysosomes. Notably, in the presence of miR-30b-5p, the number of red dots decreased, while yellow dots had no significant change. In addition, under nutrition deprivation, the number of red dots showed a decrease in miR-30b-5p-transfected cells. Similarly, treated with bafilomycin A1, miR-30b-5p repressed the number of red dots as well as yellow dots, indicating that miR-30b-5p could suppress the fusion of autophagosomes and lysosomes (Fig. 3i). Taken together, it indicated that miR-30b-5p inhibited the autophagosomes biogenesis, as well as autophagosome–lysosome fusion.
Knocking out the endogenous miR-30b-5p elevated the lysosomal biogenesis and autophagy
To study the regulatory effect of endogenous miR-30b-5p on the autophagy–lysosome process, the lysosomal biogenesis and autophagy were assessed in miR-30b-5p+/− HEK293 cells (Fig. 4) and HeLa cells (Fig. S4D–F). As expected, the protein levels of lysosomal biogenesis and autophagy genes were upregulated in miR-30b-5p knockout HEK293 cells (Fig. 4a). While the miR-30b-5p+/− cells were treated with EBSS or Bafilomycin A1, the protein levels of above genes increased more dramatically (Fig. 4b, c). Then, the results from fluorescence imaging for LAMP1 (Fig. 4d) and Lysotracker (Fig. 4e), along with the tandem EGFP-mCherry-LC3 probe (Fig. 4f) in miR-30b-5p+/− cells further confirmed the relationship between endogenous miR-30b-5p and autophagy–lysosome process. Thus, knocking out the endogenous miR-30b-5p promoted the lysosomal biogenesis and autophagy.

a–c Protein levels were measured in miR-30b-5p wild-type or knocking out HEK293 cells. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. d Wild-type or miR-30b-5p+/− HEK293 cells were incubated with EBSS for 2 h. Then, the cells were stained using LAMP1 antibody (green). Scale bar, 10 μm. The relative fluorescence densities of LAMP1 were quantified and presented as mean ± SD of three independent experiments, ***p < 0.001 and ****p < 0.0001. e Similar treatment as in (d) was performed and the cells were stained with Lysotracker and visualized. Scale bar, 10 μm. The relative fluorescence densities of Lysotracker were shown. Data were presented as mean ± SD of three independent experiments, **p < 0.01 and ****p < 0.0001. f Wild-type or miR-30b-5p+/− HEK293 cells were transfected with mCherry-EGFP-LC3 for 24 h. After being treated with EBSS for 2 h or bafilomycin A1 (100 nM) for 6 h, the fluorescence images were captured by confocal microscopy. Scale bar, 10 μm. The quantification data of yellow (autophagosomes) or red (autolysosomes) were shown. Data were presented as mean ± SD of three independent experiments, *p < 0.05 and ****p < 0.0001.
The inhibitory effects of miR-30b-5p on the lysosomal biogenesis and autophagy were TFEB dependent
We were then promoted to confirm that the inhibition of miR-30b-5p on lysosomal biogenesis and autophagy was due to reduced TFEB transcriptional activity. MiR-30b-5p, Mut-miR-30b-5p (the potential binding site of the CLEAR element was mutated), and miR-control were transfected into WT or miR-30b-5p+/− HEK293 cells, respectively. As shown in Fig. 5a, compared with miR-control, Mut-miR-30b-5p reversed the inhibition on transcriptional activity of TFEB by luciferase activities of lysosomal gene promoters in both WT and miR-30b-5p+/− cells. Furthermore, the mRNA and protein levels of lysosomal and autophagy-related genes were not affected by Mut-miR-30b-5p (Fig. 5b, c). Subsequently, immunofluorescence staining for LAMP1 was also performed to validate these results (Fig. 5d). On the other hand, knockdown TFEB by siRNA rescued the decrease of luciferase activity (Fig. 5e) and mRNA level (Fig. 5f) with overexpressed miR-30b-5p in HEK293 cells. Notably, overexpression of EGFP-TFEBS211A, a constitutively active version of TFEB could partially rescue the above inhibitory effect of miR-30b-5p (Fig. 5g). These data revealed that TFEB was indispensable for the inhibitory effects on lysosomal biogenesis and autophagy mediated by miR-30b-5p.

