
Abstract
Despite strong evidence of the neurodevelopmental origins of psychosis, current pharmacological treatment is not usually initiated until after a clinical diagnosis is made, and is focussed on antagonising striatal dopamine receptors. These drugs are only partially effective, have serious side effects, fail to alleviate the negative and cognitive symptoms of the disorder, and are not useful as a preventive treatment. In recent years, attention has turned to upstream brain regions that regulate striatal dopamine function, such as the hippocampus. This review draws together these recent data to discuss why the hippocampus may be especially vulnerable in the pathophysiology of psychosis. First, we describe the neurodevelopmental trajectory of the hippocampus and its susceptibility to dysfunction, exploring this region’s proneness to structural and functional imbalances, metabolic pressures, and oxidative stress. We then examine mechanisms of hippocampal dysfunction in psychosis and in individuals at high-risk for psychosis and discuss how and when hippocampal abnormalities may be targeted in these groups. We conclude with future directions for prospective studies to unlock the discovery of novel therapeutic strategies targeting hippocampal circuit imbalances to prevent or delay the onset of psychosis.
Similar content being viewed by others

Introduction
Hippocampal dysfunction is a robust feature in the pathophysiology of psychosis [1, 2]. In patients with a psychotic disorder such as schizophrenia, hippocampal volume is reduced [3,4,5] and function is abnormal: neuroimaging measures of resting activity including positron emission tomography (PET) [6], resting-state functional MRI (rs-fMRI) [7,8,9,10], and resting cerebral blood volume (CBV) [11,12,13], converge to suggest that the hippocampus is hyperactive in psychosis. Moreover, hippocampal alterations are already present before illness onset in people at clinical high-risk (CHR) of psychosis, including increased hippocampal resting cerebral blood flow (rCBF) [14, 15] and disrupted hippocampal-basal ganglia and hippocampal-prefrontal connectivity [16,17,18]. These human findings are broadly consistent with preclinical data demonstrating that hippocampal hyperactivity drives the development of striatal dopamine dysfunction [19] and associated psychosis-relevant behaviours [20]. If hippocampal dysfunction plays such a key role in the onset of psychosis, correcting this dysfunction may represent a promising strategy for the development of new treatments and preventive interventions. The aim of the present review is to explore recent developments in the study of hippocampal circuit dysfunction in psychosis, understand what they mean in the context of normal hippocampal neurodevelopment or under exposure to known environmental risk factors for psychosis, and how these findings can inform the development of novel treatments.
Hippocampal circuitry
The hippocampal formation comprises several distinct, densely packed, highly connected areas (Fig. 1). The circuitry of the hippocampus is inextricably linked to its functions—pattern completion and separation—which requires distinct subfield contributions [2]. Hippocampal cells also recruit distal cortical regions in hippocampal-cortical circuits, processing information involving memory, spatial navigation, emotion, and stress [21].

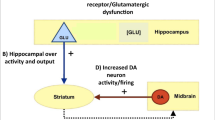
A The subdivisions of the hippocampus along its long axis—the dentate gyrus (DG) and four sections of the cornu ammonis (CA1 red; CA2, blue; CA3, green; CA4/DG, yellow; subiculum, cyan)—are demarcated early in prenatal neurodevelopment through specific genetic molecular markers. Segmentation derived from cytoarchitectonic anatomical probability map [244]. B Cross-section of left hippocampi, highlighting primary internal circuitry of the hippocampal formation. Solid lines reflect the ‘trisynaptic circuit’, dash lines reflect supporting entorhinal circuitry. Each subdivision of the hippocampus is linked to the neighbouring entorhinal cortex through the ‘trisynaptic circuit’, an excitatory projection that links hippocampal subregions via the perforant pathway to granule cells in the DG. These granule cells are linked to pyramidal cells in region CA3 via mossy fibres, which in turn project to pyramidal cells in CA1 via Schaffer collaterals, before exiting the hippocampus via the subiculum [245]. In addition to the trisynaptic circuit, there are supporting connections between the entorhinal cortex and CA1 and CA3, and subiculum, as well as projections between CA1 and CA3. The hippocampus receives input primarily from the entorhinal cortex but is also extensively connected with proximate regions, including the anterior cingulate (ACC), medial prefrontal cortex (mPFC), and amygdala [246]. The hippocampus sends direct outputs to the nucleus accumbens, hypothalamus, and thalamus, and indirect outputs to the striatum via the nucleus accumbens and ventral tegmental area [247].
Together, these hippocampal-cortical circuits form a systematic mental representation of accumulated knowledge and experiences, which can be conceptualised as a ‘cognitive map’ [22]. When a neuronal assembly relating to a particular cognitive map is cued, this triggers a sequence of oscillatory signalling which preferentially follows an encoded representation, thereby supporting the prediction of event consequences and prompting behaviour. These patterns are believed to be consolidated and integrated during rest, when hippocampal sharp-wave ripple oscillations reactivate the sequence of neural assemblies in the absence of the original stimuli—a process known as neural replay [23]. The circuitry of hippocampal subregions is crucial for processes used to distinguish between (pattern separation) and link (pattern completion) cognitive maps [2]. Cognitive maps are a useful framework for understanding psychotic symptoms. If the dynamics cueing neurons and attracting signal through a cognitive map become imbalanced, pattern separation and completion processes are compromised [2]. The resulting “shallow cognitive maps”—over-general pattern completion and reduced pattern separation—could give rise to the cognitive impairments, thought disorder, and aberrant salience in psychosis through the reinforcement of loosely associated circuits during neurodevelopment (see [24] for a review).
Neurodevelopment of hippocampus
The internal circuitry of the hippocampus develops in a complex and lengthy process. Neural cell proliferation, migration, differentiation, and synaptogenesis all begin prenatally and extend through the first years of postnatal life, with synaptogenesis continuing right through to adolescence [25]. The developmental timeframe of these processes reflects critical or sensitive periods of brain maturation—developmental windows where brain circuitry is shaped by postnatal sensory stimulation—that are essential for healthy neurodevelopment [26].
During early life, sensory information in the environment influences the organisation of the brain through effects on synaptogenesis, pruning, and myelination. Development is rapid through the first year of postnatal life; most structures are already established, and the pace of subsequent synaptogenesis slows [27]. A critical period for synaptogenesis is estimated to close in early childhood [28, 29], as the mental representations of early life experiences become encoded and distributed in neural circuits. This leads to another critical period in human development in adolescence, with synaptic pruning and myelination processes accelerating to solidify these neural patterns, before tapering off in early adulthood [25].
Specific maturational processes are difficult to characterise in vivo [30], although these are indirectly reflected through MRI-derived grey matter volume decreases and white matter volume increases between childhood and early adulthood [31]. Unlike much of the cortex, several subcortical structures including the hippocampus display a contrasting lack of hippocampal grey matter decline over adolescence and early adulthood [32,33,34], suggesting that the processes involved in synaptic pruning over neurodevelopment may be occurring over a protracted period and potentially leaving the hippocampus more vulnerable to aberrant adolescence development. Unlike other structures, there is also ongoing postnatal neurogenesis within the dentate gyrus (DG), likely contributing to protracted hippocampal development. The rate of neurogenesis appears to taper off in early adulthood [35], though there is other evidence that it persists into adulthood [36]. Overall, the role of ongoing neurogenesis is undoubtedly important for hippocampal development and the emergence of psychotic phenotypes (see [37,38,39] for reviews).
Developmental changes within the hippocampus are also not uniform. The posterior/dorsal hippocampus, most often associated with memory and spatial learning, gains in volume as a ratio to its size at age four, over 19 years of subsequent development [40]. Conversely, the anterior/ventral hippocampus, with projections to the prefrontal cortex and amygdala and associated with socio-emotional and stress response processes, decreases in relative volume over the same period. The differences in anterior/posterior specialisation and development are relevant for psychosis, as socio-emotional and stress response abnormalities in psychosis [41] are particularly accentuated in adolescence [42].
This vulnerability of the hippocampus may particularly impact normal development by its dense connectivity. The hippocampus is part of the so-called “rich club” of hub brain regions (including the superior frontal cortex, precentral gyri, thalamus, and putamen), with particularly high network centrality and vulnerability to dysfunction [43, 44]. There is diverse connectivity between hippocampal subfields and known resting-state networks in mature brains [45]; these networks are already partially established by age four and continue to consolidate over childhood [46]. Together, the protracted neurodevelopment and high connectivity of the hippocampus may leave immature neurons vulnerable to damage during a period where risk factors for psychosis may be particularly influential.
Hippocampal alterations in psychosis
Hippocampal volume deficits are evident at the first episode of a psychotic disorder (FEP), when the clinical diagnosis is first made, and are most marked in anterior cornu ammonis (CA) regions [47]. These reductions also appear to be greater in patients with relatively severe positive symptoms [48], as well as in those in whom there was a long delay before treatment was initiated [49], though hippocampal volume reductions do not appear to be progressive from FEP to chronic illness [47, 50, 51].
Abnormal metabolic activity within the hippocampus may precede and drive hippocampal volume loss [1]. Elevated CBV left anterior CA1 was found in CHR individuals who subsequently transitioned to psychosis [52]. After a 24-month follow-up, higher CBV was also evident in the subiculum in the CHR subgroup that transitioned to psychosis, overlapping with longitudinal MRI volume loss. Parallel animal model work involving the chronic administration of ketamine produced a similar pattern of hippocampal CBV increase and volume reduction, suggesting that these MRI-based changes may be driven by local glutamatergic dysfunction [52]. In humans, longitudinal neuroimaging data suggest that increased hippocampal rCBF normalises in CHR individuals who subsequently remit from the CHR state [15], whereas in CHR subjects with adverse clinical outcomes, elevated hippocampal rCBF has been associated with lower striatal dopamine synthesis capacity [53]. Small hippocampal volume reductions been reported in people at CHR for psychosis [54, 55], which may be more prominent in CA1 [56]. There is also evidence that these reductions at baseline are more pronounced in the subgroup of CHR subjects go on to develop psychosis, and that they continue to decrease longitudinally as these individuals transition to psychosis [57, 58]. However, a recent meta-analysis suggests these findings have not been consistently replicated [59]. In the most recent structural MRI study to date in CHR individuals, baseline grey matter volume in the hippocampus or other cortical regions was not predictive of remission or transition to psychosis [60]. Longitudinal studies in an early psychosis or CHR population are therefore necessary to expand on the links between increased hippocampal metabolism, glutamate dysfunction, and subsequent volume loss.
Alterations in hippocampal structure and function have also been associated with psychotic-like experiences in the general population [61,62,63] (although see [64]). Moreover, hippocampal dysfunction is shared across other neuropsychiatric disorders [65, 66], which is perhaps unsurprising given the large overlap in genetic risk and symptoms [67]. Due in part to this overlap, hippocampal circuit dysfunction specific to psychosis is often difficult to discriminate [6, 68]. Notwithstanding, there is some evidence that hippocampal alterations are most pronounced in schizophrenia, decreasing in severity along the psychiatric spectrum [4, 69, 70]. The nature and specificity of hippocampal dysfunction in psychosis is thus still being parsed, and while beyond the scope of the present review, this overlap has broad transdiagnostic implications for the pathophysiology and putative treatment of other disorders.
