
Abstract
Chloride homeostasis, the main determinant factor for the dynamic tuning of GABAergic inhibition during development, has emerged as a key element altered in a wide variety of brain disorders. Accordingly, developmental disorders such as schizophrenia, Autism Spectrum Disorder, Down syndrome, epilepsy, and tuberous sclerosis complex (TSC) have been associated with alterations in the expression of genes codifying for either of the two cotransporters involved in the excitatory-to-inhibitory GABA switch, KCC2 and NKCC1. These alterations can result from environmental insults, including prenatal stress and maternal separation which share, as common molecular denominator, the elevation of pro-inflammatory cytokines. In this review we report and systemize recent research articles indicating that different perinatal environmental perturbations affect the expression of chloride transporters, delaying the developmental switch of GABA signaling, and that inflammatory cytokines, in particular interleukin 1β, may represent a key causal factor for this phenomenon. Based on literature data, we provide therefore a unifying conceptual framework, linking environmental hits with the excitatory-to-inhibitory GABA switch in the context of brain developmental disorders.
Similar content being viewed by others

Introduction
Besides genetics, individual susceptibility to psychiatric disorders is known to be influenced by environmental factors, where the term “environmental” encompasses all those factors that are not genetically inherited. The prenatal window and the early phases of post-natal development represent critical periods, when environmental insults may have long-lasting consequences on the brain developmental trajectories1,2,3,4. Among these insults, stress and inflammation are two recognized risk factors for a wide spectrum of brain disorders, which may emerge either soon after birth or later in postnatal development5,6,7. Since the first indications of the long-lasting effects of stress occurring during pregnancy in the development of behavioral disturbances8, evidence is progressively accumulating that prenatal stress may influence birth outcomes and offspring neurodevelopment. In particular, maternal prenatal stress, regardless of whether psychological (e.g., infections or maternal restraints) or physical (e.g. cardiovascular abnormalities), increases the risk of preterm birth9,10 and psychiatric conditions such as depression and anxiety11, schizophrenia12, personality disorders13, autism, and ADHD14.
In the complex set of intertwined processes found to be defective in several neurodevelopmental disorders, including altered spine morphology and function15 and uneven synaptic maintenance/dendritic complexity16, the unbalanced excitatory/inhibitory (E/I) transmission17,18 and the altered GABAergic function19,20 represent highly investigated potential pathogenic mechanisms, possibly accounting for the lack of a balanced and flexible neuronal network typical of neuropsychiatric patients21. In the developmental sequence of events governing the inhibitory neurotransmission maturation, a central role is played by the so-called excitatory-to-inhibitory GABA switch. Discovered almost three decades ago, the GABA switch relies on the occurrence, at early developmental stages, of elevated intracellular chloride levels which progressively reduce, giving rise to an excitatory-to-inhibitory shift of GABA action. A temporal deviation from this switch leads to the persistence of immature GABAergic features in the adult brain, which perturb the operation of well-developed functional networks22. According to its functional importance during development, GABA is implicated in a number of neurodevelopmental disorders such as Autism Spectrum Disorders (ASD)23, Fragile X24, Rett syndrome25,26, Down syndrome27, and schizophrenia28. In this review, we discuss experimental evidence indicating that developmental GABA switch is largely influenced by environmental stimuli, thus possibly representing a central link that translates early-life environmental hits into behavioral changes in the adult.
Neuronal chloride homeostasis: a key factor in brain maturation
The discovery of the dichotomic action of GABAergic neurotransmission dates back to over 30 years ago29,30. The inhibitory action of GABA in mature neurons relies on its ability to open chloride (Cl−)-permeable GABAA receptor channels that allow Cl− influx and hyperpolarization. However, at early stages of development, when intracellular Cl− concentrations are high and Cl− equilibrium potential is positive, compared to the resting membrane potential, GABAergic neurotransmission results in Cl−efflux and membrane depolarization. The homeostatic regulation of intracellular ionic composition, which ends up determining the polarity of GABA action in neurons, is ensured by a variety of ion-transporters localized at the cell surface (Table 1). Cation chloride co-transporters (CCCs) represent a group of ion-transporters involved in the control of electrolyte homeostasis through the coupling of cations transport with Cl− in opposite directions31. The solute carrier 12 (SLC12) family of CCCs includes four potassium (K)-Cl co-transporters (KCCs) isoforms, named KCC1, KCC2, KCC3, and KCC4, encoded by distinct SLC12 genes (SLC12A4-7 respectively)32 and two sodium (Na)-K-Cl cotransporters, namely NKCC1 and NKCC2, encoded by SLC12A2 and SLC12A1 genes, respectively. In particular, NKCC cotransporters drive Cl- uptake fueled by Na+ 33, while Cl- extrusion is fueled by K+ through the KCC cotransporters34,35,36,37,38. All these transporters derive their energy from the Na+ and K+ gradient generated by Na-K-ATPase. NKCC2 exhibits kidney-specific expression and functions in renal salt reabsorption, whereas NKCC1 plays a key role in regulation of salt secretion and cell volume in epithelial cells and is critical for the appropriate response to GABA in neuronal cells. Both NKCC isoforms are inhibited by 5-sulfamoylbenzoic acid loop diuretics, including bumetanide. Concerning KCCs, KCC1 was found to be ubiquitously expressed by northern blot analysis34, while KCC2, initially identified only in the brain37, has been recently found also in chicken myocytes39; KCC3 and KCC4 are expressed in both the central and peripheral nervous systems (PNS)40,41,42, heart, skeletal muscle, and kidney35,36 (Table 1). KCC2 exhibits two isoforms, named KCC2a and KCC2b, which differ in their most N-terminal part43, and is barely detectable in the PNS and non-neuronal cell types as glial cells37,44,45,46. Unlike the other KCC-family members -whose levels remain stable over time- KCC2 expression follows a robust upregulation during brain development47, a process indispensable for a correct neuronal differentiation and functioning48. In particular, increases occur in the cortical expression of KCC2b isoform over KCC2a during late postnatal development, while the two isoforms display nearly comparable changes in the brain stem and spinal cord during prenatal and early postnatal stages43. KCC2a expression is therefore barely expressed in, or even lacking from, most parts of the adult cortex, hippocampus, thalamus, and cerebellar cortex, whereas both isoforms are detectable in the developing and mature hypothalamus, brainstem (except for the brainstem auditory system that lacks KCC2a immunoreactivity), and spinal cord49 (Table 1). This molecular dynamic has been also reported in other species, from reptile to humans, indicating that the developmental GABA switch is an evolutionary conserved process50,51. The progressive increase of KCC2b activity during neuronal development, together with the progressive reduction of NKCC1, causes the lowering of intracellular chloride levels and set the driving force and the reversal potential of the anion currents31. This allows the physiological transition from the excitatory to the inhibitory action of GABA51,52,53,54,55,56. Notably, the depolarizing GABA-A transmission is implicated in numerous developmental processes, including neural stem cell proliferation57, cell migration58, neurite outgrowth59, synapse formation and circuit refinement59,60,61 as well as shaping excitatory connectivity during neural circuit development62. The dual role played by GABAergic transmission represents therefore one of the fundamental pillars of brain development, ensuring a proper brain wiring and functioning at adult stages.
