
Abstract
Metastatic dissemination of solid tumors, a leading cause of cancer-related mortality, underscores the urgent need for enhanced insights into the molecular and cellular mechanisms underlying metastasis, chemoresistance, and the mechanistic backgrounds of individuals whose cancers are prone to migration. The most prevalent signaling cascade governed by multi-kinase inhibitors is the mitogen-activated protein kinase (MAPK) pathway, encompassing the RAS–RAF–MAPK kinase (MEK)–extracellular signal-related kinase (ERK) pathway. RAF kinase is a primary mediator of the MAPK pathway, responsible for the sequential activation of downstream targets, such as MEK and the transcription factor ERK, which control numerous cellular and physiological processes, including organism development, cell cycle control, cell proliferation and differentiation, cell survival, and death. Defects in this signaling cascade are associated with diseases such as cancer. RAF inhibitors (RAFi) combined with MEK blockers represent an FDA-approved therapeutic strategy for numerous RAF-mutant cancers, including melanoma, non-small cell lung carcinoma, and thyroid cancer. However, the development of therapy resistance by cancer cells remains an important barrier. Autophagy, an intracellular lysosome-dependent catabolic recycling process, plays a critical role in the development of RAFi resistance in cancer. Thus, targeting RAF and autophagy could be novel treatment strategies for RAF-mutant cancers. In this review, we delve deeper into the mechanistic insights surrounding RAF kinase signaling in tumorigenesis and RAFi-resistance. Furthermore, we explore and discuss the ongoing development of next-generation RAF inhibitors with enhanced therapeutic profiles. Additionally, this review sheds light on the functional interplay between RAF-targeted therapies and autophagy in cancer.
Similar content being viewed by others

Introduction
The mitogen-activated protein kinase (MAPK) pathway transmits extracellular signals from the membrane to intracellular destinations and is involved in various biological functions.1 The MAPK pathway is dysregulated in many RAS-associated cancers. RAS mutations result in the constitutive activation of the MAPK pathway, leading to uncontrolled cell proliferation and resistance to apoptosis-inducing drugs.2,3 Although many RAS inhibitors have been isolated and studied, the development of drugs targeting RAS is limited by a lack of well-defined druggable nooks and cavities on the RAS surface.4 However, interrupting signals between RAS and downstream effectors, such as the RAF–MAPK kinase (MEK)–extracellular signal-related kinase (ERK) pathway, could represent a new therapeutic strategy for RAS-driven cancers.5,6,7
The RAF protein family consists of three serine (Ser)/threonine (Thr) kinases (ARAF, BRAF, and CRAF) that act as mediators between membrane-bound RAS-GTPases and downstream kinases, such as MEK and ERK, in the MAPK signaling pathway.8 RAF proteins coordinate various cellular responses by regulating cytoplasmic and nuclear activities, such as cell cycle progression, proliferation, metabolism, migration, differentiation, and apoptosis.9,10 RAF is highly conserved in mammals, and RAF mutations are associated with many human cancers, including melanoma, breast cancer, ovarian cancer, colon cancer, thyroid cancer, and prostate cancer.11 Mutations in BRAF and RAS that dysregulate MAPK signaling are strongly associated with human malignancies.12 All members of the RAF family interact with RAS; however, this contact alone is insufficient to activate RAF. For example, several RAS mutants, such as RASV12Y32F and RASV12T35S are insufficient to activate RAF in vitro, suggesting that RAF kinase activation requires other factors.13 A recent study indicates that the activation of RAF necessitates dimerization, and exploring RAF activation is currently being viewed as a potential target for therapeutic intervention in several clinical contexts, including diverse cancer types.10
Numerous RAF inhibitors are considered potential therapeutic agents, eliciting high levels of responses in various RAF-mutant carcinomas.14,15 However, single-agent therapies targeting RAF have not resulted in significant long-term survival benefits due to the frequent development of drug resistance, often associated with mutational changes in MAPK components that result in the reactivation of the MAPK pathway.16,17 Combination therapeutic strategies using both RAF and MEK inhibitors may represent a more effective treatment strategy in patients with advanced or metastatic RAF-mutant carcinomas.18,19,20,21 Although this approach has demonstrated potential efficacy in preclinical studies, clinical testing has not demonstrated durable responses, and a single-arm study demonstrated that this strategy is associated with a predictable pattern of adverse effects due to the substantial inhibition of multiple paralogs.22,23 Identifying key downstream signals in the MAPK pathway is essential for minimizing paralog redundancy and cascade interactions, which may limit both the cancerous activity of RAF and drug toxicity in normal cells.
Autophagy, an intracellular catabolic process, may assist cancer cells in evading from RAFi, as many RAFi-resistant cells exhibit enhanced autophagic activity.24,25,26 Both preclinical and clinical data suggest that inhibiting both autophagy and MAPK pathway activity may serve as a novel and effective treatment strategy for BRAF and KRAS-mutant cancers.24,25,27 In particular, Chih-Shia Lee’s group showed that targeting both RAF and autophagy genes results in the best therapeutic outcomes. The inhibition of BRAF or CRAF, together with ATG7 inhibition, was found to be a viable treatment strategy for RAS-driven tumors.27 Understanding the mechanisms underlying RAFi-induced autophagy in the setting of recurrent somatic genetic alterations and RAF mutations could offer a “precision medicine” paradigm for diagnosing and treating tumors, including RAF-mutant tumors. This review focuses on potentially unique therapeutic approaches that target the basic components of RAF signaling and autophagy in RAS-dependent and RAS-independent cancers.
The discovery and major developments of RAS/RAF/MAPK in health and disease
The RAF/RAF/MEK/ERK signaling cascade is a well-established MAPK pathway in cell biology that governs several crucial cellular processes such as development, differentiation, proliferation, and death.28 With this cascade, various isoforms of RAS, RAF, MEK, and ERK exhibit differences in efficacy, function, and, notably, carcinogenic potential.