Causes of hippocampal alterations in psychosis
Excitation–inhibition imbalance
The balanced interplay between excitatory and inhibitory (E/I) neural activity is crucial for regulating brain excitability and the synchronisation of signalling between disparate cortical regions involved in cognitive functioning [71]. Psychosis may involve an imbalance between E/I signalling (Fig. 2), proposed to drive hippocampal hyperactivity and “shallow cognitive maps”, resulting in aberrant coupling between loosely associated neural assemblies [24]. One example of this is impaired hippocampal neural replay in schizophrenia, where abnormal hippocampal oscillations impair learning [72].

A Stressors lead to cascade of down-scale imbalances, including increased hippocampal metabolism and network (blue spheres) dysfunction (red spheres). Hub regions, including the hippocampus, are most likely to be impacted [43]. B Excitatory/inhibitory imbalance and hyperactivity in hippocampal subregions (enlarged arrows). C Oxidative stress damages metabolically demanding parvalbumin-positive interneurons (PVI), resulting in N-methyl-D-aspartate receptor (NMDAR) hypofunction and (1) altered pyramidal signalling; (2) reduced pyramidal input to interneurons; (3) reduced interneuron inhibition of pyramidal cells, and a cascade of up-scale imbalances These processes are accentuated in those at highest genetic risk. Diagram adapted in part, with permission [248].
In most cortical regions, ~80% of neurons are excitatory glutamatergic pyramidal cells, whereas in the hippocampus these cells represent ~90% of all neurons [73]. Because only ≈10% of hippocampal neurons are inhibitory GABAergic interneurons, this region may be more susceptible to E/I imbalance. For example, the hippocampus is a source of high-frequency gamma oscillations (~30–100 Hz range) that are implicated in memory encoding, storage, and retrieval [74, 75], and known to be disrupted in psychosis [76]. Gamma oscillations are regulated by fast-spiking GABAergic parvalbumin-positive interneurons (PVI) [77, 78]. In transgenic mice, optogenetic stimulation of PVI generates gamma oscillations and improves cognitive performance [79], while disruptions to N-methyl-D-aspartate receptor (NMDAR) input on PVI impair gamma oscillations [80]. PVI play a role in hippocampal neurogenesis [81], and constitute around 20% of hippocampal inhibitory neurons, somewhat more concentrated in CA1 and CA3 than in the DG [82]. This compares to around 40% of cortical interneurons [83].
In psychosis, reductions in hippocampal PVI density have been found in post-mortem studies [84, 85] and hippocampal PVI cell loss is evident in several animal models of psychosis [52, 78]. There is also significant post-mortem and in vivo evidence of synaptic deficits in psychotic disorders [86,87,88,89], which may be a result of PVI impairments and may drive hippocampal hyperactivity. PVI synaptogenesis is regulated by Neuregulin 1 and Erbb4 receptors, which are also implicated in neurodevelopmental and psychotic disorders [90]. Experiments in a genetic animal model involving Erbb4 deletion from PVI show lower synaptic density as indexed by synaptic vesicle glycoprotein 2A (SV2A) sampling, together with increased rCBF in the ventral hippocampus [91]. These results resemble neuroimaging findings associated with psychosis-risk [14, 86], linking PVI and SV2A deficits with hippocampal hyperactivity.
PVI dysfunction has also been linked to NMDAR hypofunction in psychosis [92, 93]. NMDAR antagonists such as phencyclidine decrease PVI count [94], thereby leading to lower PVI inhibition of pyramidal neurons. Moreover, activation of the NR2A subunit of NDMARs contributes to the maturation of PVI [95] and therefore NMDAR hypofunction may be most detrimental to PVI early in development [96].
Single-photon emission computed tomography (SPECT) and PET have been used to quantify NMDAR density in vivo in patients with psychosis. While the specificity of these radioligands for the receptor remains controversial, one SPECT study reported reduced NMDAR binding in the left hippocampus relative to the whole cortex in unmedicated patients with psychosis, but this deficit was ameliorated in antipsychotic treated patients [97]. A recent PET study replicated this finding in early psychosis; FEP patients showed lower hippocampal NMDAR binding relative to the whole cortex compared to healthy volunteers, although no differences in hippocampal NMDAR availability were observed [98]. Overall, there is some evidence that hippocampal abnormalities in psychosis may be related to NMDAR hypofunction.
Although currently PVI activity cannot be measured in vivo in humans, levels of glutamate and GABA partially reflect E/I processes [99] and can be measured with proton magnetic resonance spectroscopy (1H-MRS). Meta-analysis of 1H-MRS studies in patients with schizophrenia, FEP, and CHR individuals showed elevations in levels of glutamate + glutamine (Glx) in the basal ganglia of schizophrenia and FEP patients, regardless of whether they were medicated [100]. However, hippocampal Glx was only elevated in unmedicated patients, with no elevations in basal ganglia Glx in CHR unless combined with patient groups. In CHR individuals, one study found higher levels of hippocampal glutamate levels in the CHR subgroup that subsequently transitioned to psychosis[101], although another study did not [102]. One multimodal neuroimaging study found that hippocampal glutamate levels and striatal dopamine synthesis capacity were negatively correlated in CHR individuals who later developed psychosis [103]. This is an intriguing finding given the proposed causal link between hippocampal hyperactivity and elevated striatal dopamine [104], though important to note that Glx signal acquired through 1H-MRS captures more than hippocampal-striatal projections and includes inputs to the anterior hippocampus. If inhibitory signalling is perturbed, one would predict a Glx decrease.
In terms of GABA levels, relatively few studies have investigated these in the hippocampus in patients with psychosis, partly due to the technical difficulties in reliably positioning a GABA-edited 1H-MRS voxel in this region [105]. Nevertheless, 1H-MRS studies at both 3T and 7T have not found significant alterations in absolute concentrations of GABA in patients with psychosis compared to healthy controls [106, 107]. In a multimodal imaging study in people at CHR, GABA levels in the prefrontal cortex were correlated with hippocampal rCBF, a correlation driven by those individuals who subsequently transitioned to psychosis [108]. Some of the technical difficulties in measuring hippocampal GABA in vivo with 1H-MRS may be overcome by using PET imaging. A recent study reported lower GABAA α5 receptor availability (highly expressed in hippocampus) only in the subgroup of patients with schizophrenia that were antipsychotic-free [109], suggesting that PET measures of GABA dysfunction can be influenced by the effects of antipsychotic medications [110].
Stress sensitivity
Environmental stressors (including poverty, ethnicity/immigration, and childhood adversity) are among the strongest risk factors for psychosis [111]. The hippocampus appears to play a role in mediating their effects; preclinical studies have found increased hippocampal activity and PVI loss following environmental stressors (including maternal separation and random foot shocks) [112, 113], particularly before adulthood [114]. In children, environmental stressors such as maltreatment, poverty and neglect have deleterious effects on anterior hippocampal volume [115]. While hippocampal volume loss may not occur after a single traumatic event [116], the extended period of vulnerability allows for an accumulating cascade of maladaptive environmental influences on neurodevelopment [117]. In CHR individuals, a history of childhood trauma has been associated with reductions in hippocampal volume in early adulthood [14]. The impact of early life risk factors may vary according to sex: females may be more resilient to certain types of early maltreatment [118], but more susceptible to abuse later in life [119]. These differences in vulnerability to stressors may contribute to the later age of onset of psychosis in females [120], and raises the possibility of sex-dependent differences on the putative window for effective preventive intervention.
A possible mechanism by which early life stressors may precipitate metabolic pressures on the hippocampus is through its involvement in stress and emotion processing [121]. The hypothalamic pituitary adrenal (HPA)-axis regulates the response to stress through cortisol binding to glucocorticoid and mineralocorticoid receptors, which are present at a high density in the anterior hippocampus [122]. While this stress response is critical for normal learning [123], repeated activity burdens the system [124]. Hence, early life stressors may alter hippocampal neuronal structure [125] and potentially sensitise the HPA-axis. Environmental stressors during key developmental periods may reinforce a stress-sensitised phenotype, with aberrant synaptic pruning in adolescence leading to altered E/I balance and dysfunctional cognitive maps, restricting the deployment of alternative cognitive strategies. For instance, excess glucocorticoid activity reduces the complexity of CA3 hippocampal pyramidal cells [126] and inhibits neurogenesis [127]. The effects of these stressors on the HPA-axis may be especially pronounced in adolescence due to neurohormonal processes involved in puberty [128]. Sustained activation of glucocorticoids during vulnerable developmental periods may also impair energy metabolism in the hippocampus and lead to oxidative stress [129].
Oxidative stress
Oxidative stress results from an impairment in the redox cycle of oxygen metabolism. Redox dysregulation occurs when there is an imbalance between reactive nitrogen and oxygen species (ROS), and the capacity of the cells to detoxify the ROS with antioxidant defences, predominantly through the reduction of glutathione (GSH) [130]. ROS are deleterious by-products of aerobic metabolism, resulting from the mitochondrial electron transfer chain that converts glucose and oxygen into adenosine triphosphate [131]. High-energy demands can perturb the redox balance and result in ROS-mediated damage of surrounding molecules [130]. The hippocampal region may be particularly vulnerable to oxidative stress because it is a metabolically active region, with high degree of connectivity to distal regions and relatively fine-tuned excitatory/inhibitory cell balance.
PVIs display a high rate of firing relative to other neurons, and therefore have relatively high metabolic demands [132]. The generation of gamma oscillations, mediated by hippocampal PVIs, can require more than double the baseline oxygen consumption, making PVIs particularly vulnerable to oxidative stress [133]. Glutamate cysteine ligase (GCL) is the rate-limiting enzyme for synthesising GSH, and a GCL modulatory subunit (M) knockout model shows depleted GSH [134] and loss of PVI [135]. Indeed, oxidative stress has been shown to disrupt gamma oscillations from ventral CA3 in a mouse model of relevance to psychosis [134]. Oxidative stress is a vicious cycle whereby ROS, if not neutralised, may impair mitochondrial function, generating even more ROS.
Converging evidence links psychosis with increased oxidative stress [136,137,138]. Schizophrenia patients have increased DNA oxidation, lipid oxidation/peroxidation, and free radicals [138], as well as decreased antioxidants [137]. A meta-analysis of oxidative stress in schizophrenia found differences in 10 peripheral oxidative stress markers—from red blood cells, plasma, and serum—depending on FEP or chronic schizophrenia clinical status [137]. Though most studies focus on peripheral markers of oxidative stress, oxidative DNA damage in hippocampal tissue was found to be 10× higher in patients with chronic schizophrenia than in non-psychiatric subjects by post-mortem examination [139].
Oxidative stress is difficult to spatially localise in vivo, so instead researchers look for signs of redox dysregulation, with GSH levels as a widely used marker of an impaired antioxidant system. Animal model evidence for an association of reduced GSH levels with hippocampal abnormalities [130] implicates oxidative stress in the pathophysiology of psychosis. For instance, GSH-deficient transgenic mice show a time-dependent impairment of PVI functioning in the ventral hippocampus, linking GSH to PVI damage/loss [134]. In this same mouse model, reduced fractional anisotropy (FA) and conduction velocity of slow-conducting fibres of the fornix-fimbria were found during adolescence [140], suggesting connectivity impairments of the hippocampus to other subcortical areas. Interestingly, reduced FA in the fornix is also observed in patients at the early stage of psychosis, underscoring the potential role of oxidative stress in these alterations [141].