Modulating GABA switch through KCC2 regulation by hormonal and soluble factors: implications for neurodevelopmental diseases
The molecular processes controlling KCC2 expression are subjected to multiple regulatory mechanisms relying on both transcriptional and post-transcriptional pathways. At transcriptional level, KCC2 gene is modulated via the coordinated activity of several transcription factors, such as RE1-Silencing Transcription factor (REST)63, a neuronal gene repressor that plays a crucial role in neuronal differentiation. REST activity depends on its interaction with other cofactors, including CoREST and MeCP2, thereby forming a complex transcriptional machinery which ultimately regulates the expression of many neuronal-specific genes. Interestingly, the involvement of the transcription factor methyl-CpG-binding protein 2 (MeCP2) in the control of KCC2 expression has been recently reported, supporting the interplay between REST and MeCP2 in this process64,65.
Several soluble factors are also known to influence the process of GABA switch. The first pioneer study66, subsequently corroborated by a more recent research article67, proposed that GABA signaling per se represents the fundamental extracellular cue that allows the occurrence of the switch and the progressive upregulation of KCC2 transporter. The GABA switch process is also promoted by trophic factors including the neurotrophic factor BDNF68,69,70, which exerts a facilitatory effect on the expression of KCC2 mRNA and protein, during development. The effect of BDNF on KCC2 protein expression occurs via extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent upregulation of the transcription factor early growth response 4 (Egr4) and via Egr4-dependent activation of the KCC2b promoter70.
Hormonal factors also play a central role in GABA switch modulation. For instance, estradiol downregulates KCC2 mRNA71, while testosterone and dihydrotestosterone upregulate KCC2 mRNA72. Similarly, the thyroid hormone triiodothyronine (T3) enhances the expression of KCC2 protein, thus accelerating the developmental shift of GABA action from depolarizing to hyperpolarizing73. A special attention has been paid to oxytocin, which exerts a crucial role in the control of GABA developmental shift during labor. Shortly before delivery, the surge of maternal oxytocin triggers, in the offspring brain, a transient switch of the GABA response from excitatory to inhibitory74,75,76 that is crucial for the entire subsequent neurodevelopmental process. Oxytocin positively promotes the plasmamembrane insertion of KCC2 through a change of the phosphorylation state of the transporter77, highlighting the relevance of post-translational mechanisms in the transporter regulation. Among the kinases that control the phosphorylation of KCC2, thus regulating the transporter activity and/or its cell surface stability (for review see, 78,79), PKC is mainly involved in the phosphorylation of several serine residues, cytosolic c-Src kinase80 and BDNF-dependent TrkB receptor kinase target tyrosine residues of KCC268,81,82 whilst the WNK (with-no-lysine[K]) family of serine-threonine kinases together with SPAK, Ste20p-related proline/alanine-rich kinase modulate the phosphorylation level at threonine residues. Regarding the latter, WNK/SPAK-kinase activity is reduced during neuronal development, leading to a substantial dephosphorylation of threonine 906 and 1007 and a consequent increased of KCC2 activity83.
The key role of hormonal factors in the regulation of KCC2 is in line with the reported sex-biased functional maturation of GABA inhibitory signaling, which occurs, in a brain region-specific manner, earlier in females than in males71,84,85,86. While the male hormone testosterone or its metabolites at early post-natal period appear to be crucial for GABA switch in the substantia nigra (SNR) in both sexes, via the upregulation of KCC2 mRNA, the female hormone estradiol downregulates KCC2 mRNA in males but not in females72. In the hippocampus and entorhinal cortex of neonatal rats, KCC2 remains consistently higher in females compared to males especially in the second post-natal week, when NKCC1 peaks are detectable only in males87. Notably, however, hypothalamic cells obtained from embryos before the first testosterone peak, already display sex-dimorphism in their response to the GABAA-receptor agonist muscimol, indicating that differences in GABA switch timing can also occur independently of sex hormones exposure88.
The earlier functional maturation of GABAA inhibitory signaling in females71 could contribute in making the male GABAergic system less resilient to early environmental insults89, with possible, important implications for brain disorders, such as epilepsy and neurodevelopmental diseases. For example, reduction of KCC2 expression and function appears to contribute to the development of epileptic seizures52. Interestingly, stressors in early life accelerate epileptogenesis in a sexually dimorphic manner, with males showing a higher susceptibility than females, both in rodents90 and in humans91. It is important to note that neurodevelopmental diseases, such as ASD and schizophrenia -which display a sex-biased incidence, with ASD affecting males 3 times and schizophrenia 1.4 times more than females92,93,94- are characterized by defects in GABAergic maturation. Alterations of the GABA switch has been recently detected in neurodevelopmental disorders associated or not associated with autism-like behaviors, including Fragile-X95 and Rett syndrome26,65,96,97. Neurons from a mouse model of Rett syndrome (lacking Mecp2) or derived from induced pluripotent stem cells (iPSCs) of patients with Rett syndrome exhibit a more depolarizing GABA signaling26 that could be rescued by either KCC2 exogenous expression65 or pharmacological drugs enhancing KCC2 activity97. Consistently, reduced KCC2 expression has been observed in post-mortem brain tissues from Rett syndrome patients98. Similar phenotypes have been found in Fragile-X syndrome mouse models, where the developmental shift of GABA signaling was found to be impaired owing to KCC275,76 or NKCC195 deregulation. Moreover, in Down syndrome models, the GABA signaling in the adult brain is excitatory instead than inhibitory53. Also, impairment of the inhibitory strength of GABAergic neurotransmission consequent to altered KCC2 Thr906/Thr1007 phosphorylation, results in increased seizure susceptibility, altered ultra-sonic vocalization, and social interaction deficits, that are typical of ASD99. Finally, decreased expression of KCC2 or increased NKCC1/KCC2 ratio was observed in human cortical brain samples from Dravet syndrome patients100 and from TSC patients, respectively101.