Our understanding of oncogenic potential began with the discovery of the highly carcinogenic Harvey murine sarcoma virus29 in 1964 and the Kirsten murine sarcoma virus30 in 1967. In the late 1970s and early 1980s, groundbreaking studies by Scolnick and colleagues identified the cellular origins of viral H-RAS and K-RAS genes31, and an avian homolog MH2 retrovirus32 in 1984, and an avian homolog MH2 retrovirus32 in 1984, respectively. Later, these two oncogenes are known as the first rapidly accelerated fibrosarcoma (RAF) gene with serine/threonine kinase activity.33 Subsequently, Raf-1 gene product (named as CRAF), a cellular counterpart of v-Raf, and other cellular counterparts such as C-Raf-1 and C-Raf-2 genes were cloned and sequenced in 1985.34 ARAF and BRAF, two additional members of the RAF family, were reported in 1986 and 1988, respectively.35 In 1988, MAPK, initially named microtubule-associated protein-2 protein kinase (MAP-2 kinase), was identified in mammalian cells36, and subsequently in yeast cells.37 MEK (mitogen-activated protein kinase kinase) and ERK (mitogen-activated protein kinase), both cytoplasmic protein kinases activated by mitogens, were discovered in the 1990s38. Further, RAF protein was functionally identified as a direct MEK activator39 in 1992 and a RAS effector in 1993.40 These findings marked the beginning of the MAPK cascade era, where it become evident that MAPK kinase signaling cascades play a pivotal role in initiating proliferative and oncogenic activities.41 In 2005, the U.S. Food and Drug Administration (FDA) approved Nexavar (Sorafenib), an oral multi-kinase inhibitor targeting the MAPK pathway, for the treatment of hepatocellular carcinoma (HCC), renal cell carcinoma (RCC), and thyroid carcinoma (TC).42 Subsequently, Sorafenib was proposed as a MAPK pathway inhibitor for malignant peripheral nerve sheath tumors (MPNSTs).43 Vemurafenib (Zelboraf), a potent BRAFV600E mutant inhibitor, was synthesized in early 2005 and received FDA approval for the treatment of metastatic and late-stage melanoma in 2011.44 Following this, FDA approval were granted for two BRAF inhibitors, Dabrafenib (Tafinlar) in 2013 and Encorafenib (Braftovi) in 2018. Trametinib (Mekinist) was approved in 2013 as a single-agent oral treatment for unresectable or metastatic melanoma in adult patients with BRAFV600E or BRAFV600K mutations.45 From 2014 to 2023, Trametinib, in combination with Dabrafenib, received FDA approval for the treatment of various solid tumors, including melanoma, non-small cell lung cancer (NSCLC), anaplastic thyroid cancer (ATC), and, low-grade glioma (LGG).46 In addition, two MEK inhibitors, Cotellic (Cobimetinib) and Mektovi (Binimetinib) were approved in 2015 and 2018, respectively, for the treatment of melanoma, either as a single-agent or in combination with other MAPK inhibitors.47 Ongoing preclinical and clinical investigations underscore the potential of the RAS/RAF/MAPK pathway as a significant therapeutic target, particularly in the era of precision medicine, with a focus on combination treatments.48 For example, a CRISPR/cas9 gene deletion study in lung cancer cells revealed that the deletion of KEAP1, in the presence of specific RAS/RAF/MAPK pathway inhibitors, alters cell metabolism and enables cells to proliferate without MAPK signaling.49 These major milestones are depicted in Fig. 1.

Historical events of the discovery and development of the RAS/RAF/MAPK pathway in health and diseases. The journey of the MAPK signal cascade commenced in 1960s with the groundbreaking discovery of the viral RAS gene. Subsequently, in 1992, the identification of RAF as both an upstream kinase activator of MEK and a RAS effector marked significant milestones. These pivotal findings culminated in the comprehensive definition of the entire MAPK signaling pathway. Over time, the MAPK signal emerged as a critical components in the development of therapeutic strategies for combating cancer. Each of these major milestones in the RAS/RAF/MAPK discovery is represented within its respective box. This figure was created with BioRender.com
Beyond cancer, ongoing studies aim to develop new treatments targeting RAS/RAF/MAPK for various disorders, including neurological, developmental, and metabolic diseases. Neurological disorders such as autism spectrum disorder (ASD), Parkinson’s disease (PD), Alzheimer’s disease (AD), Cardio-Facio-cutaneous (CFC), and Noonan syndromes (NS) have been linked to abnormalities in the regulation of the MAPK signaling pathway.50 Moreover, the RAS/RAF/MAPK pathway is gaining attention as a potential target for the development of novel anti-inflammatory drugs, with implications for conditions like rheumatic arthritis (RA)51, inflammatory bowel disease (IBD)52 and pulmonary fibrosis (PF).53
MAPK signaling
MAPKs are Ser/Thr kinases that play various roles in cellular responses to stimuli, including mitogens, osmotic stress, heat shock, and proinflammatory cytokines. MAPKs are involved in many cellular processes, such as proliferation, gene expression, differentiation, mitosis, cell survival, and apoptosis.54 Mammals possess four primary MAPKs: (1) ERK1/2, (2) c-Jun N-terminal kinase (JNK)1–3, (3) p38, and (4) ERK5.55,56,57 In addition to these four primary MAPKs, numerous atypical MAPKs (e.g., ERK 3/4, ERK 7/8, and Nemo-like kinase [NLK]) have been identified with less well-defined roles and unique mechanisms of activation.58,59,60 MAPK cascades consist of a signaling relay that is partially regulated by phosphorylation and typically involves three consecutive protein kinases: MEK kinase (MAPKKK), MEK, and MAPK (Fig. 2). MAPK cascades are activated by cell-surface receptors via cytoplasmic signaling proteins, and these signaling pathways are often dysregulated in human cancers. ERK1 and ERK2 are frequently investigated by researchers worldwide due to their critical roles in cell proliferation and survival. The JNK and p38 MAPK pathways primarily play roles in responding to cellular stress and regulating apoptosis. In contrast, the most extensively studied RAS/RAF/MAPK pathway holds a central position in governing cell proliferation and differentiation, serving as a vital component of the cellular signal transduction network. Consequently, proteins involved in the RAS/RAF/MAPK cascade have frequently been targeted in cancer drug discovery, leading to the clinical development of protein kinase inhibitors.61

Mitogen-activated protein kinase (MAPK) cascades and their physiological functions. All cascades consist of three-layered core-signaling pathways in which each kinase is consecutively activated, and MAPK components are highly conserved. The first layer consists of MAPK kinase kinases (MAPKKKs or MEKKs), which are activated by stimuli and phosphorylate and activate MAPK kinases (MAPKKs or MEKs). MAPKKs are dual-specificity kinases that can phosphorylate threonine or tyrosine residues to activate the terminal serine/threonine MAPK, leading to the activation of multiple cytoplasmic and nuclear proteins involved in various biological functions. This figure was created with BioRender.com
RAS/RAF/MAPK signaling: structure, upstream activator and downstream effectors
The ERK (MAPK) kinase plays a pivotal role within the RAS/RAF/MAPK signal transduction pathway, exerting control over various facets of cellular metabolism in cancer cells.62,63 Consequently, when it comes to development of anticancer drugs, the focus largely centers on three key upstream regulators and ERK protein in the ERK pathway: RAS (upstream activator of RAF), RAF (direct effector of RAS and activator of MEK), MEK (functioning as MAPKK), and ERK (as MAPK).