Wild-type or mutated miR-30b-5p and miR-control were transfected into wild-type or miR-30b-5p+/− HEK293 cells. Subsequently, a the luciferase activity of lysosomal promoters, b lysosomal mRNA, and c protein levels were detected. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. d Similar transfection in (a–c) were performed. Then, the cells were stained using LAMP1 antibody (green). Scale bar, 10 μm. The relative fluorescence densities of LAMP1 were quantified and presented as mean ± SD of three independent experiments, ****p < 0.0001. miR-30b-5p or miR-control were co-transfected into miR-30b-5p+/− HEK293 cells with or without TFEB siRNA, then e the luciferase activity and f mRNA level of lysosomal genes were measured. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. g qPCR analysis of the mRNA from HEK293 cells co-transfected with miR-30b-5p or miR-control and EGFP-TFEBS211A plasmids. Data were presented as mean ± SD of three independent experiments, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Regulation of the autophagy–lysosome process by miR-30b-5p in mice liver
To further explore the function of miR-30b-5p in vivo, we evaluated the physiological role of miR-30b-5p in lysosomal biogenesis and autophagy of mouse liver induced by 48 h fasting, in which the LC-II protein was increased significantly in mouse liver (p < 0.01), compared with that in the control group (Fig. S5). Therefore, we cloned the primary miR-30b-5p into the PGMAAV-MA2 vector (AAV-mCherry vector), which was proved to specifically express mainly in mice livers [37]. Subsequently, the recombinant adeno-associated viruses were injected into mice via caudal vein. Nine weeks later, a 48 h fasting was performed to induce the lysosomal biogenesis and autophagy in mouse livers (Fig. 6a). The overexpressed mature miR-30b-5p was confirmed in livers from AAV-mCherry-miR-30b-5p injected mice (Fig. 6b), resulting in the decrease of enrichment of TFEB and pol II on the promoters of lysosomal biogenesis genes, including CLCN7, MCOLN1, CTSD, and GLA (Fig. 6c). We observed that overexpression of miR-30b-5p induced the significant decrease of LAMP1 protein (p < 0.01) and LC3-II protein (p < 0.05) and the increase of SQSTM1/p62 protein (p < 0.05), compared with the AAV-mCherry vector, in the starvation group (n = 7 for each group; Fig. 6d). Using immunofluorescence staining with antibodies against LAMP1 and SQSTM1/p62 on liver sections from fasted mice, the significant lower LAMP1 along with the higher SQSTM1/p62 were detected in AAV-mCherry-miR-30b-5p group than those in the vector group (p < 0.001; Fig. 6e). These results in vivo therefore showed that miR-30b-5p inhibits the starvation-induced autophagy–lysosome process.

a Schematic diagram illustrating the experimental design. b Taqman RT-PCR measurement of the level of miR-30b-5p in livers of mice treated with the indicated conditions. Data were presented as mean ± SD, **p < 0.01 and ***p < 0.001. c ChIP analysis of the pol II and TFEB enrichment in the promoters of lysosomal genes in livers of AAV-injected mice fasted for 48 h using anti-pol II and anti-TFEB, with IgG antibody as a negative control. Data were presented as mean ± SD of three independent experiments, *p < 0.05 and **p < 0.01. d LAMP1, SQSTM1/p62, and LC3-II protein levels in livers of AAV-injected mice normally raised or fasted for 48 h were measured by immunoblotting with GAPDH as an internal normalized reference. Densitometric ratios were quantified by ImageJ software. Data were presented as mean ± SD of three independent experiments, *p < 0.05 and **p < 0.01. e Immunofluorescence of liver from the indicated mice using antibodies to LAMP1 and SQSTM1/p62. The relative fluorescence densities were shown on the lower panel. Data were presented as mean ± SD of three independent experiments, *p < 0.05, ***p < 0.001, and ****p < 0.0001.
Discussion
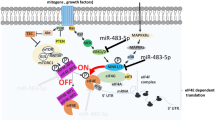
In this study, we introduced miR-30b-5p as a novel regulator on the expression of autophagy–lysosomal genes and the results were summarized in a model (Fig. 7). Through targeting the CLEAR element, the binding motif of TFEB, miR-30b-5p could downregulate almost all the expression of those TFEB-regulated genes involved in lysosomal biogenesis and autophagy.