Furthermore, a differential expression of KCC2 transcripts has been reported in schizophrenic patients50, who display a hippocampal NKCC1/KCC2 ratio that is generally higher relative to healthy subjects50. Also, KCC2 truncated splice variants are distinctly expressed in schizophrenic patients compared to healthy controls, likely reflecting the altered GABA physiology associated with the disease102. Finally, two rare functionally impairing variants in the KCC2 C-terminal regulatory domain have been detected in human ASD (R952H and R1049C) and schizophrenia (R952H). These variants significantly decrease KCC2-mediated Cl− extrusion capacity in neurons, reducing the transporter plasmalemmal expression (R952H) or lowering the intrinsic activity of transporters at the cell surface (R1049C)103.
These data clearly indicate that altered KCC2 regulation impacts the GABAergic developmental sequence in vivo, representing a risk factor for the emergence of neurological pathology, such as epilepsy, ASD, schizophrenia. The causal involvement of NKCC1 and KCC2 sexual dimorphism in these disorders is an emerging possibility that deserves further investigation.
The possible role of inflammatory pathways in KCC2 regulation
The impact of environmental stimuli in the regulation of KCC2 expression, and therefore in the timing of GABA switch, has been demonstrated in protocols of prenatal or early life stress, where an altered GABAergic signaling, leading to an E/I imbalance, occurs in the offspring (Table 2). Pregnant mice subjected to immobilization paradigms at late stages of pregnancy (GD15) give birth to offspring characterized by downregulation of KCC2 expression associated with changes of specific GABA receptor subunits104, reduction of several GABAergic markers (e.g., GABA and GAD67)105, and significant delayed migration of inhibitory progenitors106 (Table 2). Stressful conditions occurring at early post-developmental stages also impact GABAergic signaling. Indeed, animal models of maternal separation, based on daily rounds of pup separation from the mother lasting from the first postnatal day up to 2 weeks, display longer latency to play and decreased social play behavior107, and anxiety-like states108, associated with KCC2 deregulation delayed GABA developmental switch and excitatory GABA action109,110. However, it should be noted that in a different experimental setting based on 6 h daily separation from the dam at P4 for just 3 days, KCC2 immunoreactivity was found to be increased only in males and not females, indicating a gender-specific effect of early post-natal stress on KCC2 expression111. Mice subjected to acute (30 min) or chronic restraint stress (30 min/day for 14 consecutive days) display in the hippocampus112 or in the paraventricular nucleus of the hypothalamus (PVN)113 the dephosphorylation of KCC2 residue S940, which regulates KCC2 cell surface expression and function, accompanied by increased susceptibility to seizures112,113 (Table 2). Consistently, induction of repeated stress by forced water administration is associated with decreased KCC2 and increased NKCC1 membrane expression in granular and pyramidal cells in the hippocampus114. Interestingly, acute restraint stress performed in young animals is associated with loss of synaptic inhibition coupled with reduced KCC2 activity in the PVN115, a key cellular component of the hypothalamic-pituitary stress axis (HPA), leading to a reduced inhibitory constraint at HPA thus participating in the physiological control of stress hormones release115 (Table 2). Despite these lines of evidence, the research for cellular defects and signaling pathways activated by early life stress did not provide a clear molecular link yet between these early stressful events and KCC2 deregulation.
Nonetheless, it is interesting to note that chronic stress induced by maternal separation engaged inflammatory pathways in neurons. In mice subjected to maternal deprivation, increased levels of the interleukin-1 receptor (IL-1R) are detectable specifically at synapses in the male hippocampus, together with enhanced interactions with the GluN2B subunit of NMDARs116. Similarly, rats subjected to maternal separation for 3 h/day from post-natal day 1 to 14 and examined at post-natal day 15, displayed higher hippocampal levels of interleukin-1beta (IL-1β) mRNA117. The classical restraint stress paradigm during pregnancy (from E10 to E16 for 2 h/day) results in female offspring with elevated IL-1β levels in both placenta and brains118. Notably, early and prenatal stress are associated with inflammation also in humans. Social and psychological adversities in children are associated with high CRP (C-reative protein) levels119,120,121 (for a specific review see122).
A direct demonstration that a proinflammatory environment reduces the transcription of KCC2, thus delaying the excitatory to inhibitory GABA switch, has been recently provided64. Using a protocol of prenatal immune activation employing the synthetic analog of double-stranded RNA polyI:C, the enhanced binding of the transcription factors MeCP2 and REST to the KCC2 gene promoter region was demonstrated in the offspring brain. The consequent reduction of KCC2 protein was accompanied by increased susceptibility to kainate-induced seizures in the adult mouse offspring64. This was the first evidence highlighting an effect of maternal immune activation on the GABA developmental shift. A subsequent study123 reported that PolyI:C injection at later gestational day, GD12.5, abolished the GABA switch in the offspring, leading to defects that were detectable already at birth. When IL-1R knockout mice, which display silenced IL-1β pathway, were implanted in wild-type dams and prenatally exposed to poly I:C, no reduction of KCC2 could be demonstrated, pointing to a direct involvement of IL-1β in down regulating KCC2 transcription64. Accordingly, exposure of cultured neurons to IL-1β resulted in higher chloride intracellular concentrations. Therefore, inflammation-derived molecules produced during fetal development lead to a deregulation of the GABA developmental shift, which manifests at early postnatal stages and persists in the long-term period.
Interestingly, elevation of proinflammatory cytokines has been demonstrated in the pathological contexts where reduction of KCC2 has been found to be involved. IL-1β is recognized to contribute to epilepsy development124, and the therapeutic effect of the IL-1β antagonist, Anakinra, has been reported in the relapsing chronic phase of febrile infection–related epilepsy syndrome125, as well as in the super-refractory status epilepticus and febrile infection-related epilepsy syndrome126. Along the same line, a robust increase in IL-1β and IL-1 receptor were detected in post-traumatic epilepsy in children, and IL-1Ra treatment reduced seizure susceptibility 2 weeks after traumatic brain injury compared to vehicle127. In ASD, the aberrant expression of cytokines and their signaling intermediaries has been demonstrated in the brain128,129. Elevated peripheral IL-1β levels are associated with the ASD diagnosis later in childhood and vary with ASD symptom severity130,131. In high-functioning male children with ASD, the plasma levels of IL-1β, IL-1 receptor antagonist, together with IL-5, IL-8, IL-12(p70), IL-13, and IL-17, were fund to be elevated relative to age-matched controls132. These elevations may reflect a prenatal immune challenge130. Marked elevations of IL-1β in CSF of male patients with first-episode schizophrenia has been described133, while the interleukin-1 cluster was found to associate with genetic risk for schizophrenia134. Furthermore, polymorphisms of the interleukin-1 gene complex were demonstrated in schizophrenic patients135, and a gender-specific association of interleukin 1 receptor antagonist polymorphism with schizophrenia susceptibility has been detected in African male population136.