RAS/RAF/MAPK can be activate via two pathways: (1) a ligand-dependent pathway, in which ligands, such as growth factors, hormones, or cytokines, physically engage with receptors; and (2) a ligand-independent pathway, in which signaling is induced by a physical stressor, such as radiation, injury, or osmotic pressure.59 Aberrant RAF activation or mutations in upstream activators such as RAS or receptor tyrosine kinases (RTKs) can contribute to the development of malignancies in humans.64 Beyond RAS mutations, disruptions of RAS upstream components can also impact RAF activation. Receptors engaged by various growth factors, including TGF-α, EGF, VEGF, and platelet-derived growth factor-beta (PDGF-β), can instigate the canonical RAS–RAF–MEK–ERK pathway during various biological processes (Fig. 3a).65,66,67 Further investigations into RAF activation shed light on critical molecular mechanisms underlying cell proliferation, survival, and metastasis in cancer, particularly through the influence of the EGF receptor (EGFR) and small RAS-GTPases.68 Consequently, extensive research is currently underway to target RAF kinase as a promising avenue for the development of anticancer drugs.68,69

Structure and activation mechanism of RAS and RAF kinase in the RAS/RAF/MAPK signal cascade. a RAS upstream components. Various mitogens including TGF-α, EGF, VEGF, and PDGF-β bind to their own receptors and lead to RAS activation and subsequent stimulation of the MAPK pathway. b GTPase cycle. GEFs stimulates the transition of inactive RAS-GDP to active RAS-GTP, enabling to transmit the proliferation and differentiation signals through its downstream effectors. Subsequently, the active RAS can be quickly deactivated by the action of GAPs. c RAS domain. The effector lobe (1–86 a.a.), allosteric lobe (87–165 a.a.), and HVR (167–188/189 a.a.) are all parts of the structure of RAS proteins. The effector lobe contains switches I (30–40 a.a.) and II (60–76 a.a.) are involved in effector binding and GEF or GAP binding, respectively. d RAF domain structure. RAF proteins consist of three conserved regions (CR1, CR2, and CR3) or two functional domains: an N-terminal regulatory domain and a C-terminal catalytic domain. e RAF dimerization. In the absence of cellular stimulation, RAF tends to exist in the monomeric, autoinhibited state. Upon stimulation by RAS-GTP, the autoinhibitory domain is released, freeing the inactive kinase domain to form homo- or heterodimers (with kinase suppressor of RAS [KSR]). Dimerization triggers mutual phosphorylation of the dimer components, fully activating the kinase. Phosphorylation and activation of target proteins, such as MEK1 and MEK2, propagates the MAPK cascade, leading to ERK1/ERK2 activation. This figure was created with BioRender.com
RAS
RAS, a pivotal upstream protein in the RAF/MAPK pathway, holds the distinction of being the founding member of the extensive RAS superfamily of small GTPases.70 RAS is extremely prevalent, as RAS mutations are detected in approximately 30% of all tumors. RAS activity varies across different cancer types. For example, NRAS is activated in lymphoid and myeloid malignancies, whereas KRAS is highly elevated in colon and pancreatic cancers, and HRAS activity is upregulated in bladder and kidney cancers. This upregulation of RAS activity within the context of cancer leads to the dysregulation of downstream protein kinase activities.
RAS structures and activations mechanism
RAS activation is triggered by various extracellular stimuli, with the primary mechanism involving the formation of complexes comprising autophosphorylated growth factor receptors, the adapter protein GRB2 and the exchange factor SOS.71 It has been proposed that RAS dimerization plays a critical role in facilitating RAS signal transmission, directing influencing RAF activation.72
In their normal and resting states, RAS proteins exist in an inactive GDP-bound form.73 Guanine nucleotide exchange factors (GEFs), like SOS, are recruited to the plasma membrane following the stimulation of mitogenic growth factors (Fig. 3b). Once GEFs bind to RAS, the stability of nucleotide binding disrupted, leading to the release of GDP from RAS and the transient formation of a nucleotide-free state. This, in turn, activates RAF and other downstream targets recruited by RAS-GTP. The signaling from RAS is terminated by the hydrolysis of GTP, which is mediated by the intrinsic enzymatic activity of RAS. Mammalian cells typically express three GEFs that are recognized as RAS activators: SOS, RASGRF, and RASGRP.74 Some RAS-related malignancies have been associated with GAPs, including NF1, p120GAP/RASA1, SynGAP/RASA5, GAP1 family, DAB2IP, and GAPVD174. The conformational changes that accompany GTP hydrolysis are critical for RAS to function as a molecular switch in signaling pathways.75 There are four isomers of RAS genes, including KRAS4A, KRAS4B, NRAS, and HRAS. These isomers exhibit relatively consistent sequences or structures, encompassing N-terminal G-domains (1–166 a.a.) and C-terminal hypervariable regions (HVR) (166-189 a.a.).76 The G-domain conveys signals to downstream RAS effectors, featuring switch 1 (30–40 a.a.), switch 2 (60–76 a.a.), and a P loop (10–17a.a.) (Fig. 3c).77,78 The C-terminal region (final four amino acids, CAAX) undergoes posttranslational modification like iso-prenylation, proteolysis, and methylation, facilitating RAS localization and attachment to the membrane.77
RAF
RAF proteins, part of serine/threonine kinase family encoded by the RAF gene, serve as the upstream activator of MAPK and direct effector of RAS. In mammalian cells, there exist three distinct RAF proteins; ARAF, BRAF, and CRAF (also known as RAF-1). The initial member of the RAF family, CRAF, was initially characterized as an oncogene. Fusion of the CRAF catalytic domain with the retroviral Gag protein results in constitutive RAF kinase activation.79 Subsequently, two additional RAF family proteins, ARAF and BRAF, were discovered, each demonstrating similar function to that of CRAF.32,80,81,82 CRAF forms interactions with MEK, a dual-specificity kinase responsible for activating ERK.
RAF structure and activation mechanisms
The MAPK pathway is tightly regulated by several activation steps. The physical interaction between the RAF regulatory domain and membrane-bound RAS results in the attachment of RAF to the membrane, dephosphorylation, a conformation change for the kinase domain, and the subsequent phosphorylation of active sites (Ser338 and Tyr341).83 Many modulators mediate the negative or positive regulation of RAF activity through the formation of signaling complexes, which play critical roles in cancer growth and progression.83,84 To date, approximately 30 RAF-interacting proteins have been identified as putative RAF regulators.83
CRAF activation is a complex process in which Ser338, Tyr340, and Tyr341 are phosphorylated in response to oncogenes and growth factors. Many up- and downstream RAF effectors are associated with cancer transformation. Although the exact mechanisms underlying CRAF regulation remain unclear, phosphorylation of Ser338 and Tyr341 have been identified as crucial regulators of RAF kinase activity.85 In humans, all three RAF proteins are activated by phosphorylation at shared, conserved residues. Direct interaction between RAS and the N-terminal regulatory domain of CRAF is essential for RAF activation, and RAS mutations that cause constitutive RAF activation are detected in more than 30% of all human malignancies.86,87,88 However, RAS interaction alone is not sufficient to activate CRAF in vitro, indicating that other biochemical activities are necessary for RAF activation (Table 1). Some RAS mutants, including V12Y32F and V12T35S, are incapable of RAF activation but are able to interact with members of the RHO GTPase family.13 Previous studies have shown that p21-activated kinase (PAK) family members serve as molecular linkers, connecting RAS with RHO GTPases, including RAC and CDC42.89 In addition, CK2, JNK2, or SRC may be involved in CRAF activation through either RAS-dependent or RAS-independent mechanisms.90,91,92
RAF proteins do not possess inherent subcellular localization motifs, and initially, they are within the cytoplasm in an inactive monomeric form.93 The activation of RAF, transitioning it from its autoinhibited, pre-signaling, and inactive state, necessitates a series of regulatory steps. These include the relief of autoinhibition, the formation of dimers or higher-order multimers, and phosphorylation.
Autoinhibition of the pre-signaling, inactive state
All RAF family members contain three conserved regions (CR1, CR2, and CR3) and two functional domains: an N-terminal regulatory domain and a C-terminal catalytic domain. The N-terminal regulatory domain contains both CR1, composed of a RAS-binding domain (RBD) and a cysteine-rich domain (CRD), and CR2, which is enriched in Ser/Thr residues, whereas CR3 is located at the C-terminal domain (Fig. 3d). RAF activation is largely accomplished through the removal of inhibitory enforcement at the RAF catalytic domain. The N-terminal regulatory region interacts with the kinase domain, leading to RAF autoinhibition, a fundamental regulatory mechanism shared by all three RAF proteins (ARAF, BRAF, and CRAF). However, both ARAF and CRAF require additional steps to achieve maximal activity, such as the phosphorylation of activating residues and the dephosphorylation of negative regulatory residues.94
Autoinhibition relief
In the absence of cellular stimuli, RAF proteins exist in a monomeric, autoinhibited, inactive form. Activation of the RAF kinase domain requires the relief of N-terminal autoinhibition, which is accomplished through a series of events, including a change in the subcellular localization, protein–protein interactions, lipid interactions, and regulatory phosphorylation.95,96 RAF activation first requires the translocation of RAF from the cytosol to the plasma membrane, which represents a vital step. Experiments have shown that retaining RAF on the plasma membrane results in the constitutive activation of RAF in a RAS-independent manner.97,98 The RAF RBD interacts with the GTP-bound RAS effector domain by adopting a conserved, ubiquitin-like structure,99,100 and RAS binding with the RAF CRD (zinc-coordinated structure) can relocate RAF to phosphatidylserines in the plasma membrane regardless whether RAS is bound to GTP.101,102,103,104 However, both the RBD and the CRD are involved in the full activation of RAF.