In humans, recent meta-analyses of 1H-MRS studies found a persistent pattern of GSH deficits in psychosis [142, 143], with most studies focusing on the mPFC/ACC. Hippocampal GSH has not been studied extensively in psychosis, owing to similar technical limitations to the use of 1H-MRS for measuring hippocampal GABA levels, although one study found higher GSH in the medial temporal lobe of FEP patients versus healthy controls [144]. In this context, peripherally measured redox blood markers and GCLC genotyping may be more practical markers of redox status and dysfunction than brain GSH measures. A genetic polymorphism in the catalytic subunit of the GCL was found to be associated with schizophrenia and led to decreased brain [145] and blood GSH levels, as well as GCL activity [146]. One study found that mPFC GSH became uncoupled from peripheral GSH-related enzymes in early psychosis patients, but remained positively correlated in healthy participants [145], whilst another linked higher peripheral glutathione peroxidase with reduced hippocampal volume in early psychosis but not in healthy participants [147]. Moreover, this association was also found in patients that had experienced childhood trauma, linking adverse life events with increased oxidative stress and hippocampal alterations [148]. Though this evidence supports a role for redox dysregulation in psychosis, further studies are warranted to determine hippocampal GSH levels in the disorder, and the links between peripheral redox markers and hippocampal activity.
The effects of oxidative stress in relation to psychosis may be especially important in preadolescence, when they may affect perineuronal nets (PNN)—an extracellular matrix of molecules that enclose neurons and provide them with structural support [133]. Their presence around PVI is essential in closing a period of plasticity in preadolescence in which synaptic pruning and apoptosis occur [26, 149, 150]. PNN also protect PVI from oxidative stress, although they themselves are still vulnerable to oxidative damage [151]. Post-mortem analyses have found a 70% reduction of PNN in patients with psychosis in the PFC [152], however, evidence of PNN deficits in the human hippocampus is extremely limited [153, 154]. Clearly, further characterisation of PNN in the human hippocampus is required. However, a study of the methylazoxymethanol acetate (MAM) preclinical model for psychosis found a reduction in PNN, which was linked to increased firing of hippocampal pyramidal cells [155]. Furthermore, chronic stress in adolescence—but not adulthood—reduces PVI and PNN [114]. Collectively, these observations suggest that oxidative stress may damage hippocampal PVI and PNN, extending a neurodevelopmental period when there is scope for aberrant plasticity and synaptic pruning [149].
Targeting hippocampal dysfunction in psychosis
This section will consider how the aforementioned hippocampal circuit abnormalities may be targeted during neurodevelopment to reduce the cascade to a psychotic disorder (Fig. 3).

E/I imbalance
One method for correcting hippocampal hyperactivity may be through targeting glutamate. For example, in a mouse model of psychosis, an mGluR 2/3 agonist prevented ketamine-induced hyper-metabolism of the hippocampus [52]. Furthermore, peri-pubertally targeting this receptor with the mGluR 2/3 agonist pomaglumetad methionil normalised VTA dopamine and ventral hippocampal pyramidal neuron activity in the MAM model of psychosis [156]. However, to date, results from clinical trials of compounds that target glutamate function in patients with psychosis have been disappointing. Pomaglumetad methionil was inferior to aripiprazole in relieving positive symptoms [157], and no better than placebo as an adjunctive treatment for negative symptoms [158]. Bitopertin, a glycine transport inhibitor, was ineffective at improving symptoms in schizophrenia in phase-III trials [159]. Still, these studies involved patients with a chronic psychotic disorder, who had been treated with antipsychotic medications. It is possible that glutamatergic medications might be more useful if given in the early phase of psychosis, in FEP or CHR individuals, before receiving prolonged antipsychotic treatment.
Another strategy to redress E/I imbalance is to target the GABAergic system to restore inhibition within the hippocampus. For instance, in the MAM model for psychosis, the experimental transplantation of GABAergic precursor cells into the ventral hippocampus normalised hippocampal and striatal dopaminergic function [160]. Similarly, in the same model, peri-pubertal administration of diazepam prevented PVI loss and hyperdopaminergia at adulthood [161]. To date, experimental interventions with GABA-enhancing medications have mainly involved benzodiazepines, which are broad GABAA agonists [162] and there is no evidence for antipsychotic efficacy of additional benzodiazepine medication in schizophrenia [163]. Levetiracetam is an antiepileptic drug that binds to SV2A [164], consequently enhancing GABAergic signalling [165] and regulating E/I balance [166]. Several clinical trials are currently ongoing with levetiracetam in schizophrenia: NCT04317807, NCT03129360, NCT03034356, and NCT02647437. As with novel glutamatergic compounds, these trials are being conducted in patients with well-established psychosis, rather than patients in its early phase, prior to antipsychotic use. This may reduce the chances of detecting clinical effects, as antipsychotic medications can confound or block the effects of GABAergic intervention [109, 167]. Of greater concern are case reports suggesting that levetiracetam can induce psychosis in patients with epilepsy, particularly in patients with a history of psychosis, so may be harmful in some patients [168, 169]. The recent advent of more specific GABAA α5 receptor subunit modulators are of great interest, due to their relative specificity for the hippocampus and their effectiveness in animal models [170].
Sodium Valproate is another anticonvulsant drug often used to treat mania [171] which has also been used effectively to augment antipsychotic treatment [172]. Valproate’s acute benefits for mania and epileptic seizures are likely partially due to an increase of endogenous GABA levels, but valproate also inhibits histone deacetylation and may have enduring effects on gene expression [173]. Interestingly, valproate may be able to reopen certain critical periods for learning [114, 174]. This raises the possibility that valproate could be used to retrain maladaptive cognitive maps and perhaps extend the neurodevelopmental window prior to psychosis onset to allow PNN to properly develop. However, extending these critical periods without sufficient support may leave patients vulnerable to the aforementioned metabolic pressures [149] and stressors [114].
Oxidative stress
N-acetyl cysteine (NAC) is an antioxidant and a precursor to GSH. Its administration has been shown to instigate a 23% increase in mPFC GSH, improvement of cognitive symptoms and increase in white matter integrity in the fornix [175] of early psychosis patients [176]. Administration of NAC to juvenile and adolescent rats with neonatal hippocampal lesion—a well-established model for psychosis—rescued the development of behavioural phenotypes associated with psychosis in adulthood [177]. NAC also rescues the PVI/PNN maturation impairments found in GSH-deficient GCLM knockout mice [135, 178, 179]. Collectively, this preclinical evidence indicates that NAC has potential as a treatment for psychosis.
A meta-analysis of 7 studies found that in patients with psychosis, NAC improved both positive and negative symptoms after 24 weeks of treatment [180]. However, in theory, NAC could be even more useful if administered prior to the onset of psychosis. If oxidative stress damages PNN/PVI in adolescence, before they are mature, it is crucial that strategies tailored to oxidative stress are developed to target these developmental periods. In preclinical studies, NAC has been administered to juvenile and peri-pubertal animals, during PVI/PNN development. In humans, the earliest that NAC has been given is after the first episode of psychosis [180], though a clinical trial in CHR subjects is currently ongoing [181].
In addition to NAC, other food supplements may reduce the effects of oxidative stress. Sulforaphane is an extract from broccoli that increases peripheral and hippocampal GSH in healthy volunteers [182], and is being evaluated in CHR subjects [183]. Nicotinamide mononucleotide is important in the biosynthesis of nicotinamide adenine dinucleotide, which itself plays an important role in energy metabolism [184] and has reduced levels in patients experiencing FEP and their non-psychotic siblings [185], warranting further investigation. Resveratrol, another antioxidant, is promising for its neuroprotective effects in animal model studies but so far has had little success in human trials [186]. Dietary supplement of ω-3 polyunsaturated fatty acids, antioxidants that reduce inflammation, may also be beneficial [187, 188]. However, despite initial success [189], the most extensive clinical trial to date failed to replicate ω-3 reducing CHR transition to psychosis [190]. Future studies investigating antioxidant supplementation may consider including peripheral markers of redox status, to identify whether these interventions may be helpful for a subset of patients with the most compromised antioxidant defences.
Cannabidiol (CBD)
Cannabis use is a robust risk factor for psychosis [191], and this effect is attributable to its constituent delta-9-tetrahydrocannabinol (THC). However, another of its constituents, cannabidiol (CBD), is anxiolytic [192], neuroprotective [193], and appears to have antipsychotic effects [194]. CBD’s precise mechanism of action is unclear but appears to be different from that of antipsychotic medications and other potential treatments for psychosis. One plausible mechanism is through cannabinoid (CB) receptors, where CBD acts as a negative allosteric modulator and antagonises CB1&2 agonists [195], possibly in opposition to THC which is a CB1 receptor agonist [196]. The hippocampus is one of the most densely populated regions with CB1 receptors, along with the frontal cortex and basal ganglia [197]. Likewise, CB2 receptors, once only thought to be expressed peripherally, have now been found in the hippocampus and midbrain, though in much lower concentrations than CB1 [198]. CB1 activation disinhibits pyramidal cells and promotes hippocampal hyperactivity [199], as well as reducing theta, gamma, and ripple oscillations in the hippocampus [200], conceivably contributing to the detrimental effects of THC in psychosis. Conversely, CB2 receptors may function in opposition to CB1 [201]. By preventing endogenous CB1 receptor agonism and activating CB2, CBD may assist in stabilising this pathway. In support of this view, CBD increases rCBF in the hippocampus, but not in the amygdala, orbitofrontal or prefrontal cortices [202]. SPECT studies of CBD also demonstrate some hippocampal specificity: CBD reduced rCBF in the hippocampus, parahippocampal and inferior temporal gyri, and increased rCBF in the posterior cingulate gyrus [203, 204]. Several other mechanisms of CBD have been proposed including regulating the GPA axis through facilitating 5-HT1A neurotransmission [205] and inhibition of fatty acid amide hydrolase [206]. Other putative targets identified pre-clinically include GPR55 and transient receptor potential vanilloid type 1 [207, 208], though further work is needed to understand the role of these receptors in psychosis.
Preliminary work using CBD as a treatment of psychosis indicates that it is effective in reducing psychotic symptoms, both alone and when used as an adjunct to antipsychotic medications [194, 209]. In CHR individuals, a single dose of 600 mg CBD normalised brain activation in regions that showed abnormal responses under placebo, including the hippocampus [210]. One week of treatment of 600 mg/day CBD significantly reduced cortisol reactivity in healthy control participants compared with a placebo-administered CHR group, while the CBD-administered CHR group experienced an intermediate but non-significant reduction in cortisol reactivity [211], which may support its use as a stress-desensitisation treatment. Longitudinal studies in CHR individuals are needed to ascertain whether CBD reduces symptoms or likelihood of transition to psychosis.
Behavioural interventions
In theory, stress sensitivity could be targeted through interventions that reduce the risk of exposure to early life stressors, or that provide individuals with strategies to cope with stressors when they are active. Environmental enrichment prevented hippocampal hyperactivity in the MAM model for psychosis [212], and may be particularly effective when combined following NAC treatment [178]. In a longitudinal community sample, an environmental enrichment programme focussing on nutrition, education, and exercise from 3 to 5 years of age was associated with lower schizotypal personality scores at 17 years old [213]. Stress-coping skills interventions on children or adolescents, particularly targeted to vulnerable subgroups, may therefore be a feasible strategy. However, many of the environmental stressors that increase psychosis risk, such as poverty, social isolation, and childhood adversity, are difficult to modify by clinical intervention, and can only be changed through social and political action.