The reports outlined above converge on the evidence that inflammatory mediators, and in particular IL-1β, may be central in the control of KCC2 expression. Of note, both environmentally- or genetically-induced elevations of this proinflammatory cytokines associate with pathological conditions, that frequently manifest in a gender-specific manner, characterized by altered KCC2 expression.
Through which processes do these environmental stimuli induce the described changes? Although the exact molecular mechanisms are far from being defined, few lines of evidence point to the possible occurrence of epigenetic modifications of the Kcc2 gene. For example, the early exposure to environmental xenobiotics (i.e. Bisphenol) can delay the developmental expression of KCC2, by increasing MeCP2 binding to the Kcc2 promoter, similarly to what observed in paradigms of prenatal immune activation64. Interestingly, the inhibition of DNA methyltransferase or Histone Deacetylase (HDAC) rescues the delay, suggesting the involvement of epigenetic factors in this process. Similarly, persistent inflammatory pain results in KCC2 suppression through HDAC-mediated histone hypoacetylation137, while the suppression of HDAC2 upon spinal cord mechanical hyperalgesia restores KCC2 expression and alleviates symptoms138. Interestingly, during infections, proinflammatory cytokines support the immune response through chromatin modifiers and epigenetic regulators, which contribute to the altered gene transcription patterns139. The possibility of a link among environmental cues, inflammation-derived molecules and epigenetic regulation of genes, including KCC2, is therefore very likely.
Reducing inflammation to enhance KCC2 expression?
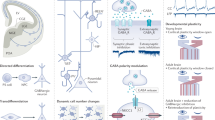
The above literature reports open the exciting scenario that inflammation and inflammatory cytokines contribute in controlling the expression of KCC2, a central actor in the developmental sequence at the basis of the inhibitory neurotransmission maturation. As demonstrated, IL-1β could reduce KCC2 expression via enhanced binding of the transcription factors MeCP2 and REST to its gene promoter region64. However, other mechanisms may contribute, such as the capacity of the proinflammatory cytokines to influence the release of soluble factors, like hormones, which are known to affect the GABA developmental shift71. Of note, immune hits, like stress and inflammation, could amplify the physiological differences of KCC2 detected among sexes, thus differently contributing to alterations of the GABAergic system maturation in males and females (Fig. 1).

KCC2 expression levels are regulated through the transcription factors REST and MeCP2, while KCC2 post-translational modifications control the transporter functionality and its insertion into the plasmamembrane (see text for details). Hormones and trophic factors are recognized soluble regulators of KCC2 expression. Stress occurring at either prenatal or early post-natal stages is a main environmental stimulus that reduces KCC2 expression, while exercise/environmental enrichment increase the transporter expression. Stress and exercise/EE likely control KCC2 expression through the modulation of inflammatory pathways. Inflammation, and in particular IL-1β -which is elevated by stress and reduced by exercise/EE-, lowers KCC2 expression levels, by acting through transcription factors or neurotrophic factors. REST (RE1-Silencing Transcription factor); MeCP2 (methyl-CpG-binding protein 2); DHT (dihydrotestosterone); T3 (triiodothyronine); WNK (with-no-lysine[K] family of serine-threonine kinases); SPAK (Ste20p-related proline/alanine-rich kinase). The figures were adapted and modified from Servier Medical Art (http://smart.servier.com/).
These findings could be exploited in the search of paradigms aimed to accelerate the GABA switch under pathological contexts. So far only bumetanide, a FDA-approved loop diuretic that acts by antagonizing NKCC1 and NKCC2, has been tested as a potential chloride homeostasis-restoring drug in the context of several diseases, including neonatal seizures, temporal lobe epilepsy, autism and schizophrenia, in preclinical rodent studies and in off-label clinical studies (for a review see140), as well as in adult mouse models of Down syndrome53 and DiGeorge syndrome (22q11.2 microdeletion)141. The evidence reported above, however, suggest that the acceleration of GABA switch could be achieved also through the exploitation of either pharmacological or not pharmacological strategies aimed at reducing inflammation. Although conceptually consistent, pharmacological approaches aimed at inhibiting cytokine-dependent signaling in the attempt to restore a normal KCC2 expression should be considered with caution. Indeed, it is known that several immune molecules, besides acting as inflammatory mediators, have crucial roles in physiological brain development142,143, hence their pharmacological modulation, especially during brain development, might result in untoward effects. Unlike pharmacological-based approach, exercise and environmental enrichment, may bring important contributions as non-invasive strategies aimed to restore GABAergic maturation (Fig. 1). Exercise modulates chloride homeostasis after spinal cord injury, returning toward normal KCC2 levels144,145 whose downregulation contribute to the development of spasticity and dysfunctions82,146. Similarly, environmental enrichment paradigms -i.e., the addition of social, physical and somatosensory stimulation able to increase synapse formation, plasticity, and neurogenesis147- enhance the GABAergic neurotransmission at one week after birth by accelerating the transition of GABA action from excitation to inhibition148.
Although the molecular processes by which exercise and environmental enrichment increase KCC2 levels are still to be defined, neurotrophins are likely to play a major role in this process. Exercise is recognized to upregulate expression of BDNF (for a review see149), which is a major determinant of KCC2 upregulation. Similarly, IGF-1, that is elevated in the visual cortex upon environmental enrichment, has been found to decrease the ratio between the expression of NKCC1 and KCC2, promoting the developmental switch of GABA polarity from excitation to inhibition150. Of note, physical exercise and environmental enrichment are also linked to the ability of preventing and modulating inflammatory conditions. Voluntary wheel-running in mice reduces the expression of key drivers of the cytokine cascade, including IL-1β151,152,153, while environmental enrichment reduces IL-1β and CD68 expression154, also modifying microglia toward a resting phenotype155. Interestingly, early environmental interventions normalize the immunological dysfunctions produced by prenatal restraint stress, reverting most of immunological alterations and reducing IL-1β levels156. Whether this paradigm may also rescue KCC2 expression under the same conditions is still to be defined.
Conclusions
Besides being genetically defined, KCC2 expression can be regulated by environmental stimuli, such as stress and inflammation (Fig. 1). The recent evidence that the chloride transporter expression is controlled by proinflammatory cytokines, alongside the engagement of inflammatory molecules upon stressful events, open the possibility that these immune molecules might represent the common molecular link between these stimuli and KCC2 alteration. IL-1β, that increases upon infection but also during prenatal stress and maternal deprivation, appears to reduce the transporter expression, delaying the GABA switch and resulting in abnormal brain development. Under this perspective, pharmacological or environmental paradigms aimed to reduce the inflammatory load could represent promising strategies to normalize KCC2 levels. This approach could be possibly attempted even in the context of developmental disorders consequent to prenatal or early postnatal environmental stressors, a medical situation which still completely lacks preventative pharmacological approaches.