Dimerization and activating phosphorylation
RAS engagement on the membrane increases the phosphorylation of the RAF kinase domain and RAF dimerization. Recent work indicates that RAF dimerization is necessary for RAS-dependent RAF kinase activity and correlates with the pathogenic role of disease-associated mutant RAF, which displays strong intrinsic kinase activity.105 The formation of the side-by-side RAF dimer involves a structural association between the N- and C-terminal regions of the kinase domain.106 Following the release of inhibitory domain from the complex, the RAF kinase domain readily forms RAF–RAF homodimers, subsequently leading in kinase activation.107 The RAF-related pseudo-kinase KSR (kinase suppressor of RAS) also participates in forming side-to-side heterodimers with RAF (RAF-KSR heterodimer). Activated RAF kinase phosphorylates target proteins, such as MEK1 and MEK2, leading to the subsequent activation of ERK1 and ERK2 (Fig. 3e). By contrast, inhibitory phosphorylation of the RAF hinge region can disrupt and inactivate dimeric structures. Hyperphosphorylated RAF proteins are recycled to an inactive state, ready to receive a new round of activating signals.108,109,110
RAF and MEK1 activity can also be regulated by activated ERK via a feedback loop. Phosphorylation regulates the activities of RAF, MEK1, and ERK depending on the phosphorylation site.111,112,113 Upon signal engagement, active RAS promotes the exchange capacity of son of sevenless (SOS) through a positive feedback loop, eventually activating ERK1/2. By contrast, ERK-dependent SOS phosphorylation and disassociation of the SOS-Grb2 complex prevents RAS activation through a negative feedback loop.114,115 Therefore, the phosphorylation of target proteins in the ERK pathway can regulate associated signaling pathways based on the functional location of target proteins.116,117 Improved understanding of the RAS–RAF axis and RAF dimerization has revealed the roles played by RAF in many cellular conditions. In patients with cancer, RAF homo- and heterodimers likely mediate cellular responses to ATP-competitive inhibitors and cancer progression, suggesting that RAF dimers may represent potential therapeutic targets.
MEK and ERK
MEK represents a family of protein kinases that possess dual-specificity for tyrosine and serine/threonine residues, facilitating the activation of ERK by phosphorylating regulatory Tyr and Thr sites. Upon interaction of the catalytic VIII sub-region of RAF with MEK via its C-terminal catalytic domain, a serine residue becomes phosphorylated, thereby initiating MEK activation. The primary targets for phosphorylation by activated RAF are dual-specificity kinases, such as MEK1 and MEK2, with molecular weights of 44 and 45 kDa, respectively.83 Subsequent to MEK-dependent phosphorylation, ERK is set into action, triggering a range of functional responses in cells in response to growth factors or stressors. These responses are mediated by various cytoplasmic and nuclear substrates, including transcription factors.118,119,120
ERK (MAPK), a Ser/Thr protein kinase, occupies a crucial position in the cellular signal transduction network, and any aberrations in its activation faults can significantly impact cellular functions. When activated, MEK directly interacts with ERKs via its N-terminal region. In situations where multiple kinases are at work, they catalyze the bispecific phosphorylation of Tyr and Thr residues within the 8 “TEY box” of the sub-functional region of ERK, thereby activating ERK. Activated ERKs subsequently translocate to the nucleus, where they increase the phosphorylation of target proteins in the cytoplasm or regulate the activity of other protein kinases. This occurs before further phosphorylation and dimerization of ERK in response to signals that promote ERK activation.121
ERK downstream signals
Several ERK1/2 target proteins are ubiquitously found in cells.60 Including the cytoplasmic substrates death-associated protein kinase (DAPK), tuberous sclerosis complex 2, RSK, and MNK, and the nuclear transcription factor substrates nuclear factor of activated T cells (NF-AT), Elk-1, myocyte enhancer factor 2 (MEF2), c-Fos, c-Myc, and signal transducer and activator of transcription (STAT3). Some membrane-associated proteins (e.g., CD120a, Syk, and Calnexin) and cytoskeleton proteins (e.g., Neurofilaments and Paxillin) are also directly phosphorylated by ERK1/2.
Other MAPK pathway
As summarized in Fig. 2, other several classical and atypical pathways and their related proteins are regulated by MAPKs.
The p38 signaling pathway
The p38 is reliably activated by a wide range of environmental stressors and inflammation and, in some cell types, by insulin and growth hormones. The p38 pathway is regulated by apoptosis-related receptors and physical sensors, including CDC42, RAC1, and mammalian Ste20-like kinases (MSTs), which also regulate JNK, resulting in the phosphorylation of the activation loop of MEK3/6. In particular, RAC1, a small G protein, controls the activation of p38 MAPK by a retinoic acid–induced beta1 integrin. p38 isoforms phosphorylate a wide range of cytoplasmic (e.g., cPLA2, MNK1/2, MK2/3, HuR, Bax, and Tau) and nuclear proteins (e.g., ATF1/2/6, MEF2, Elk-1, GADD153, Ets1, p53, and MSK1/2).122 p38 signaling is involved in immunological and inflammatory responses,123 cell fate determinants, and other stress responses124. Three anticancer compounds (e.g., SB203580, SB202190, and BIRB0796) specifically inhibit p38 isoforms (p38α and p38β) by competing with ATP in the binding pocket.125
The JNK pathway
The JNK pathway responds strongly to cytokines, growth factor deprivation, intracellular stimuli (e.g., DNA damage, cytoskeletal changes, oxidative, and ER stress), and extracellular stressors (e.g., UV radiation and osmotic stress). The JNK cascade is activated by adapter proteins in the TNF receptor-associated factor (TRAF) family, such as TRAF6, which is involved in the IL-1–induced activation of JNK.126 Two ATP-competitive JNK inhibitors, SP600125 (also known as JNK inhibitor II)127 and AS601245 (JNK inhibitor V),128 have been widely employed in the cancer research, although they exhibit low specificity. Many transcriptional factors (e.g., c-Jun, p53, ATF-2, NF-ATc1, Elk-1, HSF-1, STAT3, c-Myc, and JunB) are also regulated by JNK-directed phosphorylation.129 JNK plays an essential role in cell proliferation by modulating cell cycle genes130 and is involved in the differentiation of hematopoietic populations and the apoptotic response to cellular stressors.131
The ERK5 pathway
The ERK5 Pathway is activated by growth factors (e.g., epidermal growth factor [EGF], nerve growth factor, fibroblast growth factor [FGF]-2, and brain-derived neurotrophic factor); some cytokines, including leukemia inhibitory factor; and stressors, such as osmotic stress and hydrogen peroxide. ERK5 can be activated by several upstream factors, such as c-SRC, RAS, LAD1 adapter protein, and WNK Ser/Thr kinases132 In addition, ERK5 is activated by dual phosphorylation with a unique MAPK/ERK kinase 5 and MEK5,133,134 and activated ERK5 phosphorylates several cellular proteins, including the MEF2 transcription factor family, Sap1a (ETS domain transcription factor), c-MYC, serum and glucocorticoid inducible protein kinase, Connexin 43, and Bcl-2 agonist of cell death (BAD).132 Similar to ERK1/2, ERK5 is involved in cell survival and proliferation, increasing cyclin D1 expression during the G1/S transition.135 ERK5 is also necessary for vascular endothelial growth factor (VEGF)-mediated survival and tubular morphogenesis in primary human microvascular endothelial cells and the MEK inhibitors PD98059 and U0126 effectively inhibit ERK5.136
ERK3/4, ERK 7/8, and NLK
The ERK3/4 and ERK7 pathways are poorly characterized, although these proteins autophosphorylate the activating loop residues in vitro and in vivo.137,138,139 Serum and hydrogen peroxide stimulate ERK8 phosphorylation via conventional MAPKs. The MAPKAPK MK5 is the only known target of ERK3/4 according to several previous studies.137,140,141,142,143,144 ERK7/8 directly controls a number of proteins (e.g., Myelin basic protein [MBP], c-FOS, and c-MYC) in vitro, although their cellular functions are not clear.139,145,146,147 Many cytokines, including interleukin (IL)-6, granulocyte colony-stimulating factor, and transforming growth factor-beta (TGF-β), are associated with the activation of NLK, which is a key regulator of cell fate determination. NLK is triggered by Wnt pathway stimulation (Wnt-1 and Wnt-5a) and TGF-β.148 Last, NLK targets T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors and STAT3.148 ERK3, ERK7, and NLK are involved in cell proliferation, cell cycle progression, migration, invasion, apoptosis, and cell differentiation. However, few inhibitors for atypical MAPKs have been validated as anticancer drugs, although GPLG0259, an inhibitor of MK5, is currently under clinical study for use in obesity and diabetes.