Aerobic exercise (AE) is one promising candidate intervention that exerts both antidepressant and anxiolytic effects, and improves resilience to stress [214]. Patients with schizophrenia and CHR individuals show poorer aerobic fitness than healthy volunteers [215, 216] and FEP with lower physical activity have greater reductions in hippocampal volume compared to FEP with higher physical activity [217]. A longitudinal population study found that self-reported physical activity is lower in youth 9–18 years old that went on to develop a psychotic disorder, with one unit lower on their physical activity index associated with a 26% higher risk for developing psychosis, but not affective or substance use disorders [218]. AE specifically targets the hippocampus, selectively triggering immediate rCBF increases in healthy adults [219], and longer-term training is associated with an increase in hippocampal volume [220], particularly in the anterior hippocampus [221].
AE is effective at reducing symptoms in schizophrenia patients [222]. However, a meta-analysis of four AE studies revealed no significant hippocampal volume increases in schizophrenia or FEP patients [220], though a subsequent study found increased left-CA1 volume in treatment-resistant schizophrenia patients [223]. Plausibly, the benefit of AE may be most marked earlier in the stages of increased vulnerability, coinciding with hippocampal development. In a recent clinical trial, AE improved positive symptoms in individuals at CHR [224]. Moreover, the AE group had stable subiculum volume and increased hippocampal-occipital functional connectivity over the intervention, whereas subiculum volume decreased in the non-AE group. While positive symptom improvements were no longer significant at the 12-month follow-up, these results are encouraging. Large-scale, extended interventions are now needed to determine the effective window for intervention and prevention.
Interestingly, acute exercise induces redox imbalance [225]. However, this temporary imbalance may be essential for triggering repair processes and increasing antioxidant efficiencies, gained through regular training. This double-edged sword of acute versus regular exercise should be considered when implementing aerobic interventions in redox imbalance prone CHR individuals, who are often unreliable reporters of activity [215], and given unsupervised individual interventions are least efficacious [222]. One strategy to mitigate engagement issues may be the use of exercise-oriented videogames [226], which would come at reduced costs, and could be integrated into other computerised interventions such as virtual reality tasks [227].
Implementing treatments
Psychosis is heterogeneous and multiple aetiologies may contribute to a common pathophysiology. Individual differences in hippocampal dysfunction in psychosis likely arise from different genetic risk, the extent of environmental stressors and protective factors, recreational drug or medication use, as well as the neurodevelopmental timing. Moreover, symptoms overlap with other psychiatric disorders. The aforementioned causes of dysfunction—stress sensitivity, oxidative stress and E/I imbalance—are therefore likely to play a role in the aetiology of other disorders [228, 229], so some treatments may work transdiagnostically. Accordingly, it may be more useful to focus on clusters of overlapping symptoms [230], such as thought disorder, or specific socio-emotional and cognitive deficits. For instance, one study found that thought disorder positively correlated with left amygdala-hippocampus volume loss across major depressive, bipolar, and schizophrenia disorders, irrespective of formal diagnosis [231].
The prospect of prophylactic treatments is tantalising but attempting to correct for imbalances in immature brains should be approached cautiously to avoid unintended consequences on neurodevelopment. Preventive interventions could be provided at different stages of neurodevelopment (Fig. 3), with varying intensity according to stage. The choice of treatment could be tailored to the underlying biology, identified as a deviation from normative markers derived from longitudinal population studies [232] (Fig. 3A). For example, non-specific support could be offered to children and adolescents at increased risk, along with stress-coping skills training, while more specific pharmacological interventions might be appropriate for young adults at CHR, ideally the subgroup of CHR individuals that are most likely to transition to psychosis. The latter might be identified through the assessment of hippocampal and redox dysfunction, with neuroimaging and peripheral blood measures serving as biomarkers [101, 233]. The promise of targeting hippocampal circuit dysfunction lies in reducing the likelihood of transition to psychosis while also addressing underlying transdiagnostic symptoms, such as cognitive deficits.
Future directions
Longitudinal studies in CHR and other at-risk populations, as well as large community samples, will be critical to mapping maladaptive hippocampal neurodevelopment leading to adverse clinical outcomes. Moreover, hippocampal circuit abnormalities are often first detectable in anterior hippocampal subregions, before dysfunction spreads to surrounding circuits [1]. Consequently, measuring dysfunction within specific hippocampal subregions may be important for understanding the neurodevelopment of psychosis and time-appropriate treatments. Accordingly, it is crucial that any MRI-based segmentation method should also be approached cautiously due to the small size of the hippocampus. A typical voxel size of ~1 mm [3] may be insufficient to segment the hippocampus reliably [234].
Segmentation and functional imaging of the hippocampus will improve as higher field-strength (7T+) scanners become more available. Higher field-strengths also afford the possibility of cortical layer and column analysis [235], allowing for the delineation of hippocampal layering and more specific imbalance localisation. Still, several techniques can boost signal-to-noise ratio (SNR) without new hardware. For instance, reduced-field-of-view imaging around basal ganglia structures rather than whole-brain imaging facilitates sub-1mm [3] voxels without the use of dedicated hardware or invasive imaging contrasts [236].
We have discussed many neuroimaging techniques used to capture early hippocampal abnormalities; these and several emerging technologies, such as optically-pumped magnetoencephalography [237], or chemical exchange saturation transfer [238], may eventually lead to new hippocampal biomarkers for clinical staging in psychosis (Table 1). In addition, other innovative indicators such as maximal oxygen consumption (a measure of aerobic fitness), or gut bacteria diversity—which also impact healthy neurodevelopment and hippocampal processes [239]—could potentially be used as markers of dysfunction and targets for treatment [240, 241].
To detect potential markers, the complexity and heterogeneity of psychosis-risk, and how these patterns are divergent from other mental health disorders, it is critical that this multidimensional information is integrated across scales (Fig. 2). This will include not only multi-modal imaging but also the integration of genetic [242] and other neurobiological information [243] in the modelling of the dysfunction. Though the hippocampus is a core hub in the pathology of psychosis, hippocampal abnormalities across scales—genetic, cellular/molecular, whole-brain network dysconnectivity—must be integrated through large-scale collaborative and integrative computational models.
Conclusions
Improving our understanding of the role of the hippocampus as a central hub of abnormality in the pathophysiology of psychosis may unlock the development of novel treatments and much-needed preventive interventions. Preclinical models indicate that hippocampal changes that occur before the onset of frank psychosis can be reversible, suggesting that clinical interventions at this premorbid stage in humans might be able to reduce the risk of illness onset.
Change history
12 September 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41398-022-02154-y
References
Lieberman JA, Girgis RR, Brucato G, Moore H, Provenzano F, Kegeles L, et al. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry. 2018;23:1764–72.
Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167:1178–93.
Roeske MJ, Konradi C, Heckers S, Lewis AS. Hippocampal volume and hippocampal neuron density, number and size in schizophrenia: a systematic review and meta-analysis of postmortem studies. Mol Psychiatry. 2021;26:3524–35.
van Erp TG, Hibar DP, Rasmussen JM, Glahn DC, Pearlson GD, Andreassen OA, et al. Subcortical brain volume abnormalities in 2028 individuals with schizophrenia and 2540 healthy controls via the ENIGMA consortium. Mol Psychiatry. 2016;21:547–53.
Haijma SV, Van Haren N, Cahn W, Koolschijn PC, Hulshoff Pol HE, Kahn RS. Brain volumes in schizophrenia: a meta-analysis in over 18 000 subjects. Schizophr Bull. 2013;39:1129–38.
Mitelman SA, Bralet MC, Mehmet Haznedar M, Hollander E, Shihabuddin L, Hazlett EA, et al. Positron emission tomography assessment of cerebral glucose metabolic rates in autism spectrum disorder and schizophrenia. Brain Imaging Behav. 2018;12:532–46.
Edmiston EK, Song Y, Chang M, Yin Z, Zhou Q, Zhou Y, et al. Hippocampal resting state functional connectivity in patients with schizophrenia and unaffected family members. Front Psychiatry. 2020;11:278.
Gangadin SS, Cahn W, Scheewe TW, Hulshoff Pol HE, Bossong MG. Reduced resting state functional connectivity in the hippocampus-midbrain-striatum network of schizophrenia patients. J Psychiatr Res. 2021;138:83–8.
Samudra N, Ivleva EI, Hubbard NA, Rypma B, Sweeney JA, Clementz BA, et al. Alterations in hippocampal connectivity across the psychosis dimension. Psychiatry Res. 2015;233:148–57.
Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E, et al. Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. Am J Psychiatry. 2014;171:549–56.
McHugo M, Talati P, Armstrong K, Vandekar SN, Blackford JU, Woodward ND, et al. Hyperactivity and reduced activation of anterior hippocampus in early psychosis. Am J Psychiatry. 2019;176:1030–8.
Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, et al. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–46.
Talati P, Rane S, Skinner J, Gore J, Heckers S. Increased hippocampal blood volume and normal blood flow in schizophrenia. Psychiatry Res. 2015;232:219–25.
Allen P, Azis M, Modinos G, Bossong MG, Bonoldi I, Samson C, et al. Increased resting hippocampal and basal ganglia perfusion in people at ultra high risk for psychosis: replication in a second cohort. Schizophr Bull. 2018;44:1323–31.
Allen P, Chaddock CA, Egerton A, Howes OD, Bonoldi I, Zelaya F, et al. Resting hyperperfusion of the hippocampus, midbrain, and basal ganglia in people at high risk for psychosis. Am J Psychiatry. 2016;173:392–9.
Modinos G, Kempton MJ, Tognin S, Calem M, Porffy L, Antoniades M, et al. Association of adverse outcomes with emotion processing and its neural substrate in individuals at clinical high risk for psychosis. JAMA Psychiatry. 2020;77:190–200.
Winton-Brown T, Schmidt A, Roiser JP, Howes OD, Egerton A, Fusar-Poli P, et al. Altered activation and connectivity in a hippocampal-basal ganglia-midbrain circuit during salience processing in subjects at ultra high risk for psychosis. Transl Psychiatry. 2017;7:e1245.
Allen P, Seal ML, Valli I, Fusar-Poli P, Perlini C, Day F, et al. Altered prefrontal and hippocampal function during verbal encoding and recognition in people with prodromal symptoms of psychosis. Schizophr Bull. 2011;37:746–56.
Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharm Sci. 2011;32:507–13.
Grace AA, Gomes FV. The circuitry of dopamine system regulation and its disruption in schizophrenia: insights into treatment and prevention. Schizophr Bull. 2019;45:148–57.
Buzsaki G, Tingley D. Space and time: the hippocampus as a sequence generator. Trends Cogn Sci. 2018;22:853–69.
Behrens TEJ, Muller TH, Whittington JCR, Mark S, Baram AB, Stachenfeld KL, et al. What is a cognitive map? Organizing knowledge for flexible behavior. Neuron. 2018;100:490–509.
Liu Y, Dolan RJ, Kurth-Nelson Z, Behrens TEJ. Human replay spontaneously reorganizes experience. Cell. 2019;178:640–52.e614.
Musa A, Khan S, Mujahid M, El-Gaby M. The shallow cognitive map hypothesis: A hippocampal framework for thought disorder in schizophrenia. Schizophrenia. 2022;8:34.
Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. 2016;89:248–68.
Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–88.
Gilmore JH, Knickmeyer RC, Gao W. Imaging structural and functional brain development in early childhood. Nat Rev Neurosci. 2018;19:123–37.
Petanjek Z, Judas M, Simic G, Rasin MR, Uylings HB, Rakic P, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci USA. 2011;108:13281–6.
Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387:167–78.
Lavenex P, Banta Lavenex P, Amaral DG. Postnatal development of the primate hippocampal formation. Dev Neurosci. 2007;29:179–92.