Schematic diagram showing that nuclear miR-30b-5p inhibits the transcriptional activity of TFEB by binding to its binding sites to suppress the autophagy–lysosomal process.
As a master regulator of lysosomal biogenesis and autophagy, TFEB is associated with many physiological and pathological processes. In diet-induced or genetic mouse models of obesity, TFEB regulates lipid metabolism to prevent weight gain and metabolic syndrome by directly targeting PGC1α and PPARα [3]. Nigral dopamine neurons with impaired TFEB function were identified to show defects in the clearance of α-synuclein and contribute to Parkinson’s disease (PD) [38]. Lysosomal storage disorders (LSD) are a group of hereditary metabolic diseases. Due to the defect of lysosomal acid hydrolases, the corresponding biological macromolecules cannot be degraded normally and stored in lysosomes which lead to the dysfunction of cells, tissues, and organs [39,40,41]. Recently, TFEB is reported to be a fatal therapeutic strategy for LSD. Stimulation of endogenous TFEB activity or overexpression of exogenous TFEB rescues the abnormalities of the lysosomal cysteine levels to be used in the treatment of cystinosis [42, 43]. In addition, in Pompe disease (glycogen storage disease type II), overexpression of TFEB reduces glycogen load and the excessive accumulation of autophagy vacuoles [44]. The activation of lysosomal biogenesis and autophagy requires TFEB nuclear localization, which is regulated by mTOR. When nutrients are scarce, mTOR is inactivated and TFEB transports into the nucleus to activate the transcription of target genes [7, 45,46,47]. In addition to mTOR, some noncoding RNA such as lncRNA could also affect TFEB regulation. As reported, metastasis-associated lung adenocarcinoma transcript 1, the first lncRNA identified to be highly expressed in a variety of cancers, triggers TFEB expression and may be required for the translocation of TFEB, which promotes cell death in human cutaneous squamous cell carcinoma [48]. Herein, we found that miR-30b-5p, a kind of small noncoding RNA, did not interrupt the nucleoplasmic shuttling of TFEB, nor affected its protein level, but inhibited the transcription of autophagy–lysosomal genes in the nucleus, which led to a decrease in lysosomal biogenesis and autophagy in vitro and in vivo.
Autophagy regulated by miRNA is associated with many diseases. It was found to be a double-edged sword for tumorigenesis. Some miRNAs promote tumorigenesis by regulating autophagy. In Nb4 acute myeloid leukemia cells, miR-106a downregulates autophagy and promotes tumorigenesis by directly targeting ULK1, mATG13, and FIP200 in ULK1 complex. Meanwhile, mTORC1, a negative regulator of autophagy, is also the target of miRNA-106a. MiR-106a destroys the binding of mATG13 to ULK1 via regulating their phosphorylation by mTORC1 to interrupt the stability of the ULK1 complex and inhibit autophagy [49]. Most of the autophagy mediated by miRNA is found to inhibit tumorigenesis. MiR-517c suppresses the state of EMT in human glioblastomas by inhibiting autophagy as well as plays an important role in inhibiting tumor metastasis and invasion [50]. In addition to tumors, autophagy mediated by miRNA also plays an important role in other diseases. Many studies have shown that ATG16L is closely related to the pathogenesis of Crohn’s disease. Coincidentally, miR-142-3p downregulates autophagy induced by starvation or L18-MDP through ATG16L in HCT116 cells, which plays an important role in the treatment of Crohn’s disease [51]. In human myocardial fibroblasts, hsa-miR-19a-3p/19b-3p inhibits autophagy by targeting TGFβ-R II to inhibit cardiomyocyte fibrosis induced by TGF-β1 [52]. ULK1 is closely related to PD and recently, it has been found that miR-4487 and miR-595, the regulatory factors of ULK1 can be used as potential biomarkers of PD therapy [53]. In most of these studies, miRNA functions by targeting one or two autophagy-related factors. Unlike this, we found that miR-30b-5p regulated the expression of almost all autophagy-lysosome-related genes to regulate the whole autophagy–lysosome pathway.