Accessory proteins in the RAS/RAF/MAPK cascade
MAPK signaling is linked to various malignancies in humans, and its activation is associated with many extracellular signals and intracellular proteins.149 Therefore, targeting the constituents of this signaling cascade frequently results in severe toxicity, activation of backup mechanisms, and reduced drug efficacy, often associated with an increase in therapy burden. To avoid these undesirable effects, other approaches targeting RAF modulators must be developed. The spatiotemporal characteristics of MAPK pathway constituents may offer an alternative strategy MAPK accessory proteins are spatially assembled to promote cooperation during signaling150 and can be divided into four categories: (1) anchoring proteins, (2) docking proteins, (3) adapter proteins, and (4) scaffold proteins (Fig. 4a).

RAF signaling regulation by accessory proteins. a Accessory proteins consist of anchoring proteins, docking proteins, adapter proteins, and scaffold proteins. Anchoring proteins bind to membrane kinases and other effectors, whereas adapter proteins link receptor kinases with guanine exchange factors (GEFs). Docking proteins connect active receptors with multiple effectors. Scaffold proteins offer a signaling platform for the spatial regulation of the mitogen-activated protein kinase (MAPK) pathway. b CRAF kinase inhibitor protein (RKIP) is a tumor suppressor. RKIP, an intrinsic RAF kinase inhibitor, is associated with many malignant features, including metastasis and chemotherapy resistance, through the regulation of oncogenic mediators and signaling axes, such as NF-kB, YY1, MAPK28, STAT3, NRF2, and AKT29. Arrows and bars indicate stimulating and inhibiting signals, respectively. This figure was created with BioRender.com
Anchoring proteins
Anchoring proteins are bound to the membrane and connect and associate with effector proteins, which primarily consist of kinases. For example, the A-kinase-anchoring protein (AKAP-Lbc) and the scaffolding protein (KSR-1) constitute a signaling network that efficiently relays signals from RAF to MEK to ERK1/2.151,152 Many anchoring proteins have been discovered in other signaling pathways, including connector enhancer of KSR (CNK)1,153 Flotillin ½,154 linker for activation of T cells,155 non–T cell activation linker (NTAL),156 progestin and adipoQ receptor family members (PAQR10/11),157 and Sef1.158
RAF docking proteins
Docking proteins play a crucial role in cellular signaling by binding to receptors like RTKs and GPCRs, subsequently recruiting effector molecules. These specialized proteins typically possess PTB domains that enable them to selectively interaction with activated RTKs, as well as PH domains that serve to extend their presence at the cell membrane.159 The FGF receptor substrate 2 (FRS2) was initially identified as a substrate for the FGF receptor160 but was later found to be a key docking protein for various RTKs.161 Other identified docking proteins include docking protein 1/2 and GRB2-associated binding protein GAB1/2.162
RAF adapter proteins
Adapter proteins connect two functional components (e.g., receptor and GEF), providing additional docking sites for signaling proteins and promoting signal transduction from one-way receptors. Phosphorylation at Ser residues generates protein–protein interaction sites mediated by adapter proteins, such as the 14-3-3 family, facilitating associations with various signaling modulators, including CRAF, KSR, B-cell receptor (BCR), and phosphoinositide 3-kinase (PI3K).163 Many adapter proteins play important roles in signaling pathways, including CRK proto-oncogene (CRK and CRKL), Casitas B-lineage lymphoma, GRB2,164 SHC, SH2-containing protein tyrosine phosphatase 2 (SHP2) and SOS.165
RAF scaffolding proteins
Scaffolding proteins bind two or more partners to provide a signaling platform able to regulate the MAPK pathway both spatially and temporally.166 MAPK signaling components that exist as freely diffuse cytosolic forms are unable to effectively transmit signals to corresponding partners. Scaffolding proteins offer a platform at which many components can associate, allowing the efficient propagation of signals. Scaffolds also facilitate tighter control of MAPK signaling.150 KSR is a scaffold protein in the MAPK pathway that assembles B/CRAF, MEK1/2, and ERK1/2.167 In addition, both KSR and MEK partner-1 (MP1) retain ERK proteins in the proximity of critical cellular effectors.168 Several scaffolding proteins are involved in cellular functions169, including IQGAP1/2/3,170 Paxillin,171 β-arrestin,172 apoptosis stimulating proteins of p53 1/2 (ASPP1/2),173 SPRED1/2/3,174 SPRY1/2,175 RAF kinase inhibitory protein (RKIP), Merlin,176 Nucleolin and Nucleophosmin,177,178 PEA15,179 Regulator of G-protein signaling 14 (RGS14),180 MAPK organizer 1 (MORG1),181 Galectin 1/3,182 GIT1,183 Calmodulin,184 Erbin,185 and FHL1/2.186
RKIP is ubiquitously expressed in a broad range of cells and serves as an integral scaffolding protein187 and a negative modulator of the RAF–MEK–ERK signaling pathway188. RKIP directly binds CRAF and inhibits the MEK-dependent phosphorylation of CRAF by interfering with the formation of a kinase–substrate complex between CRAF and MEK.189
RKIP as a tumor suppressor
RKIP belongs to the phosphatidylethanolamine-binding protein family, which functions in lipid metabolism and phospholipid membrane biogenesis.190 RKIP is a highly dynamic protein with a malleable pocket loop that exists in a variety of states, serving as a functional switch. This protein has pleiotropic roles in several signaling pathways involved in physiological processes As a MAPK signaling modulator, RKIP can inhibit the metastatic process by modulating RAF activation and may represent a new avenue for therapeutic intervention (Fig. 4b). RKIP also regulates cancer development and progression, and191 its expression is severely downregulated in many cancer tissues, including breast cancer192, prostate cancer193, gastric cancer,194 lung cancer,195 esophageal squamous cell carcinoma,196 colorectal cancer,197,198 and nasopharyngeal carcinoma.199 Low RKIP expression levels are generally associated with malignant features, such as metastasis and chemotherapy resistance, promoting oncogenic signaling axes, including nuclear factor kappa B (NF-κB),200 YY1,201 MAPK28,202 and AKT29.203 RKIP levels are regulated by STAT3 activation during metastasis in NSCLC cells,204 and RKIP downregulation leads to nuclear factor erythroid 2-related factor 2 (NRF2) hyperactivation, which is responsible for the development of chemotherapeutic resistance in colorectal cancer cells.205 Reduced RKIP levels stimulate invasion, metastasis, and radio-resistance in nasopharyngeal cancer cells.206
Physiological functions of RAS/RAF/MAPK
The RAS/RAF/MAPK pathway involves signal transmission from membrane-based receptors, which interact with mitogens, to various destinations with cells, including the nuclear, cytoplasmic, and cell. These signals play a pivotal role in orchestrating a diverse range of physiological responses, encompassing cell proliferation, tumor invasion and metastasis, cellular metabolism, cell cycle progression, and ultimately cell survival or death.207 Consequently, any disruption or dysregulation of the RAS/RAF/MAPK pathway is closely linked to numerous human disorders, most notably cancer.