Mills KL, Goddings AL, Herting MM, Meuwese R, Blakemore SJ, Crone EA, et al. Structural brain development between childhood and adulthood: Convergence across four longitudinal samples. Neuroimage. 2016;141:273–81.
Backhausen LL, Fröhner JH, Lemaître H, Artiges E, Palillère Martinot M-L, Herting MM, et al. Adolescent to young adult longitudinal development of subcortical volumes in two European sites with four waves. bioRxiv. 2021; https://doi.org/10.1101/2021.06.09.447677.
Herting MM, Johnson C, Mills KL, Vijayakumar N, Dennison M, Liu C, et al. Development of subcortical volumes across adolescence in males and females: A multisample study of longitudinal changes. Neuroimage. 2018;172:194–205.
Wierenga L, Langen M, Ambrosino S, van Dijk S, Oranje B, Durston S. Typical development of basal ganglia, hippocampus, amygdala and cerebellum from age 7 to 24. Neuroimage. 2014;96:67–72.
Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature. 2018;555:377–81.
Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, et al. Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell. 2018;22:589–99.e585.
Kempermann G, Gage FH, Aigner L, Song H, Curtis MA, Thuret S, et al. Human adult neurogenesis: evidence and remaining questions. Cell Stem Cell. 2018;23:25–30.
Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006;11:514–22.
Kozareva DA, Cryan JF, Nolan YM. Born this way: Hippocampal neurogenesis across the lifespan. Aging Cell. 2019;18:e13007.
Gogtay N, Nugent TF 3rd, Herman DH, Ordonez A, Greenstein D, Hayashi KM, et al. Dynamic mapping of normal human hippocampal development. Hippocampus. 2006;16:664–72.
van Winkel R, Stefanis NC, Myin-Germeys I. Psychosocial stress and psychosis. A review of the neurobiological mechanisms and the evidence for gene-stress interaction. Schizophr Bull. 2008;34:1095–105.
Walker EF, Trotman HD, Pearce BD, Addington J, Cadenhead KS, Cornblatt BA, et al. Cortisol levels and risk for psychosis: initial findings from the North American prodrome longitudinal study. Biol Psychiatry. 2013;74:410–7.
Crossley NA, Mechelli A, Scott J, Carletti F, Fox PT, McGuire P, et al. The hubs of the human connectome are generally implicated in the anatomy of brain disorders. Brain. 2014;137:2382–95.
van den Heuvel MP, Sporns O. Rich-club organization of the human connectome. J Neurosci. 2011;31:15775–86.
Ezama L, Hernandez-Cabrera JA, Seoane S, Pereda E, Janssen N. Functional connectivity of the hippocampus and its subfields in resting-state networks. Eur J Neurosci. 2021;53:3378–93.
Blankenship SL, Redcay E, Dougherty LR, Riggins T. Development of hippocampal functional connectivity during childhood. Hum Brain Mapp. 2017;38:182–201.
McHugo M, Armstrong K, Roeske MJ, Woodward ND, Blackford JU, Heckers S. Hippocampal volume in early psychosis: a 2-year longitudinal study. Transl Psychiatry. 2020;10:306.
Vargas T, Dean DJ, Osborne KJ, Gupta T, Ristanovic I, Ozturk S, et al. Hippocampal subregions across the psychosis spectrum. Schizophr Bull. 2018;44:1091–9.
Hyza M, Kuhn M, Ceskova E, Ustohal L, Kasparek T. Hippocampal volume in first-episode schizophrenia and longitudinal course of the illness. World J Biol Psychiatry. 2016;17:429–38.
Kochunov P, Fan F, Ryan MC, Hatch KS, Tan S, Jahanshad N, et al. Translating ENIGMA schizophrenia findings using the regional vulnerability index: association with cognition, symptoms, and disease trajectory. Hum Brain Mapp. 2022;43:566–75.
Adriano F, Caltagirone C, Spalletta G. Hippocampal volume reduction in first-episode and chronic schizophrenia: a review and meta-analysis. Neuroscientist. 2012;18:180–200.
Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78:81–93.
Modinos G, Richter A, Egerton A, Bonoldi I, Azis M, Antoniades M, et al. Interactions between hippocampal activity and striatal dopamine in people at clinical high risk for psychosis: relationship to adverse outcomes. Neuropsychopharmacology. 2021;46:1468–74.
Walter A, Suenderhauf C, Harrisberger F, Lenz C, Smieskova R, Chung Y, et al. Hippocampal volume in subjects at clinical high-risk for psychosis: A systematic review and meta-analysis. Neurosci Biobehav Rev. 2016;71:680–90.
Group ECHRfPW, Jalbrzikowski M, Hayes RA, Wood SJ, Nordholm D, Zhou JH, et al. Association of structural magnetic resonance imaging measures with psychosis onset in individuals at clinical high risk for developing psychosis: an ENIGMA Working Group Mega-analysis. JAMA Psychiatry. 2021;78:753–66.
Sasabayashi D, Yoshimura R, Takahashi T, Takayanagi Y, Nishiyama S, Higuchi Y, et al. Reduced hippocampal subfield volume in schizophrenia and clinical high-risk state for psychosis. Front Psychiatry. 2021;12:642048.
Mechelli A, Riecher-Rössler A, Meisenzahl EM, Tognin S, Wood SJ, Borgwardt SJ, et al. Neuroanatomical abnormalities that predate the onset of psychosis: a multicenter study. Arch Gen Psychiatry. 2011;68:489–95.
Pantelis C, Velakoulis D, McGorry PD, Wood SJ, Suckling J, Phillips LJ, et al. Neuroanatomical abnormalities before and after onset of psychosis: a cross-sectional and longitudinal MRI comparison. Lancet. 2003;361:281–8.
Hinney B, Walter A, Aghlmandi S, Andreou C, Borgwardt S. Does hippocampal volume predict transition to psychosis in a high-risk group? A Meta-Analysis. Front Psychiatry. 2020;11:614659.
Tognin S, Richter A, Kempton MJ, Modinos G, Antoniades M, Azis M, et al. The relationship between grey matter volume and clinical and functional outcomes in people at clinical high risk for psychosis. Schizophrenia Bulletin Open. 2022;sgac040.
Calvo A, Roddy DW, Coughlan H, Kelleher I, Healy C, Harley M, et al. Reduced hippocampal volume in adolescents with psychotic experiences: A longitudinal population-based study. PLoS ONE. 2020;15:e0233670.
Satterthwaite TD, Wolf DH, Calkins ME, Vandekar SN, Erus G, Ruparel K, et al. Structural brain abnormalities in youth with psychosis spectrum symptoms. JAMA Psychiatry. 2016;73:515–24.
Modinos G, Egerton A, McMullen K, McLaughlin A, Kumari V, Barker GJ, et al. Increased resting perfusion of the hippocampus in high positive schizotypy: a pseudocontinuous arterial spin labeling study. Hum Brain Mapp. 2018;39:4055–64.
Schoorl J, Barbu MC, Shen X, Harris MR, Adams MJ, Whalley HC, et al. Grey and white matter associations of psychotic-like experiences in a general population sample (UK Biobank). Transl Psychiatry. 2021;11:21.
Brosch K, Stein F, Schmitt S, Pfarr JK, Ringwald KG, Thomas-Odenthal F, et al. Reduced hippocampal gray matter volume is a common feature of patients with major depression, bipolar disorder, and schizophrenia spectrum disorders. Mol Psychiatry. 2022; https://doi.org/10.1038/s41380-022-01687-4.
Goodkind M, Eickhoff SB, Oathes DJ, Jiang Y, Chang A, Jones-Hagata LB, et al. Identification of a common neurobiological substrate for mental illness. JAMA Psychiatry. 2015;72:305–15.
Radonjic NV, Hess JL, Rovira P, Andreassen O, Buitelaar JK, Ching CRK, et al. Structural brain imaging studies offer clues about the effects of the shared genetic etiology among neuropsychiatric disorders. Mol Psychiatry. 2021;26:2101–10.
Chen F, Bertelsen AB, Holm IE, Nyengaard JR, Rosenberg R, Dorph-Petersen KA. Hippocampal volume and cell number in depression, schizophrenia, and suicide subjects. Brain Res. 2020;1727:146546.
Hibar DP, Westlye LT, van Erp TG, Rasmussen J, Leonardo CD, Faskowitz J, et al. Subcortical volumetric abnormalities in bipolar disorder. Mol Psychiatry. 2016;21:1710–6.
Schmaal L, Veltman DJ, van Erp TG, Samann PG, Frodl T, Jahanshad N, et al. Subcortical brain alterations in major depressive disorder: findings from the ENIGMA Major Depressive Disorder working group. Mol Psychiatry. 2016;21:806–12.
Uhlhaas PJ. Dysconnectivity, large-scale networks and neuronal dynamics in schizophrenia. Curr Opin Neurobiol. 2013;23:283–90.
Nour MM, Liu Y, Arumuham A, Kurth-Nelson Z, Dolan RJ. Impaired neural replay of inferred relationships in schizophrenia. Cell. 2021;184:4315–28.e4317.
Olbrich H-G, Braak H. Ratio of pyramidal cells versus non-pyramidal cells in sector CA1 of the human Ammon’s horn. Anat Embryol. 1985;173:105–10.
Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56.
Fries P, Nikolic D, Singer W. The gamma cycle. Trends Neurosci. 2007;30:309–16.
McNally JM, McCarley RW, Brown RE. Impaired GABAergic neurotransmission in schizophrenia underlies impairments in cortical gamma band oscillations. Curr Psychiatry Rep. 2013;15:346.
Antonoudiou P, Tan YL, Kontou G, Upton AL, Mann EO. Parvalbumin and somatostatin interneurons contribute to the generation of hippocampal gamma oscillations. J Neurosci. 2020;40:7668–87.
Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. 2009;29:2344–54.
Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702.
Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17:537–48.
Song J, Sun J, Moss J, Wen Z, Sun GJ, Hsu D, et al. Parvalbumin interneurons mediate neuronal circuitry-neurogenesis coupling in the adult hippocampus. Nat Neurosci. 2013;16:1728–30.
Freund TF, Buzsáki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470.
Rudy B, Fishell G, Lee S, Hjerling-Leffler J. Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol. 2011;71:45–61.
Wang AY, Lohmann KM, Yang CK, Zimmerman EI, Pantazopoulos H, Herring N, et al. Bipolar disorder type 1 and schizophrenia are accompanied by decreased density of parvalbumin- and somatostatin-positive interneurons in the parahippocampal region. Acta Neuropathol. 2011;122:615–26.
Zhang Z, Sun J, Reynolds GP. A selective reduction in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia patients. Chin Med J. 2002;115:819–23.
Onwordi EC, Halff EF, Whitehurst T, Mansur A, Cotel MC, Wells L, et al. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat Commun. 2020;11:246.
Onwordi EC, Whitehurst T, Mansur A, Statton B, Berry A, Quinlan M, et al. The relationship between synaptic density marker SV2A, glutamate and N-acetyl aspartate levels in healthy volunteers and schizophrenia: a multimodal PET and magnetic resonance spectroscopy brain imaging study. Transl Psychiatry. 2021;11:393.
Osimo EF, Beck K, Reis Marques T, Howes OD. Synaptic loss in schizophrenia: a meta-analysis and systematic review of synaptic protein and mRNA measures. Mol Psychiatry. 2019;24:549–61.
Radhakrishnan R, Skosnik PD, Ranganathan M, Naganawa M, Toyonaga T, Finnema S, et al. In vivo evidence of lower synaptic vesicle density in schizophrenia. Mol Psychiatry. 2021;26:7690–8.