In conclusion, our study revealed as follows: (1) It may be a general model that noncoding RNAs transport into the nucleus to regulate transcription factors. In fact, besides miR-30b-5p, we also found other miRNAs that could bind to the CLEAR elements and regulate the transcriptional activity of TFEB in the nucleus. (2) Besides proteins, noncoding RNAs such as miR-30b-5p could regulate the activity of transcription factors through targeting their binding motif suggesting a model of the comprehensive elucidation of the molecular mechanisms underlying noncoding RNA mediated regulation. (3) It expands the molecular mechanism of miRNA regulation, which brings a hint for the exploration of miRNAs in the treatment of TFEB-related diseases.
References
Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–48.
Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–73.
Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647–58.
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33.
Xia Q, Wang H, Hao Z, Fu C, Hu Q, Gao F, et al. TDP-43 loss of function increases TFEB activity and blocks autophagosome-lysosome fusion. EMBO J. 2016;35:121–42.
Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–81.
Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–14.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108.
Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–66.
Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7.
Castanotto D, Lingeman R, Riggs AD, Rossi JJ. CRM1 mediates nuclear-cytoplasmic shuttling of mature microRNAs. Proc Natl Acad Sci USA. 2009;106:21655–9.
Khudayberdiev SA, Zampa F, Rajman M, Schratt G. A comprehensive characterization of the nuclear microRNA repertoire of post-mitotic neurons. Front Mol Neurosci. 2013;6:43.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350.
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415.
Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–70.
Lund E, Güttinger S, Calado A, Dahlberg JE, Kutay U. Nuclear export of microRNA precursors. Science. 2004;303:95–8.
Kim VN. MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol. 2004;14:156–9.
Kim DH, Saetrom P, Snove O Jr, Rossi JJ. MicroRNA-directed transcriptional gene silencing in mammalian cells. Proc Natl Acad Sci USA. 2008;105:16230–5.
Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci. 2003;100:9779–84.
Filipowicz W, Jaskiewicz L, Kolb FA, Pillai RS. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr Opin Struct Biol. 2005;15:331–41.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33.
Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci USA. 2008;105:1608–13.
Meister G, Landthaler M, Patkaniowska A, Dorsett Y, Teng G, Tuschl T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol Cell. 2004;15:185–97.
Hwang H-W, Wentzel EA, Mendell JT. A hexanucleotide element directs microRNA nuclear import. Science. 2007;315:97–100.
Foldes-Papp Z, Konig K, Studier H, Buckle R, Breunig HG, Uchugonova A. et al. Trafficking of mature miRNA-122 into the nucleus of live liver cells. Curr Pharm Biotechnol. 2009;10:569–78.
Politz JCR, Hogan EM, Pederson T. MicroRNAs with a nucleolar location. RNA. 2009;15:1705–15.
Liao J-Y, Ma L-M, Guo Y-H, Zhang Y-C, Zhou H, Shao P, et al. Deep sequencing of human nuclear and cytoplasmic small RNAs reveals an unexpectedly complex subcellular distribution of miRNAs and tRNA 3′ trailers. PloS ONE. 2010;5:e10563.
Jeffries CD, Fried HM, Perkins DO. Nuclear and cytoplasmic localization of neural stem cell microRNAs. RNA. 2011;17:675–86.
Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circulation Res. 2012;110:1596–603.
Zardo G, Ciolfi A, Vian L, Starnes LM, Billi M, Racanicchi S, et al. Polycombs and microRNA-223 regulate human granulopoiesis by transcriptional control of target gene expression. Blood. 2012;119:4034–46.
Weinmann L, Hock J, Ivacevic T, Ohrt T, Mutze J, Schwille P, et al. Importin 8 is a gene silencing factor that targets argonaute proteins to distinct mRNAs. Cell. 2009;136:496–507.
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–9.
Washburn MP, Wolters D, Yates III JR. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242.
Pathan M, Keerthikumar S, Chisanga D, Alessandro R, Ang CS, Askenase P, et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J Extracell Vesicles. 2017;6:1321455.
Wei Y, Li L, Wang D, Zhang CY, Zen K. Importin 8 regulates the transport of mature microRNAs into the cell nucleus. J Biol Chem. 2014;289:10270–5.
Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18:43–52.
Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci USA. 2013;110:E1817–26.
Platt FM, Boland B, van der Spoel AC. The cell biology of disease: lysosomal storage disorders: the cellular impact of lysosomal dysfunction. J Cell Biol. 2012;199:723–34.