Role of ERK/MAPK in tumorigenesis
While tumorigenesis and the metastatic spread of cancer involve multiple cooperative cellular signals, the significance of the ERK/MAPK signaling pathway cannot be overstated when it comes to cancer invasion and metastasis. Notably, a heightened activation of ERK is evident across a spectrum of human cancer types, including ovarian, colon, breast, lung cancer, and others.208 Furthermore, in vitro experiments have revealed that microRNA-508 effectively inhibits EMT, migration, and invasion in ovarian cancer cells by modulating the ERK/MAPK1 signaling system.208 Meanwhile, in vivo studies have demonstrated that blocking the MAP kinase pathway leads to the suppression of growth of colon cancer cells.68 Additionally, Emodin has been found to inhibit the proliferation of non-small-cell lung cancer (NSCLC) cells by inducing PPARs and subsequently reducing Sp1 levels through the activation of ERK and AMPK.209
Cell proliferation and cell apoptosis
The ERK/MAPK signaling pathway primarily plays a role in promoting cell proliferation and exerts an anti-apoptotic influence. Specifically, under hypoxia conditions, it facilitates the survival of nutrient-starved tumor cell by reducing their susceptibility to apoptosis.210 Moreover, in the context of large B-lymphoma cells, microRNA-101 exerts control over cell proliferation and apoptosis by directly modulating MEK1 in the RAS/RAF/MAPK pathway.211 Furthermore, both ERK1 and ERK2 contribute to cell proliferation in a manner that depends on their expression levels, as they integrate signals from RAS, RAF, and MEK.212 The constitutive activation of the RAS/RAF/MAPK pathway contributes to tumorigenesis by inhibiting Caspase-9 through MAPK-dependent phosphorylation at Thr125.213
Cell cycle progression
Numerous downstream effectors of the RAS/RAF/MAPK pathway have a multifaceted impact. They not only drive cell cycle progression by instigating the production of cyclins and cell cycle-dependent protein kinases (CDKs) through the regulation of MYC and E2F but also orchestrate an early G1 cell cycle arrest. This arrest is achieved by influencing the expression of various CDK inhibitor proteins, including p16Ink4a, p15Ink4b, and p21Cip. Furthermore, the RAS/RAF/MAPK pathway is closely linked to the induction of cellular senescence, which is mediated by these CDK inhibitors, resulting in a premature G1 arrest.214
Tumor ECM Degradation and angiogenesis
Furthermore, the RAS/RAF/MAPK pathway plays a vital role in degradation of extracellular matrix proteins, a crucial process for cancer metastasis and angiogenesis. For instance, Mesothelin, a secretory protein, stimulates the production of MMP-7 by activating the MAPK/ERK and JNK signaling pathways in ovarian cancers215. Additionally, p70S6K, a downstream target of the RAS/RAF/MAPK pathway, exerts control over tumor growth and angiogenesis by promoting the activation of HIF-1alpha and the production of VEGF in ovarian cancer cells.216
It is fair to state that as we understand more about the specifics of RAF (ARAF, BRAF, and CRAF), we found that there are more unanswered questions about how they work and how they specifically affect physiological functions.
Cell regulatory pathways mediated by MAPK-independent RAF kinase
Generally, RAF activation leads to ERK1/2 activation via MEK1/2 phosphorylation217. MEK1/2 was thought to be the only RAF kinase substrate prior to the discovery of MEK1/2-independent RAF functions.218,219 Although only a few MEK-independent RAF targets have been defined, these MEK-independent RAF activities are thought to be important for carcinogenic regulation (Fig. 5).

Kinase-independent regulation of RAF-interacting signaling. Three RAF effector proteins, Bcl-2 agonist of cell death (BAD), apoptosis signal-regulating kinase 1 (ASK1), and mammalian Ste20-like kinase 2 (MST2), are kinase-independent negative regulators of apoptosis. RAF can enhance cell cycle progression via the extracellular signal-related kinase (ERK) pathway, and RAF can regulate the cell cycle in a kinase-independent manner. RAF interacts with polo-like kinase 1 (PLK1) and Aurora kinase A (Aurora-A). During cell migration, RAF also functions as a spatial regulator of Rho-associated kinase (ROK)-α, a downstream effector of RHO, in a kinase-independent manner by inhibiting ROK-α activity. Several additional RAF substrates, such as NF-κB, Vimentin, Snail, and Keratin, are associated with cytoskeleton organization. This figure was created with BioRender.com
RAF as an apoptosis regulator
MEK-independent RAF is a negative regulator of apoptosis. Three RAF effector proteins, BAD, apoptosis signal-regulating kinase 1 (ASK1), and MST2, play critical roles in the regulation of apoptosis. Interactions between RAF and these targets occur at the outer mitochondrial membrane, unlike classical RAF signaling, which is localized to the plasma membrane.220
BAD promotes apoptosis by inhibiting the pro-survival effects of BCL2 proteins,221 and RAF increases cell survival through the direct phosphorylation of BAD.222 Raf also acts as an adapter protein that stimulates the binding of BAD with protein kinase C, which phosphorylates BAD and inactivates downstream signals.223
ASK1, also referred to as MAP3K5, is a Ser/Thr kinase able to activate the SAPK, JNK, and p38 pathways to trigger apoptosis under oxidative conditions.217,224 FGF receptor activation increases interactions between RAF and ASK1 in the mitochondria, preventing the activation of the p38 MAPK pathway in an ERK- and PI3K-independent fashion.225,226 RAF modulates ASK1 activity, which is necessary for JNK- and p38-induced apoptosis, and the loss of RAF expression increases ASK1 activity, followed by increased JNK and p38 activation. ASK1 knockout reverses the effects of RAF loss.227
RAF can inhibit the MST2-associated tumor suppression pathway in various cancers. RAF binds MST2 independent of kinase activity, preventing apoptosis in cancer cells via a two-pronged mechanism.228,229,230,231 First, RAF binding to MST2 blocks MST2 dimerization, a critical step for MST activation. Second, RAF recruitment of a phosphatase can prevent MST2 autophosphorylation at the Thr180 residue in the activation loop.228,232
RAF as a cell cycle and mitosis checkpoint mediator
CRAF can promote cell cycle progression in a MEK/ERK-independent manner233. CRAF directly interacts with key regulators of mitotic progression, polo-like kinase 1 (PLK1) and Aurora kinase A (Aurora-A).233,234 At the G2/M transition during mitosis, CRAF phosphorylation (Ser338) induces protein localization to centrosomes and mitotic spindle poles, where CRAF interacts with and activates Aurora-A and PLK1, leading to mitosis and tumor growth.