References
Ben-Ari, Y. Is birth a critical period in the pathogenesis of autism spectrum disorders? Nat. Rev. Neurosci. 16, 498–505 (2015).
Depino, A. M. Perinatal inflammation and adult psychopathology: from preclinical models to humans. Seminars Cell Dev. Biol. 77, 104–114 (2018).
Hagberg, H. et al. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 11, 192–208 (2015).
McAdams, R. M. & Juul, S. E. The role of cytokines and inflammatory cells in perinatal brain injury. Neurol. Res. Int. 2012, 561494 (2012).
Knuesel, I. et al. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 10, 643–660 (2014).
Estes, M. L. & McAllister, A. K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 353, 772–777 (2016).
Boulanger-Bertolus, J., Pancaro, C. & Mashour, G. A. Increasing role of maternal immune activation in neurodevelopmental disorders. Front. Behav. Neurosci. 12, 230 (2018).
Stott, D. H. Follow-up study from birth of the effects of prenatal stresses. Dev. Med. Child Neurol. 15, 770–787 (1973).
Lautarescu, A. et al. Maternal prenatal stress is associated with altered uncinate fasciculus microstructure in premature neonates. Biol. Psychiatry 87, 559–569 (2020).
Walsh, K. et al. Maternal prenatal stress phenotypes associate with fetal neurodevelopment and birth outcomes. Proc. Natl. Acad. Sci. USA 116, 23996–24005 (2019).
Nugent, N. R., Tyrka, A. R., Carpenter, L. L. & Price, L. H. Gene-environment interactions: early life stress and risk for depressive and anxiety disorders. Psychopharmacology 214, 175–196 (2011).
King, S., Laplante, D. & Joober, R. Understanding putative risk factors for schizophrenia: retrospective and prospective studies. J. Psychiatry Neurosci. 30, 342–348 (2005).
Brannigan, R. et al. The role of prenatal stress as a pathway to personality disorder: longitudinal birth cohort study. Br J. Psychiatry 216, 85–89 (2020).
Ronald, A., Pennell, C. E. & Whitehouse, A. J. Prenatal maternal stress associated with ADHD and autistic traits in early childhood. Front. Psychol. 1, 223 (2010).
Penzes, P., Cahill, M. E., Jones, K. A., VanLeeuwen, J. E. & Woolfrey, K. M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293 (2011).
Bowling, H. & Klann, E. Shaping dendritic spines in autism spectrum disorder: mTORC1-dependent macroautophagy. Neuron 83, 994–996 (2014).
Bateup, H. S. et al. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522 (2013).
Nelson, S. B. & Valakh, V. Excitatory/inhibitory balance and circuit homeostasis in autism spectrum disorders. Neuron 87, 684–698 (2015).
Chattopadhyaya, B. & Cristo, G. D. GABAergic circuit dysfunctions in neurodevelopmental disorders. Front. psychiatry 3, 51 (2012).
Braat, S. & Kooy, R. F. The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86, 1119–1130 (2015).
Fine, R., Zhang, J. & Stevens, H. E. Prenatal stress and inhibitory neuron systems: implications for neuropsychiatric disorders. Mol. Psychiatry 19, 641–651 (2014).
Ben-Ari, Y. The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience 279, 187–219 (2014).
Pizzarelli, R. & Cherubini, E. Alterations of GABAergic signaling in autism spectrum disorders. Neural plasticity 2011, 297153 (2011).
Gao, F. et al. Impaired GABA neural circuits are critical for fragile X syndrome. Neural Plast. 2018, 8423420 (2018).
Chao, H. T. et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 468, 263–269 (2010).
Lozovaya, N. et al. Early alterations in a mouse model of Rett syndrome: the GABA developmental shift is abolished at birth. Sci. Rep. 9, 9276 (2019).
Contestabile, A., Magara, S. & Cancedda, L. The GABAergic hypothesis for cognitive disabilities in down syndrome. Front. Cell. Neurosci. 11, 54 (2017).
Schmidt, M. J. & Mirnics, K. Neurodevelopment, GABA system dysfunction, and schizophrenia. Neuropsychopharmacol 40, 190–206 (2015).
Ben-Ari, Y., Cherubini, E., Corradetti, R. & Gaiarsa, J. L. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J. Physiol. 416, 303–325 (1989).
Mueller, A. L., Taube, J. S. & Schwartzkroin, P. A. Development of hyperpolarizing inhibitory postsynaptic potentials and hyperpolarizing response to gamma-aminobutyric acid in rabbit hippocampus studied in vitro. J. Neurosci. 4, 860–867 (1984).
Blaesse, P., Airaksinen, M. S., Rivera, C. & Kaila, K. Cation-chloride cotransporters and neuronal function. Neuron 61, 820–838 (2009).
Kaila, K., Price, T. J., Payne, J. A., Puskarjov, M. & Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 15, 637–654 (2014).
Haas, M. The Na-K-Cl cotransporters. Am. J. Physiol. 267(4 Pt 1), C869–C885 (1994).
Gillen, C. M., Brill, S., Payne, J. A. & Forbush, B. Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J. Biol. Chem. 271, 16237–16244 (1996).
Hiki, K. et al. Cloning, characterization, and chromosomal location of a novel human K+-Cl- cotransporter. J. Biol. Chem. 274, 10661–10667 (1999).
Mount, D. B. et al. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J. Biol. Chem. 274, 16355–16362 (1999).
Payne, J. A., Stevenson, T. J. & Donaldson, L. F. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J. Biol. Chem. 271, 16245–16252 (1996).
Race, J. E. et al. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am. J. Physiol. 277, C1210–C1219 (1999).
Antrobus, S. P. & Lytle, C. & Payne, JA. K. +-Cl- cotransporter-2 KCC2 in chicken cardiomyocytes. Am. J. Physiol. Cell Physiol. 303, C1180–C1191 (2012).
Pearson, M. M., Lu, J., Mount, D. B. & Delpire, E. Localization of the K(+)-Cl(-) cotransporter, KCC3, in the central and peripheral nervous systems: expression in the choroid plexus, large neurons and white matter tracts. Neuroscience 103, 481–491 (2001).
Kanaka, C. et al. The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KCC1,2) and Na-K-2Cl cotransporter (NKCC1) in the rat nervous system. Neuroscience 104, 933–946 (2001).
Karadsheh, M. F., Byun, N., Mount, D. B. & Delpire, E. Localization of the KCC4 potassium-chloride cotransporter in the nervous system. Neuroscience 123, 381–391 (2004).