Del Pino I, Garcia-Frigola C, Dehorter N, Brotons-Mas JR, Alvarez-Salvado E, Martinez de Lagran M, et al. Erbb4 deletion from fast-spiking interneurons causes schizophrenia-like phenotypes. Neuron. 2013;79:1152–68.
Kiemes A, Serrano Navacerrada ME, Kim E, Randall K, Simmons C, Rojo Gonzalez L, et al. Erbb4 deletion from fast-spiking interneurons causes psychosis-relevant neuroimaging phenotypes. bioRxiv. 2022; https://doi.org/10.1101/2022.03.07.483347.
Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31:234–42.
Nakazawa K, Sapkota K. The origin of NMDA receptor hypofunction in schizophrenia. Pharm Ther. 2020;205:107426.
Abdul-Monim Z, Neill JC, Reynolds GP. Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol. 2007;21:198–205.
Zhang Z, Sun QQ. Development of NMDA NR2 subunits and their roles in critical period maturation of neocortical GABAergic interneurons. Dev Neurobiol. 2011;71:221–45.
Honeycutt JA, Chrobak JJ. Parvalbumin loss following chronic sub-anesthetic NMDA antagonist treatment is age-dependent in the hippocampus: implications for modeling NMDA hypofunction. Neuroscience. 2018;393:73–82.
Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry. 2006;11:118–9.
Beck K, Arumuham A, Veronese M, Santangelo B, McGinnity CJ, Dunn J, et al. N-methyl-D-aspartate receptor availability in first-episode psychosis: a PET-MR brain imaging study. Transl Psychiatry. 2021;11:425.
Takado Y, Takuwa H, Sampei K, Urushihata T, Takahashi M, Shimojo M, et al. MRS-measured glutamate versus GABA reflects excitatory versus inhibitory neural activities in awake mice. J Cereb Blood Flow Metab. 2022;42:197–212.
Nakahara T, Tsugawa S, Noda Y, Ueno F, Honda S, Kinjo M, et al. Glutamatergic and GABAergic metabolite levels in schizophrenia-spectrum disorders: a meta-analysis of (1)H-magnetic resonance spectroscopy studies. Mol Psychiatry. 2022;27:744–57.
Bossong MG, Antoniades M, Azis M, Samson C, Quinn B, Bonoldi I, et al. Association of hippocampal glutamate levels with adverse outcomes in individuals at clinical high risk for psychosis. JAMA Psychiatry. 2019;76:199–207.
Provenzano FA, Guo J, Wall MM, Feng X, Sigmon HC, Brucato G, et al. Hippocampal pathology in clinical high-risk patients and the onset of schizophrenia. Biol Psychiatry. 2020;87:234–42.
Stone JM, Howes OD, Egerton A, Kambeitz J, Allen P, Lythgoe DJ, et al. Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biol Psychiatry. 2010;68:599–602.
Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–30.
Venkatraman TN, Hamer RM, Perkins DO, Song AW, Lieberman JA, Steen RG. Single-voxel 1H PRESS at 4.0 T: precision and variability of measurements in anterior cingulate and hippocampus. NMR Biomed. 2006;19:484–91.
Stan AD, Ghose S, Zhao C, Hulsey K, Mihalakos P, Yanagi M, et al. Magnetic resonance spectroscopy and tissue protein concentrations together suggest lower glutamate signaling in dentate gyrus in schizophrenia. Mol Psychiatry. 2015;20:433–9.
Wijtenburg SA, Wang M, Korenic SA, Chen S, Barker PB, Rowland LM. Metabolite alterations in adults with schizophrenia, first degree relatives, and healthy controls: a multi-region 7T MRS study. Front Psychiatry. 2021;12:656459.
Modinos G, Simsek F, Azis M, Bossong M, Bonoldi I, Samson C, et al. Prefrontal GABA levels, hippocampal resting perfusion and the risk of psychosis. Neuropsychopharmacology. 2018;43:2652–9.
Marques TR, Ashok AH, Angelescu I, Borgan F, Myers J, Lingford-Hughes A, et al. GABA-A receptor differences in schizophrenia: a positron emission tomography study using [(11)C]Ro154513. Mol Psychiatry. 2021;26:2616–25.
Peris-Yague A, Kiemes A, Cash D, Cotel MC, Singh N, Vernon AC, et al. Region-specific and dose-specific effects of chronic haloperidol exposure on [(3)H]-flumazenil and [(3)H]-Ro15-4513 GABAA receptor binding sites in the rat brain. Eur Neuropsychopharmacol. 2020;41:106–17.
Marsman A, Pries L-K, ten Have M, de Graaf R, van Dorsselaer S, Bak M, et al. Do Current Measures of Polygenic Risk for Mental Disorders Contribute to Population Variance in Mental Health? Schizophrenia Bull. 2020;46:1353–62.
Giovanoli S, Weber L, Meyer U. Single and combined effects of prenatal immune activation and peripubertal stress on parvalbumin and reelin expression in the hippocampal formation. Brain Behav Immun. 2014;40:48–54.
Murthy S, Kane GA, Katchur NJ, Lara Mejia PS, Obiofuma G, Buschman TJ, et al. Perineuronal nets, inhibitory interneurons, and anxiety-related ventral hippocampal neuronal oscillations are altered by early life adversity. Biol Psychiatry. 2019;85:1011–20.
Gomes FV, Zhu X, Grace AA. The pathophysiological impact of stress on the dopamine system is dependent on the state of the critical period of vulnerability. Mol Psychiatry. 2020;25:3278–91.
Decker AL, Duncan K, Finn AS, Mabbott DJ. Children’s family income is associated with cognitive function and volume of anterior not posterior hippocampus. Nat Commun. 2020;11:4040.
Bonne O, Brandes D, Gilboa A, Gomori JM, Shenton ME, Pitman RK, et al. Longitudinal MRI study of hippocampal volume in trauma survivors with PTSD. Am J Psychiatry. 2001;158:1248–51.
Brown AS. The environment and susceptibility to schizophrenia. Prog Neurobiol. 2011;93:23–58.
Samplin E, Ikuta T, Malhotra AK, Szeszko PR, DeRosse P. Sex differences in resilience to childhood maltreatment: effects of trauma history on hippocampal volume, general cognition and subclinical psychosis in healthy adults. J Psychiatr Res. 2013;47:1174–9.
Teicher MH, Anderson CM, Ohashi K, Khan A, McGreenery CE, Bolger EA, et al. Differential effects of childhood neglect and abuse during sensitive exposure periods on male and female hippocampus. Neuroimage. 2018;169:443–52.
Eranti SV, MacCabe JH, Bundy H, Murray RM. Gender difference in age at onset of schizophrenia: a meta-analysis. Psychological Med. 2013;43:155–67.
Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–62.
Phillips LJ, McGorry PD, Garner B, Thompson KN, Pantelis C, Wood SJ, et al. Stress, the hippocampus and the hypothalamic-pituitary-adrenal axis: implications for the development of psychotic disorders. Aust N. Z J Psychiatry. 2006;40:725–41.
Joëls M, Pu Z, Wiegert O, Oitzl MS, Krugers HJ. Learning under stress: how does it work? Trends Cogn Sci. 2006;10:152–8.
McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–22.
Sandi C. Stress, cognitive impairment and cell adhesion molecules. Nat Rev Neurosci. 2004;5:917–30.
Woolley CS, Gould E, McEwen BS. Exposure to excess glucocorticoids alters dendritic morphology of adult hippocampal pyramidal neurons. Brain Res. 1990;531:225–31.
Anacker C, Hen R. Adult hippocampal neurogenesis and cognitive flexibility—linking memory and mood. Nat Rev Neurosci. 2017;18:335–46.
Walker E, Mittal V, Tessner K. Stress and the hypothalamic pituitary adrenal axis in the developmental course of schizophrenia. Annu Rev Clin Psychol. 2008;4:189–216.
Spiers JG, Chen HJ, Sernia C, Lavidis NA. Activation of the hypothalamic-pituitary-adrenal stress axis induces cellular oxidative stress. Front Neurosci. 2014;8:456.
Perkins DO, Jeffries CD, Do KQ. Potential roles of redox dysregulation in the development of schizophrenia. Biol Psychiatry. 2020;88:326–36.
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84.
Kann O, Papageorgiou IE, Draguhn A. Highly energized inhibitory interneurons are a central element for information processing in cortical networks. J Cereb Blood Flow Metab. 2014;34:1270–82.
Cabungcal JH, Steullet P, Morishita H, Kraftsik R, Cuenod M, Hensch TK, et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci USA. 2013;110:9130–5.
Steullet P, Cabungcal JH, Kulak A, Kraftsik R, Chen Y, Dalton TP, et al. Redox dysregulation affects the ventral but not dorsal hippocampus: impairment of parvalbumin neurons, gamma oscillations, and related behaviors. J Neurosci. 2010;30:2547–58.
Cabungcal JH, Steullet P, Kraftsik R, Cuenod M, Do KQ. Early-life insults impair parvalbumin interneurons via oxidative stress: reversal by N-acetylcysteine. Biol Psychiatry. 2013;73:574–82.
Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 2009;19:220–30.
Flatow J, Buckley P, Miller BJ. Meta-analysis of oxidative stress in schizophrenia. Biol Psychiatry. 2013;74:400–9.
Koga M, Serritella AV, Sawa A, Sedlak TW. Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr Res. 2016;176:52–71.
Nishioka N, Arnold SE. Evidence for oxidative DNA damage in the hippocampus of elderly patients with chronic schizophrenia. Am J Geriatr Psychiatry. 2004;12:167–75.
Corcoba A, Steullet P, Duarte JM, Van de Looij Y, Monin A, Cuenod M, et al. Glutathione deficit affects the integrity and function of the fimbria/fornix and anterior commissure in mice: relevance for schizophrenia. Int J Neuropsychopharmacol. 2015;19:pyv110.
Fitzsimmons J, Hamoda HM, Swisher T, Terry D, Rosenberger G, Seidman LJ, et al. Diffusion tensor imaging study of the fornix in first episode schizophrenia and in healthy controls. Schizophr Res. 2014;156:157–60.
Das TK, Javadzadeh A, Dey A, Sabesan P, Theberge J, Radua J, et al. Antioxidant defense in schizophrenia and bipolar disorder: a meta-analysis of MRS studies of anterior cingulate glutathione. Prog Neuropsychopharmacol Biol Psychiatry. 2019;91:94–102.
Tsugawa S, Noda Y, Tarumi R, Mimura Y, Yoshida K, Iwata Y, et al. Glutathione levels and activities of glutathione metabolism enzymes in patients with schizophrenia: a systematic review and meta-analysis. J Psychopharmacol. 2019;33:1199–214.
Wood SJ, Berger GE, Wellard RM, Proffitt TM, McConchie M, Berk M, et al. Medial temporal lobe glutathione concentration in first episode psychosis: a 1H-MRS investigation. Neurobiol Dis. 2009;33:354–7.
Xin L, Mekle R, Fournier M, Baumann PS, Ferrari C, Alameda L, et al. Genetic polymorphism associated prefrontal glutathione and its coupling with brain glutamate and peripheral redox status in early psychosis. Schizophr Bull. 2016;42:1185–96.
Gysin R, Kraftsik R, Sandell J, Bovet P, Chappuis C, Conus P, et al. Impaired glutathione synthesis in schizophrenia: convergent genetic and functional evidence. Proc Natl Acad Sci USA. 2007;104:16621–6.