In addition, RAF regulates checkpoint kinase 2 (CHK2) activity during cell cycle progression. Upon DNA damage, PAK1 induces the formation of a RAF–CHK2 complex to stimulate the DNA repair system.235 RAF phosphorylation at Ser338 promotes the RAF–CHK2 interaction, which is associated with radiation resistance and CHK2 activation. In addition, RAF interacts with CDC25 phosphatase, which links mitogenic signaling with cell cycle activation.236,237
RAF as a regulator of the EMT and cytoskeletal organization
Cancer metastasis has been linked to the dysregulation of various signal pathways, including the constitutive activation of MAPK, NF-κB, and PI3K in melanoma.238 RAF-dependent AKT3 inhibition has also been associated with the promotion of cell proliferation, survival, and metastasis and the inhibition of cellular defense mechanisms and cellular senescence.239,240 NF-κB activation promotes metastasis by increasing the expression of many metastatic genes, including cyclooxygenase 2 (COX2), genes encoding metalloproteinases, VEGF, and SNAIL.241,242 Jacqueline’s group demonstrated that RAS can activate NF-κB transcriptional activity via either RAF-dependent or RAF-independent mechanisms, both of which are dependent on SAPKs, such as p38.243 According to another study, RAF-dependent NF-κB activation involves MEKK1 rather than the traditional mitogenic cytoplasmic kinase pathway.244 RAF stimulates the expression of atrial natriuretic factor (ANF) in cardiac myocytes, whereas MEK1/2 inhibits ANF expression.245
RAF-dependent RHO signaling is related to cell migration. During cell migration, RAF functions as a spatial regulator of RHO-associated kinase (ROK)-α, an effector downstream of RHO, in a kinase-independent manner.246 In a conditional knockout study, RAF was found to be necessary for proper wound healing in vivo and for keratinocyte and fibroblast cell migration in vitro. This study also indicated that RAF-mediated ROK-α inhibition is necessary for RAS-dependent carcinogenesis.247 Functionally, the interaction between RAF and ROK-α may be associated with a RAF-induced anti-apoptotic signal, as stimulation of the FAS death receptor increases RAF–ROK-α complex formation.248 In addition, ROK-α kinase activity is inhibited by the RAF regulatory domain.249 However, ROK-α is likely not regulated by RAF in RAS-mutant tumors.250 Moreover, several additional RAF substrates are involved in cytoskeleton organization, including Vimentin251 and Keratin 8.252
Crosstalk between RAS/RAF/MAPK and PI3K/AKT/mTOR or MST2/Hippo signaling pathways
The PI3K-mTOR (mammalian target of rapamycin) pathway is a crucial mechanism governing cell survival, division, and metabolism. Growth factors can engage the pathway by either directly recruiting PI3K to their receptors or indirectly involving it through docking proteins like IRS (insulin receptor substrate) or GAB (GRB2-associated binder). This activation of PI3K leads to the generation of the secondary messenger phosphatidyl inositol 3,4,5-triphosphate (PIP3), which in turn recruits the protein kinase AKT to the plasma membrane. Subsequent AKT activation, dependent on PDK1, initiates the phosphorylation of numerous factors related to survival, proliferation, motility, and the TSC2 GAP (GTPase Activating Protein). AKT-dependent TSC2 phosphorylation releases TSC1/2 inhibition by the GTPase RHEB (RAS homolog abundant in brain), ultimately activating mTORC1, which regulates cell growth. Both the RAS/RAF/MAPK and PI3K/AKT/mTOR pathways frequently experience dysregulation in many human cancers, often due to genetic alterations in their components or upstream regulators.253 The intricate network of positive feedforward and negative feedback loops in these pathways significantly influences signal dynamics. Notably, the GAB docking proteins, forming the GRB2-SOS complex upon activation of RTKs, are key players in positive loops. This complex, which includes RAS-GAP, SHP2, PI3K, and PIP3 (a protein-tyrosine phosphatase with a Src homology two domain), contributes to RAS activation. Dephosphorylation of RAS-GAP docking sites on GAB1 by SHP2 reduces RAS activation and enhances RAS-ERK signaling. Additionally, GAB2-mediated PI3K recruitment generates local PIP3, further stimulating PI3K signaling. Furthermore, SOS, RAF, and MEK1 can be phosphorylated by ERK, which creates a negative feedback loop by dampening ERK activity (Fig. 6a). Indeed, comprehensive cancer genome analyses have revealed that over-activation or mutations that enhance the MAPK and PI3K pathways are characteristic features of many human cancer types. Importantly, the interplay between RAS/RAF/MAPK and PI3K/AKT pathways is tightly controlled in response to ligands in a dose-dependent manner. For Instance, elevated levels of insulin-like growth factor I (IGF-I) induce a rapid and potent phosphorylation of AKT at the serine 259 residue, effectively restraining RAF kinase activity. Conversely, low concentration of IGH-1 fail to induce such crosstalk, but still exert mitogenic effects254

The crosstalk of RAS/RAF/MAPK with other pathways. a Crosstalk between the RAS/RAF/MAPK, PI3K/mTOR or MAT2/Hippo signaling pathways. The RAS/RAF/MAPK signal collaborates within its own cascade and interfaces with the PI3K/mTOR pathway, where its influence is MAPK-dependent. Conversely, the MST-2/Hippo pathway operates independently of MAPK activity but relies on the presence of RAF for its functionality. b Functional interactions between autophagy and mitogen-activated protein kinase (MAPK) signaling during the epithelial-to-mesenchymal transition (EMT). The MAPK pathway can be activated by canonical receptor tyrosine kinases (RTKs) and through Smad-independent activation by transforming growth factor-beta (TGF-β). Both signals activate a typical RAS–RAF–MAPK cascade, stimulating the EMT process. During the metastasis process, cancer cells migrate from a primary site to a secondary site facing many stressors, and therefore metabolic and cellular alterations are necessary to overcome these stressors. c The role of autophagy in tumorigenesis. During early tumorigenesis, autophagy acts as a tumor suppressor. However, autophagy drives tumor growth, progression, and metastasis by enhancing migration, invasion, EMT, and metabolic tolerance during advanced stages of cancer, allowing cancer cells to evade RAFi therapy. Arrows and bars indicate stimulating and inhibiting signals, respectively. This figure was created with BioRender.com
The MST2 protein family, characterized by serine/threonine kinases, becomes activated in response to stress signals in mammalian cells, and their overexpression has been observed to trigger apoptosis.232 The MST2/Hippo pathway is intricately linked to the RAS/RAF/MAPK pathway (Fig. 6a). Specifically, the MST-CRAF complex, induced by mitogenic and apoptotic signals, acts as a safeguard against unchecked cell proliferation.229 A previous study has shed light on the dynamic changes in protein-protein interactions (PPIs) arising from the function association between the kinases MST2 and CRAF kinases, along with the modulation of their respective upstream activators RASSF1A and RAS231. The interaction between CRAF and MST2 inhibits the binding of the scaffold protein RASSF1A to MST2, leading to MST2 dimerization and activation. Interestingly, activated AKT promotes the binding of MST2 to RAF and subsequently preventing MST2 activation.255 Additionally, MST2 can be inhibited by a phosphatase, likely PP2A, associated with RAF.256 Conversely, MST2 interferes with RAS-dependent RAF activation by blocking the RAS-binding domain (RBD) domain in CRAF. RASSF1A can rerelease MST2 from its inhibitory complex with CRAF, activating LATS1. This activation of LATS1, in turn, induces the formation of the YAP1-p73 transcriptional complex, ultimately leading to apoptosis.257 Furthermore, the RAS/RAF/MAPK pathway engages in crosstalk with several other signal pathways, including those involved in DNA repair process or cell cycle regulation, as depicted in Fig. 5.