Uvarov, P. et al. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J. Biol. Chem. 282, 30570–30576 (2007).
Li, H., Tornberg, J., Kaila, K., Airaksinen, M. S. & Rivera, C. Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur. J. Neurosci. 16, 2358–2370 (2002).
Le Rouzic, P. et al. KCC3 and KCC4 expression in rat adult forebrain. Brain Res. 1110, 39–45 (2006).
Gagnon, K. B., Adragna, N. C., Fyffe, R. E. & Lauf, P. K. Characterization of glial cell K-Cl cotransport. Cell. Physiol. Biochem. 20, 121–130 (2007).
Rivera, C. et al. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397, 251–255 (1999).
Fiumelli, H. & Woodin, M. A. Role of activity-dependent regulation of neuronal chloride homeostasis in development. Curr. Opin. Neurobiol. 17, 81–86 (2007).
Markkanen, M. et al. Distribution of neuronal KCC2a and KCC2b isoforms in mouse CNS. J. Comp. Neurol. 522, 1897–1914 (2014).
Hyde, T. M. et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J. Neurosci. 31, 11088–11095 (2011).
Ben-Ari, Y. Excitatory actions of gaba during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739 (2002).
Chen, L. et al. KCC2 downregulation facilitates epileptic seizures. Sci. Rep. 7, 156 (2017).
Deidda, G. et al. Reversing excitatory GABAAR signaling restores synaptic plasticity and memory in a mouse model of Down syndrome. Nat. Med. 21, 318–326 (2015).
Moore, Y. E., Deeb, T. Z., Chadchankar, H., Brandon, N. J. & Moss, S. J. Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl Acad. Sci. USA 115, 10166–10171 (2018).
Represa, A. & Ben-Ari, Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 28, 278–283 (2005).
Schulte, J. T., Wierenga, C. J. & Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 90, 260–271 (2018).
Liu, X., Wang, Q., Haydar, T. F. & Bordey, A. Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci. 8, 1179–1187 (2005).
Behar, T. N., Schaffner, A. E., Scott, C. A., Greene, C. L. & Barker, J. L. GABA receptor antagonists modulate postmitotic cell migration in slice cultures of embryonic rat cortex. Cereb. Cortex 10, 899–909 (2000).
Cancedda, L., Fiumelli, H., Chen, K. & Poo, M. M. Excitatory GABA action is essential for morphological maturation of cortical neurons in vivo. J. Neurosci. 27, 5224–5235 (2007).
Akerman, C. J. & Cline, H. T. Refining the roles of GABAergic signaling during neural circuit formation. Trends Neurosci. 30, 382–389 (2007).
Wang, D. D. & Kriegstein, A. R. GABA regulates excitatory synapse formation in the neocortex via NMDA receptor activation. J. Neurosci. 28, 5547–5558 (2008).
Salmon, C. K. et al. Depolarizing GABA transmission restrains activity-dependent glutamatergic synapse formation in the developing hippocampal circuit. Front. Cell. Neurosci. 14, 36 (2020).
Yeo, M., Berglund, K., Augustine, G. & Liedtke, W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J. Neurosci. 29, 14652–14662 (2009).
Corradini, I. et al. Maternal immune activation delays excitatory-to-inhibitory gamma-aminobutyric acid switch in offspring. Biol. Psychiatry 83, 680–691 (2018).
Tang, X. et al. KCC2 rescues functional deficits in human neurons derived from patients with Rett syndrome. Proc. Natl Acad. Sci. USA 113, 751–756 (2016).
Ganguly, K., Schinder, A. F., Wong, S. T. & Poo, M. GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105, 521–532 (2001).
Heubl, M. et al. GABAA receptor dependent synaptic inhibition rapidly tunes KCC2 activity via the Cl(-)-sensitive WNK1 kinase. Nat. Commun. 8, 1776 (2017).
Rivera, C. et al. BDNF-induced TrkB activation down-regulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J. Cell Biol. 159, 747–752 (2002).
Rivera, C. et al. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J. Neurosci. 24, 4683–4691 (2004).
Ludwig, A. et al. Early growth response 4 mediates BDNF induction of potassium chloride cotransporter 2 transcription. J. Neurosci. 31, 644–649 (2011).
Galanopoulou, A. S. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res. 80, 99–113 (2008).
Galanopoulou, A. S. & Moshe, S. L. Role of sex hormones in the sexually dimorphic expression of KCC2 in rat substantia nigra. Exp. Neurol. 184, 1003–1009 (2003).
Westerholz, S., de Lima, A. D. & Voigt, T. Thyroid hormone-dependent development of early cortical networks: temporal specificity and the contribution of trkB and mTOR pathways. Front. Cell. Neurosci. 7, 121 (2013).
Tyzio, R. et al. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science 314, 1788–1792 (2006).
Tyzio, R. et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science 343, 675–679 (2014).
Eftekhari, S. et al. Response to Comment on “Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring”. Science 346, 176 (2014).
Leonzino, M. et al. The timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 15, 96–103 (2016).
Medina, I. et al. Current view on the functional regulation of the neuronal K(+)-Cl(-) cotransporter KCC2. Front. Cell. Neurosci. 8, 27 (2014).
Rinehart, J. et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell 138, 525–536 (2009).
Kelsch, W. et al. Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J. Neurosci. 21, 8339–8347 (2001).
Coull, J. A. et al. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438, 1017–1021 (2005).
Boulenguez, P. et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat. Med. 16, 302–307 (2010).
Watanabe, M. et al. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 12, eaaw9315 (2019).
Nunez, J. L., Bambrick, L. L., Krueger, B. K. & McCarthy, M. M. Prolongation and enhancement of gamma-aminobutyric acid receptor mediated excitation by chronic treatment with estradiol in developing rat hippocampal neurons. Eur. J. Neurosci. 21, 3251–3261 (2005).
Nunez, J. L. & McCarthy, M. M. Evidence for an extended duration of GABA-mediated excitation in the developing male versus female hippocampus. Dev. Neurobiol. 67, 1879–1890 (2007).
Veliskova, J. & Moshe, S. L. Sexual dimorphism and developmental regulation of substantia nigra function. Ann. Neurol. 50, 596–601 (2001).
Murguia-Castillo, J., Beas-Zarate, C., Rivera-Cervantes, M. C., Feria-Velasco, A. I. & Urena-Guerrero, M. E. NKCC1 and KCC2 protein expression is sexually dimorphic in the hippocampus and entorhinal cortex of neonatal rats. Neurosci. Lett. 552, 52–57 (2013).
Mir, F. R., Carrer, H. F. & Cambiasso, M. J. Sex differences in depolarizing actions of GABAA receptor activation in rat embryonic hypothalamic neurons. Eur. J. Neurosci. 45, 521–527 (2017).