Baumann PS, Griffa A, Fournier M, Golay P, Ferrari C, Alameda L, et al. Impaired fornix-hippocampus integrity is linked to peripheral glutathione peroxidase in early psychosis. Transl Psychiatry. 2016;6:e859.
Alameda L, Fournier M, Khadimallah I, Griffa A, Cleusix M, Jenni R, et al. Redox dysregulation as a link between childhood trauma and psychopathological and neurocognitive profile in patients with early psychosis. Proc Natl Acad Sci USA. 2018;115:12495–500.
Do KQ, Cuenod M, Hensch TK. Targeting oxidative stress and aberrant critical period plasticity in the developmental trajectory to schizophrenia. Schizophr Bull. 2015;41:835–46.
Rogers SL, Rankin-Gee E, Risbud RM, Porter BE, Marsh ED. Normal development of the perineuronal net in humans; in patients with and without epilepsy. Neuroscience. 2018;384:350–60.
Berretta S, Pantazopoulos H, Markota M, Brown C, Batzianouli ET. Losing the sugar coating: potential impact of perineuronal net abnormalities on interneurons in schizophrenia. Schizophr Res. 2015;167:18–27.
Mauney SA, Athanas KM, Pantazopoulos H, Shaskan N, Passeri E, Berretta S, et al. Developmental pattern of perineuronal nets in the human prefrontal cortex and their deficit in schizophrenia. Biol Psychiatry. 2013;74:427–35.
Pantazopoulos H, Sawyer C, Heckers S, Berretta S, Markota M. Chondroitin sulfate proteoglycan abnormalities in the hippocampus of subjects with schizophrenia. Neuropsychopharmacology. 2014;39:S298–S299.
Yukawa T, Iwakura Y, Takei N, Saito M, Watanabe Y, Toyooka K, et al. Pathological alterations of chondroitin sulfate moiety in postmortem hippocampus of patients with schizophrenia. Psychiatry Res. 2018;270:940–6.
Shah A, Lodge DJ. A loss of hippocampal perineuronal nets produces deficits in dopamine system function: relevance to the positive symptoms of schizophrenia. Transl Psychiatry. 2013;3:e215.
Sonnenschein SF, Grace AA. Peripubertal mGluR2/3 agonist treatment prevents hippocampal dysfunction and dopamine system hyperactivity in adulthood in MAM model of schizophrenia. Schizophr Bull. 2021;47:1806–14.
Adams DH, Zhang L, Millen BA, Kinon BJ, Gomez JC. Pomaglumetad methionil (LY2140023 monohydrate) and aripiprazole in patients with schizophrenia: a phase 3, multicenter, double-blind comparison. Schizophr Res Treat. 2014;2014:758212.
Stauffer VL, Millen BA, Andersen S, Kinon BJ, LaGrandeur L, Lindenmayer JP, et al. Pomaglumetad methionil: No significant difference as an adjunctive treatment for patients with prominent negative symptoms of schizophrenia compared to placebo. Schizophrenia Res. 2013;150:434–41.
Bugarski-Kirola D, Blaettler T, Arango C, Fleischhacker WW, Garibaldi G, Wang A, et al. Bitopertin in Negative Symptoms of Schizophrenia—Results from the phase III FlashLyte and DayLyte studies. Biol Psychiatry. 2017;82:8–16.
Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry. 2013;18:1193–8.
Du Y, Grace AA. Peripubertal diazepam administration prevents the emergence of dopamine system hyperresponsivity in the MAM developmental disruption model of schizophrenia. Neuropsychopharmacology. 2013;38:1881–8.
Rudolph U, Mohler H. GABAA receptor subtypes: therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharm Toxicol. 2014;54:483–507.
Dold M, Li C, Gillies D, Leucht S. Benzodiazepine augmentation of antipsychotic drugs in schizophrenia: a meta-analysis and Cochrane review of randomized controlled trials. Eur Neuropsychopharmacol. 2013;23:1023–33.
Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA. 2004;101:9861–6.
Wakita M, Kotani N, Kogure K, Akaike N. Inhibition of excitatory synaptic transmission in hippocampal neurons by levetiracetam involves Zn2+-dependent GABA type a receptor-mediated presynaptic modulation. J Pharmacol Exp Therapeutics. 2014;348:246–59.
Vanoye-Carlo A, Gomez-Lira G. Differential expression of SV2A in hippocampal glutamatergic and GABAergic terminals during postnatal development. Brain Res. 2019;1715:73–83.
Gill KM, Cook JM, Poe MM, Grace AA. Prior antipsychotic drug treatment prevents response to novel antipsychotic agent in the methylazoxymethanol acetate model of schizophrenia. Schizophrenia Bull. 2014;40:341–50.
Chen B, Choi H, Hirsch LJ, Katz A, Legge A, Buchsbaum R, et al. Psychiatric and behavioral side effects of antiepileptic drugs in adults with epilepsy. Epilepsy Behav. 2017;76:24–31.
Pinckaers FME, Boon ME, Majoie MHJM. Risk factors predisposing to psychotic symptoms during levetiracetam therapy: a retrospective study. Epilepsy Behav. 2019;100.
Jacob TC. Neurobiology and therapeutic potential of alpha5-GABA type A receptors. Front Mol Neurosci. 2019;12:179.
Bowden CL. Valproate. Bipolar Disord. 2003;5:189–202.
Tseng PT, Chen YW, Chung W, Tu KY, Wang HY, Wu CK, et al. Significant effect of valproate augmentation therapy in patients with schizophrenia: a meta-analysis study. Medicine. 2016;95:e2475.
Rosenberg G. The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees? Cell Mol Life Sci. 2007;64:2090–103.
Gervain J, Vines BW, Chen LM, Seo RJ, Hensch TK, Werker JF, et al. Valproate reopens critical-period learning of absolute pitch. Front Syst Neurosci. 2013;7:102.
Klauser P, Xin L, Fournier M, Griffa A, Cleusix M, Jenni R, et al. N-acetylcysteine add-on treatment leads to an improvement of fornix white matter integrity in early psychosis: a double-blind randomized placebo-controlled trial. Transl Psychiatry. 2018;8:220.
Conus P, Seidman LJ, Fournier M, Xin L, Cleusix M, Baumann PS, et al. N-acetylcysteine in a double-blind randomized placebo-controlled trial: toward biomarker-guided treatment in early psychosis. Schizophr Bull. 2018;44:317–27.
Cabungcal JH, Counotte DS, Lewis E, Tejeda HA, Piantadosi P, Pollock C, et al. Juvenile antioxidant treatment prevents adult deficits in a developmental model of schizophrenia. Neuron. 2014;83:1073–84.
Dwir D, Cabungcal JH, Xin L, Giangreco B, Parietti E, Cleusix M, et al. Timely N-acetyl-cysteine and environmental enrichment rescue oxidative stress-induced parvalbumin interneuron impairments via MMP9/RAGE pathway: a translational approach for early intervention in psychosis. Schizophr Bull. 2021;47:1782–94.
Cuenod M, Steullet P, Cabungcal JH, Dwir D, Khadimallah I, Klauser P, et al. Caught in vicious circles: a perspective on dynamic feed-forward loops driving oxidative stress in schizophrenia. Mol Psychiatry. 2022;27:1886–97.
Yolland CO, Hanratty D, Neill E, Rossell SL, Berk M, Dean OM, et al. Meta-analysis of randomised controlled trials with N-acetylcysteine in the treatment of schizophrenia. Aust NZ J Psychiatry. 2020;54:453–66.
Schmidt SJ, Hurlemann R, Schultz J, Wasserthal S, Kloss C, Maier W, et al. Multimodal prevention of first psychotic episode through N-acetyl-l-cysteine and integrated preventive psychological intervention in individuals clinically at high risk for psychosis: Protocol of a randomized, placebo-controlled, parallel-group trial. Early Intervention Psychiatry. 2019;13:1404–15.
Sedlak TW, Paul BD, Parker GM, Hester LD, Snowman AM, Taniguchi Y, et al. The glutathione cycle shapes synaptic glutamate activity. Proc Natl Acad Sci USA. 2019;116:2701–6.
Li Z, Zhang T, Xu L, Wei Y, Tang Y, Hu Q, et al. Decreasing risk of psychosis by sulforaphane study protocol for a randomized, double-blind, placebo-controlled, clinical multi-centre trial. Early Inter Psychiatry. 2021;15:585–94.
Yoshino J, Baur JA, Imai S-i. NAD+ intermediates: the biology and therapeutic potential of NMN and NR. Cell Metab. 2018;27:513–28.
Chouinard VA, Kim SY, Valeri L, Yuksel C, Ryan KP, Chouinard G, et al. Brain bioenergetics and redox state measured by (31)P magnetic resonance spectroscopy in unaffected siblings of patients with psychotic disorders. Schizophr Res. 2017;187:11–16.
Shayganfard M. Molecular and biological functions of resveratrol in psychiatric disorders: a review of recent evidence. Cell Biosci. 2020;10:128.
Susai SR, Sabherwal S, Mongan D, Focking M, Cotter DR. Omega-3 fatty acid in ultra-high-risk psychosis: a systematic review based on functional outcome. Early Inter Psychiatry. 2022;16:3–16.
McLaverty A, Allott KA, Berger M, Hester R, McGorry PD, Nelson B, et al. Omega-3 fatty acids and neurocognitive ability in young people at ultra-high risk for psychosis. Early Intervention Psychiatry. 2021;15:874–81.
Amminger GP, Chanen AM, Ohmann S, Klier CM, Mossaheb N, Bechdolf A, et al. Omega-3 fatty acid supplementation in adolescents with borderline personality disorder and ultra-high risk criteria for psychosis: a post hoc subgroup analysis of a double-blind, randomized controlled trial. Can J Psychiatry. 2013;58:402–8.
Lavoie S, Berger M, Schlogelhofer M, Schafer MR, Rice S, Kim SW, et al. Erythrocyte glutathione levels as long-term predictor of transition to psychosis. Transl Psychiatry. 2017;7:e1064.
Marconi A, Di Forti M, Lewis CM, Murray RM, Vassos E. Meta-analysis of the association between the level of cannabis use and risk of psychosis. Schizophr Bull. 2016;42:1262–9.
ElBatsh MM, Assareh N, Marsden CA, Kendall DA. Anxiogenic-like effects of chronic cannabidiol administration in rats. Psychopharmacology. 2012;221:239–47.
Demirakca T, Sartorius A, Ende G, Meyer N, Welzel H, Skopp G, et al. Diminished gray matter in the hippocampus of cannabis users: possible protective effects of cannabidiol. Drug Alcohol Depend. 2011;114:242–5.
McGuire P, Robson P, Cubala WJ, Vasile D, Morrison PD, Barron R, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicenter randomized controlled trial. Am J Psychiatry. 2018;175:225–31.
Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150:613–23.
Bhattacharyya S, Morrison PD, Fusar-Poli P, Martin-Santos R, Borgwardt S, Winton-Brown T, et al. Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology. 2010;35:764–74.
Glass M, Dragunow M, Faull RL. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77:299–318.
Stempel AV, Stumpf A, Zhang HY, Ozdogan T, Pannasch U, Theis AK, et al. Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the hippocampus. Neuron. 2016;90:795–809.
Hájos N, Katona I, Naiem SS, Mackie K, Ledent C, Mody I, et al. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–49.
Robbe D, Montgomery SM, Thome A, Rueda-Orozco PE, McNaughton BL, Buzsaki G. Cannabinoids reveal importance of spike timing coordination in hippocampal function. Nat Neurosci. 2006;9:1526–33.