Futerman AH, van Meer G. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 2004;5:554–65.
Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005;128:413–31.
Zhang JZ, He J, Johnson JL, Napolitano G, Ramadass M, Rahman F, et al. Cross-regulation of defective endolysosome trafficking and enhanced autophagy through TFEB in UNC13D deficiency. Autophagy. 2019;15:1738–56.
Rega LR, Polishchuk E, Montefusco S, Napolitano G, Tozzi G, Zhang J, et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016;89:862–73.
Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med. 2013;5:691–706.
Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42.
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, et al. A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095–108.
Peña‐Llopis S, Vega‐Rubin‐de‐Celis S, Schwartz JC, Wolff NC, Tran TAT, Zou L, et al. Regulation of TFEB and V‐ATPases by mTORC1. EMBO J. 2011;30:3242–58.
Tan M, Jiang B, Wang H, Ouyang W, Chen X, Wang T, et al. Dihydromyricetin induced lncRNA MALAT1-TFEB-dependent autophagic cell death in cutaneous squamous cell carcinoma. J Cancer. 2019;10:4245–55.
Seto AG. The road toward microRNA therapeutics. Int J Biochem Cell Biol. 2010;42:1298–305.
Lu Y, Xiao L, Liu Y, Wang H, Li H, Zhou Q, et al. MIR517C inhibits autophagy and the epithelial-to-mesenchymal (-like) transition phenotype in human glioblastoma through KPNA2-dependent disruption of TP53 nuclear translocation. Autophagy. 2015;11:2213–32.
Zhai Z, Wu F, Dong F, Chuang AY, Messer JS, Boone DL, et al. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy. 2014;10:468–79.
Zou M, Wang F, Gao R, Wu J, Ou Y, Chen X, et al. Autophagy inhibition of hsa-miR-19a-3p/19b-3p by targeting TGF-beta R II during TGF-beta1-induced fibrogenesis in human cardiac fibroblasts. Sci Rep. 2016;6:24747.
Chen Y, Wang S, Zhang L, Xie T, Song S, Huang J, et al. Identification of ULK1 as a novel biomarker involved in miR-4487 and miR-595 regulation in neuroblastoma SH-SY5Y cell autophagy. Sci Rep. 2015;5:11035.
Acknowledgements
We thank Yue Yin and Ping Wu (National Center for Protein Science—Shanghai) for mass spectrometry, Yizheng Wang (The Brain Science Center, Beijing Institute of Basic Medical Sciences) and Kai Fu (Xiangya Hospital Central South University) for helpful comments. This work was supported by the National Natural Science Foundation of China (81701261 and 81771890), the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program,” China (2018ZX09101001-003-007, 2018ZX09201017-004, 2019ZX09732002-013, and 2018ZX09711002-010-001), the Open Research Fund of State Key Laboratory of Transient Optics and Photonics (SKLST201806), the Fundamental and Open Research Funds from the State Key Laboratory of Drug Research (SIMM1903ZZ-05 and SIMM2004KF-02), Shanghai Committee of Science and Technology of China under Grant No.18DZ2290200.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by H.-U. Simon
Rights and permissions
About this article

Cite this article
Guo, H., Pu, M., Tai, Y. et al. Nuclear miR-30b-5p suppresses TFEB-mediated lysosomal biogenesis and autophagy. Cell Death Differ 28, 320–336 (2021). https://doi.org/10.1038/s41418-020-0602-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-0602-4
This article is cited by
-
From the regulatory mechanism of TFEB to its therapeutic implications
Cell Death Discovery (2024)
-
PRKAA2, MTOR, and TFEB in the regulation of lysosomal damage response and autophagy
Journal of Molecular Medicine (2024)
-
Ferroptosis contributes to ethanol-induced hepatic cell death via labile iron accumulation and GPx4 inactivation
Cell Death Discovery (2023)
-
CCL20/CCR6 axis mediates macrophages to promote proliferation and migration of ESCs by blocking autophagic flux in endometriosis
Stem Cell Research & Therapy (2022)
-
Preparation of a miR-155-activating nucleic acid nanoflower to study the molecular mechanism of miR-155 in inflammation
Molecular Medicine (2022)