Bale, T. L. et al. Early life programming and neurodevelopmental disorders. Biol. psychiatry 68, 314–319 (2010).
Huang, L. T. Early-life stress impacts the developing hippocampus and primes seizure occurrence: cellular, molecular, and epigenetic mechanisms. Front. Mol. Neurosci. 7, 8 (2014).
Gholipoor, P. et al. Prenatal stress potentiates febrile seizure and leads to long-lasting increase in cortisol blood levels in children under 2years old. Epilepsy Behav. 72, 22–27 (2017).
Aleman, A., Kahn, R. S. & Selten, J. P. Sex differences in the risk of schizophrenia: evidence from meta-analysis. Arch. Gen. Psychiatry 60, 565–571 (2003).
McCarthy, M. M., Nugent, B. M. & Lenz, K. M. Neuroimmunology and neuroepigenetics in the establishment of sex differences in the brain. Nat. Rev. Neurosci. 18, 471–484 (2017).
Loomes, R., Hull, L. & Mandy, W. P. L. What is the male-to-female ratio in autism spectrum disorder? a systematic review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 56, 466–474 (2017).
He, Q., Nomura, T., Xu, J. & Contractor, A. The developmental switch in GABA polarity is delayed in fragile X mice. J. Neurosci. 34, 446–450 (2014).
Duarte, S. T. et al. Abnormal expression of cerebrospinal fluid cation chloride cotransporters in patients with Rett syndrome. PloS ONE 8, e68851 (2013).
Tang, X. et al. Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 11, eaau0164 (2019).
Hinz, L., Torrella Barrufet, J. & Heine, V. M. KCC2 expression levels are reduced in post mortem brain tissue of Rett syndrome patients. Acta Neuropathol Commun. 7, 196 (2019).
Pisella, L. I. et al. Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal. 2, eaay0300 (2019).
Ruffolo, G. et al. A novel GABAergic dysfunction in human Dravet syndrome. Epilepsia 59, 2106–2117 (2018).
Ruffolo, G. et al. Functional aspects of early brain development are preserved in tuberous sclerosis complex (TSC) epileptogenic lesions. Neurobiol. Dis. 95, 93–101 (2016).
Tao, R. et al. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J. Neurosci. 32, 5216–5222 (2012).
Merner, N. D. et al. Regulatory domain or CpG site variation in SLC12A5, encoding the chloride transporter KCC2, in human autism and schizophrenia. Front. Cell. Neurosci. 9, 386 (2015).
Veerawatananan, B., Surakul, P. & Chutabhakdikul, N. Maternal restraint stress delays maturation of cation-chloride cotransporters and GABAA receptor subunits in the hippocampus of rat pups at puberty. Neurobiol. stress 3, 1–7 (2016).
Baek, H. et al. Altered expression of KCC2 in GABAergic interneuron contributes prenatal stress-induced epileptic spasms in infant rat. Neurochemistry Int. 97, 57–64 (2016).
Stevens, H. E., Su, T., Yanagawa, Y. & Vaccarino, F. M. Prenatal stress delays inhibitory neuron progenitor migration in the developing neocortex. Psychoneuroendocrinology 38, 509–521 (2013).
Kentrop, J. et al. Effects of maternal deprivation and complex housing on rat social behavior in adolescence and adulthood. Front. Behav. Neurosci. 12, 193 (2018).
Dalle Molle, R. et al. Associations between parenting behavior and anxiety in a rodent model and a clinical sample: relationship to peripheral BDNF levels. Transl. Psychiatry 2, e195 (2012).
Furukawa, M. et al. Neonatal maternal separation delays the GABA excitatory-to-inhibitory functional switch by inhibiting KCC2 expression. Biochemical Biophys. Res. Commun. 493, 1243–1249 (2017).
Hu, D. et al. Bumetanide treatment during early development rescues maternal separation-induced susceptibility to stress. Sci. Rep. 7, 11878 (2017).
Galanopoulou, A. S. Dissociated gender-specific effects of recurrent seizures on GABA signaling in CA1 pyramidal neurons: role of GABA(A) receptors. J. Neurosci. 28, 1557–1567 (2008).
MacKenzie, G. & Maguire, J. Chronic stress shifts the GABA reversal potential in the hippocampus and increases seizure susceptibility. Epilepsy Res. 109, 13–27 (2015).
Sarkar, J., Wakefield, S., MacKenzie, G., Moss, S. J. & Maguire, J. Neurosteroidogenesis is required for the physiological response to stress: role of neurosteroid-sensitive GABAA receptors. J. Neurosci. 31, 18198–18210 (2011).
Tsukahara, T., Masuhara, M., Iwai, H., Sonomura, T. & Sato, T. Repeated stress-induced expression pattern alterations of the hippocampal chloride transporters KCC2 and NKCC1 associated with behavioral abnormalities in female mice. Biochem. Biophys. Res. Commun. 465, 145–151 (2015).
Hewitt, S. A., Wamsteeker, J. I., Kurz, E. U. & Bains, J. S. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat. Neurosci. 12, 438–443 (2009).
Viviani, B. et al. Early maternal deprivation immunologically primes hippocampal synapses by redistributing interleukin-1 receptor type I in a sex dependent manner. Brain Behav. Immun. 35, 135–143 (2014).
Roque, A., Ochoa-Zarzosa, A. & Torner, L. Maternal separation activates microglial cells and induces an inflammatory response in the hippocampus of male rat pups, independently of hypothalamic and peripheral cytokine levels. Brain Behav. Immun. 55, 39–48 (2016).
Gur, T. L. et al. Prenatal stress affects placental cytokines and neurotrophins, commensal microbes, and anxiety-like behavior in adult female offspring. Brain Behav. Immun. 64, 50–58 (2017).
Danese, A., Pariante, C. M., Caspi, A., Taylor, A. & Poulton, R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc. Natl Acad. Sci. USA 104, 1319–1324 (2007).
Slopen, N. et al. Early origins of inflammation: an examination of prenatal and childhood social adversity in a prospective cohort study. Psychoneuroendocrinology 51, 403–413 (2015).
Andersson, N. W. et al. Influence of prenatal maternal stress on umbilical cord blood cytokine levels. Arch. Women’s Ment. Health 19, 761–767 (2016).
Hantsoo, L., Kornfield, S., Anguera, M. C. & Epperson, C. N. Inflammation: a proposed intermediary between maternal stress and offspring neuropsychiatric risk. Biol. Psychiatry 85, 97–106 (2019).
Fernandez, A. et al. The GABA developmental shift is abolished by maternal immune activation already at birth. Cereb. Cortex 29, 3982–3992 (2019).