Chen DJ, Gao M, Gao FF, Su QX, Wu J. Brain cannabinoid receptor 2: expression, function and modulation. Acta Pharm Sin. 2017;38:312–6.
Bloomfield MAP, Green SF, Hindocha C, Yamamori Y, Yim JLL, Jones APM, et al. The effects of acute cannabidiol on cerebral blood flow and its relationship to memory: an arterial spin labelling magnetic resonance imaging study. J Psychopharmacol. 2020;34:981–9.
Crippa JA, Derenusson GN, Ferrari TB, Wichert-Ana L, Duran FL, Martin-Santos R, et al. Neural basis of anxiolytic effects of cannabidiol (CBD) in generalized social anxiety disorder: a preliminary report. J Psychopharmacol. 2011;25:121–30.
Crippa JADS, Zuardi AW, Garrido GEJ, Wichert-Ana L, Guarnieri R, Ferrari L, et al. Effects of cannabidiol (CBD) on regional cerebral blood flow. Neuropsychopharmacology. 2004;29:417–26.
Viudez-Martínez A, García-Gutiérrez MS, Manzanares J. Cannabidiol regulates the expression of hypothalamus-pituitary-adrenal axis-related genes in response to acute restraint stress. J Psychopharmacol. 2018;32:1379–84.
Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94–e94.
Tzavara ET, Li DL, Moutsimilli L, Bisogno T, Di Marzo V, Phebus LA, et al. Endocannabinoids activate transient receptor potential vanilloid 1 receptors to reduce hyperdopaminergia-related hyperactivity: therapeutic implications. Biol Psychiatry. 2006;59:508–15.
Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson N-O, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–101.
O’Neill A, Annibale L, Blest-Hopley G, Wilson R, Giampietro V, Bhattacharyya S. Cannabidiol modulation of hippocampal glutamate in early psychosis. J Psychopharmacol. 2021;35:814–22.
Bhattacharyya S, Wilson R, Appiah-Kusi E, O’Neill A, Brammer M, Perez J, et al. Effect of cannabidiol on medial temporal, midbrain, and striatal dysfunction in people at clinical high risk of psychosis: a randomized clinical trial. JAMA Psychiatry. 2018;75:1107–17.
Appiah-Kusi E, Petros N, Wilson R, Colizzi M, Bossong MG, Valmaggia L, et al. Effects of short-term cannabidiol treatment on response to social stress in subjects at clinical high risk of developing psychosis. Psychopharmacology. 2020;237:1121–30.
Zhu X, Cabungcal JH, Cuenod M, Uliana DL, Do KQ, Grace AA. Thalamic reticular nucleus impairments and abnormal prefrontal control of dopamine system in a developmental model of schizophrenia: prevention by N-acetylcysteine. Mol Psychiatry. 2021;26:7679–89.
Adrian R, Kjetil M, Jianghong L, Peter V, Sarnoff A, Mednick. Effects of environmental enrichment at ages 3–5 years on schizotypal personality and antisocial behavior at ages 17 and 23 years. Am J Psychiatry. 2003;160:1627–35.
Salmon P. Effects of physical exercise on anxiety, depression, and sensitivity to stress: a unifying theory. Clin Psychol Rev. 2001;21:33–61.
Damme KSF, Sloan RP, Bartels MN, Ozsan A, Ospina LH, Kimhy D, et al. Psychosis risk individuals show poor fitness and discrepancies with objective and subjective measures. Sci Rep. 2021;11:9851.
Kimhy D, Vakhrusheva J, Bartels MN, Armstrong HF, Ballon JS, Khan S, et al. Aerobic fitness and body mass index in individuals with schizophrenia: implications for neurocognition and daily functioning. Psychiatry Res. 2014;220:784–91.
Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TG, et al. Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry. 2015;77:147–57.
Sormunen E, Saarinen MM, Salokangas RKR, Telama R, Hutri-Kähönen N, Tammelin T, et al. Effects of childhood and adolescence physical activity patterns on psychosis risk—a general population cohort study. npj Schizophrenia. 2017;3:5.
Steventon JJ, Foster C, Furby H, Helme D, Wise RG, Murphy K. Hippocampal blood flow is increased after 20 min of moderate-intensity exercise. Cereb Cortex. 2019;30:525–33.
Firth J, Rosenbaum S, Ward PB, Curtis J, Teasdale SB, Yung AR, et al. Adjunctive nutrients in first-episode psychosis: a systematic review of efficacy, tolerability and neurobiological mechanisms. Early Inter Psychiatry. 2018;12:774–83.
Erickson KI, Voss MW, Prakash RS, Basak C, Szabo A, Chaddock L, et al. Exercise training increases size of hippocampus and improves memory. Proc Natl Acad Sci USA. 2011;108:3017–22.
Firth J, Cotter J, Elliott R, French P, Yung AR. A systematic review and meta-analysis of exercise interventions in schizophrenia patients. Psychological Med. 2015;45:1343–61.
McHugo M, Talati P, Woodward ND, Armstrong K, Blackford JU, Heckers S. Regionally specific volume deficits along the hippocampal long axis in early and chronic psychosis. Neuroimage Clin. 2018;20:1106–14.
Damme KSF, Gupta T, Ristanovic I, Kimhy D, Bryan AD, Mittal VA. Exercise intervention in individuals at clinical high risk for psychosis: benefits to fitness, symptoms, hippocampal volumes, and functional connectivity. Schizophr Bull. 2022;sbac084.
Radak Z, Radak ZZ, Koltai E, Ohno H, Atalay M. Oxygen consumption and usage during physical exercise: the balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid Redox Signal. 2013;18:1208–46.
Bang-Kittilsen G, Egeland J, Holmen TL, Bigseth TT, Andersen E, Mordal J, et al. High-intensity interval training and active video gaming improve neurocognition in schizophrenia: a randomized controlled trial. Eur Arch Psychiatry Clin Neurosci. 2021;271:339–53.
Rus-Calafell M, Garety P, Sason E, Craig TJK, Valmaggia LR. Virtual reality in the assessment and treatment of psychosis: a systematic review of its utility, acceptability and effectiveness. Psychological Med. 2018;48:362–91.
Marin O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–20.
Govindpani K, Turner C, Waldvogel HJ, Faull RLM, Kwakowsky A. Impaired expression of GABA signaling components in the Alzheimer's disease middle temporal gyrus. Int J Mol Sci. 2020;21:22.
Dwyer DB, Buciuman MO, Ruef A, Kambeitz J, Sen Dong M, Stinson C, et al. Clinical, brain, and multilevel clustering in early psychosis and affective stages. JAMA Psychiatry. 2022;79:677–89.
Stein F, Meller T, Brosch K, Schmitt S, Ringwald K, Pfarr JK, et al. Psychopathological syndromes across affective and psychotic disorders correlate with gray matter volumes. Schizophr Bull. 2021;47:1740–50.
Marquand AF, Kia SM, Zabihi M, Wolfers T, Buitelaar JK, Beckmann CF. Conceptualizing mental disorders as deviations from normative functioning. Mol Psychiatry. 2019;24:1415–24.
Dickens AM, Sen P, Kempton MJ, Barrantes-Vidal N, Iyegbe C, Nordentoft M, et al. Dysregulated lipid metabolism precedes onset of psychosis. Biol Psychiatry. 2021;89:288–97.
Wisse LEM, Chetelat G, Daugherty AM, de Flores R, la Joie R, Mueller SG, et al. Hippocampal subfield volumetry from structural isotropic 1 mm(3) MRI scans: a note of caution. Hum Brain Mapp. 2021;42:539–50.
De Martino F, Zimmermann J, Muckli L, Ugurbil K, Yacoub E, Goebel R. Cortical depth dependent functional responses in humans at 7T: improved specificity with 3D GRASE. PLoS One. 2013;8:e60514.
Heidemann RM, Ivanov D, Trampel R, Fasano F, Meyer H, Pfeuffer J, et al. Isotropic submillimeter fMRI in the human brain at 7 T: combining reduced field-of-view imaging and partially parallel acquisitions. Magn Reson Med. 2012;68:1506–16.
Barry DN, Tierney TM, Holmes N, Boto E, Roberts G, Leggett J, et al. Imaging the human hippocampus with optically-pumped magnetoencephalography. Neuroimage. 2019;203:116192.
Wu B, Warnock G, Zaiss M, Lin C, Chen M, Zhou Z, et al. An overview of CEST MRI for non-MR physicists. EJNMMI Phys. 2016;3:19.
Tang W, Meng Z, Li N, Liu Y, Li L, Chen D, et al. Roles of gut microbiota in the regulation of hippocampal plasticity, inflammation, and hippocampus-dependent behaviors. Front Cell Infect Microbiol. 2020;10:611014.
Olivo G, Nilsson J, Garzon B, Lebedev A, Wahlin A, Tarassova O, et al. Higher VO2max is associated with thicker cortex and lower grey matter blood flow in older adults. Sci Rep. 2021;11:16724.
Nauer RK, Dunne MF, Stern CE, Storer TW, Schon K. Improving fitness increases dentate gyrus/CA3 volume in the hippocampal head and enhances memory in young adults. Hippocampus. 2020;30:488–504.
Ding S-L, Royall JJ, Sunkin SM, Ng L, Facer BAC, Lesnar P, et al. Comprehensive cellular-resolution atlas of the adult human brain. J Comp Neurol. 2016;524:3127–481.
Norgaard M, Beliveau V, Ganz M, Svarer C, Pinborg LH, Keller SH, et al. A high-resolution in vivo atlas of the human brain’s benzodiazepine binding site of GABAA receptors. Neuroimage. 2021;232:117878.
Amunts K, Kedo O, Kindler M, Pieperhoff P, Mohlberg H, Shah NJ, et al. Cytoarchitectonic mapping of the human amygdala, hippocampal region and entorhinal cortex: intersubject variability and probability maps. Anat Embryol. 2005;210:343–52.
Khalaf-Nazzal R, Francis F. Hippocampal development—old and new findings. Neuroscience. 2013;248:225–42.
Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19.
Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47.
McCutcheon RA, Reis Marques T, Howes OD. Schizophrenia—an overview. JAMA Psychiatry. 2020;77:201–10.
Acknowledgements
SK is supported by a grant from Mental Health Research UK and the Schizophrenia Research Fund. RM is funded by a NIHR clinical lectureship. This research was funded in whole, or in part, by the Wellcome Trust [Sir Henry Dale Fellowship 202397/Z/16/Z to GM]. For the purpose of open access, the author has applied a CC-BY public copyright licence to any Author Accepted Manuscript version arising from this submission.
Author information
Authors and Affiliations
Contributions
GM, OOD, PM and SK conceived the project. SK wrote the first draft of the manuscript. GM obtained the funding. RM, DD, AAG, OOD, PM and GM all provided critical input into this first draft and approved the final version of this article.
Corresponding author
Ethics declarations
Competing interests
AAG has received consulting fees from Alkermes, Lundbeck, Takeda, Roche, Lyra, Concert, and SynAgile. RM has received honoraria from Janssen and Otsuka. The other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Knight, S., McCutcheon, R., Dwir, D. et al. Hippocampal circuit dysfunction in psychosis. Transl Psychiatry 12, 344 (2022). https://doi.org/10.1038/s41398-022-02115-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-022-02115-5
This article is cited by
-
Effects of diazepam on hippocampal blood flow in people at clinical high risk for psychosis
Neuropsychopharmacology (2024)