Vezzani, A. & Baram, T. Z. New roles for interleukin-1 Beta in the mechanisms of epilepsy. Epilepsy Curr. 7, 45–50 (2007).
Dilena, R. et al. Therapeutic effect of Anakinra in the relapsing chronic phase of febrile infection-related epilepsy syndrome. Epilepsia Open 4, 344–350 (2019).
Kenney-Jung, D. L. et al. Febrile infection-related epilepsy syndrome treated with anakinra. Ann. Neurol. 80, 939–945 (2016).
Semple, B. D. et al. Interleukin-1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J. Neurosci. 37, 7864–7877 (2017).
Vargas, D. L., Nascimbene, C., Krishnan, C., Zimmerman, A. W. & Pardo, C. A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 57, 67–81 (2005).
Ziats, M. N. & Rennert, O. M. Sex-biased gene expression in the developing brain: implications for autism spectrum disorders. Mol. Autism 4, 10 (2013).
Krakowiak, P. et al. Neonatal cytokine profiles associated with autism spectrum disorder. Biol. psychiatry 81, 442–451 (2017).
Ashwood, P. et al. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 25, 40–45 (2011).
Suzuki, K. et al. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PloS one 6, e20470 (2011).
Soderlund, J. et al. Activation of brain interleukin-1beta in schizophrenia. Mol. Psychiatry 14, 1069–1071 (2009).
Papiol, S. et al. Interleukin-1 cluster is associated with genetic risk for schizophrenia and bipolar disorder. J. Med. Genet. 41, 219–223 (2004).
Katila, H., Hanninen, K. & Hurme, M. Polymorphisms of the interleukin-1 gene complex in schizophrenia. Mol. Psychiatry 4, 179–181 (1999).
Ben Nejma, M. et al. A gender-specific association of interleukin 1 receptor antagonist polymorphism with schizophrenia susceptibility. Acta Neuropsychiatrica 25, 349–355 (2013).
Lin, C. R., Cheng, J. K., Wu, C. H., Chen, K. H. & Liu, C. K. Epigenetic suppression of potassium-chloride co-transporter 2 expression in inflammatory pain induced by complete Freund’s adjuvant (CFA). Eur. J. Pain. 21, 309–321 (2017).
Stodberg, T. et al. Mutations in SLC12A5 in epilepsy of infancy with migrating focal seizures. Nat. Commun. 6, 8038 (2015).
Zhang, Q. & Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 19, 417–432 (2019).
Kharod, S. C., Kang, S. K. & Kadam, S. D. Off-label use of bumetanide for brain disorders: an overview. Front. Neurosci. 13, 310 (2019).
Amin, H., Marinaro, F., De Pietri Tonelli, D. & Berdondini, L. Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics. Sci. Rep. 7, 15752 (2017).
Bauer, S., Kerr, B. J. & Patterson, P. H. The neuropoietic cytokine family in development, plasticity, disease and injury. Nat. Rev. Neurosci. 8, 221–232 (2007).
Boulanger, L. M. Immune proteins in brain development and synaptic plasticity. Neuron 64, 93–109 (2009).
Beverungen, H., Klaszky, S. C., Klaszky, M. & Cote, M. P. Rehabilitation decreases spasticity by restoring chloride homeostasis through the brain-derived neurotrophic factor-KCC2 pathway after spinal cord injury. J. Neurotrauma 37, 846–859 (2020).
Cote, M. P., Gandhi, S., Zambrotta, M. & Houle, J. D. Exercise modulates chloride homeostasis after spinal cord injury. J. Neurosci. 34, 8976–8987 (2014).
Chen, B. et al. Reactivation of dormant relay pathways in injured spinal cord by KCC2 manipulations. Cell 174, 521–35. e13 (2018).
Sale, A., Berardi, N. & Maffei, L. Enrich the environment to empower the brain. Trends Neurosci. 32, 233–239 (2009).
He, S., Ma, J., Liu, N. & Yu, X. Early enriched environment promotes neonatal GABAergic neurotransmission and accelerates synapse maturation. J. Neurosci. 30, 7910–7916 (2010).
Zoladz, J. A. & Pilc, A. The effect of physical activity on the brain derived neurotrophic factor: from animal to human studies. J. Physiol. Pharmacol. 61, 533–541 (2010).
Baroncelli, L. et al. Early IGF-1 primes visual cortex maturation and accelerates developmental switch between NKCC1 and KCC2 chloride transporters in enriched animals. Neuropharmacology 113, 167–177 (2017).
Nilsson, M. I. et al. Lifelong aerobic exercise protects against inflammaging and cancer. PloS ONE 14, e0210863 (2019).
Dallagnol, K. M. C. et al. Running for REST: physical activity attenuates neuroinflammation in the hippocampus of aged mice. Brain Behav. Immun. 61, 31–35 (2017).
Gomes da Silva, S. et al. Maternal exercise during pregnancy increases BDNF levels and cell numbers in the hippocampal formation but not in the cerebral cortex of adult rat offspring. PloS ONE 11, e0147200 (2016).
Birch, A. M. & Kelly, A. M. Lifelong environmental enrichment in the absence of exercise protects the brain from age-related cognitive decline. Neuropharmacology 145, 59–74 (2019).
Xu, H. et al. Environmental enrichment potently prevents microglia-mediated neuroinflammation by human amyloid beta-protein oligomers. J. Neurosci. 36, 9041–9056 (2016).
Laviola, G. et al. Beneficial effects of enriched environment on adolescent rats from stressed pregnancies. Eur. J. Neurosci. 20, 1655–1664 (2004).
Acknowledgements
Work in our lab is supported by the following organizations: PRIN (Progetti di Rilevante Interesse Nazionale) 2017A9MK4R, FISM (Fondazione dell’Associazione Italiana Sclerosi Multipla), Regione Lombardia “NeOn” ID239047, CARIPLO Foundation 2017-0886 and 2019-1973 and Telethon Foundation GGP19226A.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pozzi, D., Rasile, M., Corradini, I. et al. Environmental regulation of the chloride transporter KCC2: switching inflammation off to switch the GABA on?. Transl Psychiatry 10, 349 (2020). https://doi.org/10.1038/s41398-020-01027-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41398-020-01027-6
This article is cited by
-
Preservation of KCC2 expression in axotomized abducens motoneurons and its enhancement by VEGF
Brain Structure and Function (2023)
-
Severe inflammation in new-borns induces long-term cognitive impairment by activation of IL-1β/KCC2 signaling during early development
BMC Medicine (2022)
-
The immuno-behavioural covariation associated with the treatment response to bumetanide in young children with autism spectrum disorder
Translational Psychiatry (2022)