The link between autophagy and RAF signaling in cancer metastasis
Autophagy is an essential process that maintains cellular homeostasis via the lysosome-dependent degradation of cellular components, such as proteins and organelles, allowing cells to recycle macromolecules258,259. Irregular autophagy activation can lead to cellular dysfunction, as observed in many human diseases, including neurodegenerative diseases, heart disease, infectious diseases intervertebral disc degeneration, liver disorders, and cancers.260,261,262 Autophagy is also involved in various stress responses, developmental processes, and aging.263,264
Although autophagy can be either tumor-suppressive or tumor-promoting, depending on the context, the precise mechanisms that determine the role of autophagy are not well-defined. In general, increased catabolism driven by autophagy promotes cell survival, and autophagic abnormalities induce cell death in cancer cells. Cancer cells exhibit more microenvironmental and metabolic dependencies than normal cells, and targeting the double-edged process of autophagy represents an appealing option for the development of future therapeutic agents.265
The MAPK pathway drives the expression of autophagy-related-8 (Atg8) in cancer cells.266 Studies of RAF mutations suggest a functional association between autophagy and V600E-mutant BRAF. Autophagy accelerates the growth of V600E-mutant melanoma in mice by allowing the bypass of the senescence process.267 The reduced expression of autophagy-related ATG genes in patients with BRAFV600E-mutant melanoma appears to suppress carcinogenesis.268
Cancers can spread to secondary organs through a complicated, efficient, and deadly process called metastasis, which represents a substantial contributor to mortality.269 Metastasis requires coordination between the genetic programs that promote and prevent metastasis and the tumor microenvironment to allow cancer cells to transition from their initial locations and develop in secondary organs. Metastasis suppressor genes prevent metastasis at secondary sites with no impact on the primary tumor.
During metastasis, cancer cells dissociate from the primary sites, travel through the circulation, and deposit in a secondary site, overcoming many obstacles, including nutrient limitations and cellular stress.270 In metastatic cells, cellular metabolism undergoes dynamic alterations that promote a shift toward the metastatic cascade.271 Metabolic plasticity and flexibility in cancer cells play important roles in the metastatic process.272 Metabolic plasticity allows a single metabolite to meet multiple metabolic needs during metastasis, whereas metabolic flexibility allows several metabolites to fulfill the same metabolic need.273,274 The metabolites sapienate, generated by fatty acid desaturase 2, and palmitoleate, generated by stearoyl-CoA desaturase-1, are mono-unsaturated fatty acids involved in the metabolic flexibility of primary tumors.275,276 The presence of these metabolites in the bloodstream is essential for the invasion, migration, and survival of metastatic cells. These metabolites are also associated with the EMT,277 which represents a necessary step through which cancer cells acquire metastatic properties and plays important roles in cancer progression, metastasis, and drug resistance.278
Based on previous studies, MAPK signaling, autophagy, and EMT are physically and functionally interconnected during cancer metastasis (Fig. 6b). EMT is a multidimensional process that involves the remodeling of the cytoskeleton, cell membrane, and cell–cell junctions, resulting in the loss of epithelial characteristics and the acquisition of mesenchymal properties facilitated by MAPK activation.279 Autophagy supplies the energy required for TGF-β–induced EMT and cancer metastasis. TGF-β is involved in many cellular processes, including tissue fibrosis, growth inhibition, and EMT. TGF-β activates Smad-dependent and Smad-independent signaling pathways, including the ERK1/2 pathway, which is involved in cytoskeletal organization, cell growth, survival, migration, and invasion.280 TGF-β–directed MAPK signaling also activates common downstream signaling molecules induced by RTKs. In response to TGF-β, RAS triggers ERK1 and ERK2 activation, leading to the activation of RAF and MEK1/2, as shown in Fig. 2.281,282 Following TGF-β-induced Ser or tyrosine phosphorylation of the type I TGF-β receptor, ShcA recruits GRB2 and SOS to activate ERK1/2 through RAS, RAF, and MEK1/2.281,283 Other signaling molecules, such as integrin, Notch, Wnt, TNF-α, long non-coding (lnc) RNA, and EGF, synergize with TGF-β signaling to promote tumor invasion and metastasis.284,285,286,287,288,289,290 Many studies suggest the existence of functional connections between TGF-β and RAS–RAF signaling in tumorigenesis. TGF-β–induced EMT is enhanced by increased RAS–ERK291 signaling, and MEK1/2 pharmacological inhibition prevents TGF-β–induced EMT.292
EMT and autophagy are connected through multiple pathways, although the exact role of autophagy in EMT remains unclear. Autophagy inhibition promotes EMT, invasion, and metastasis in many cancer cells, including gastric, colorectal, melanoma, and pancreatic cancer cells, mouse embryonic fibroblasts, and keratinocytes.293,294,295 By contrast, TGF-β–induced autophagy promotes cancer cell migration via MAPK–ERK activation in NSCLC and SMAD4-negative pancreatic cancer cells.296,297 However, autophagy inhibits metastasis in HCC298 and prevents EMT in breast and SMAD4-positive pancreatic cancer cells.297,299
Autophagy plays a significant role in RAF-driven tumorigenesis, functioning as a tumor suppressor during early stages and as a tumor promotor during advanced stages (Fig. 6c). BRAF and CRAF activate autophagy to promote tumor cell survival.27 Atg7-knockout mice with RAF-mutant melanoma display reduced tumor growth and significantly increased survival compared to wild-type mice.267 Interestingly, autophagy exhibits both tumor-promoting and tumor-suppressive roles in the same mouse model of BRAFV600E–mutant lung cancer.300 However, autophagy promotes tumor growth and metabolism in BRAFV600E–mutant lung cancer,301 and BRAFV600E–mutant cancers promote autophagy to maintain mitochondrial function and glutamine metabolism.302
The fact that human cancer frequently exhibits dysregulation of the autophagy and RAS/RAF/MAPK pathways makes the components of these signaling cascades intriguing candidates for therapeutic intervention. Recent research has shown the existence of positive and negative feedback loops in these pathways, which activate one pathway when the other signaling cascade is blocked. Therefore, blocking both pathways with a combination of signaling inhibitors may have a stronger antitumor effect than using a single medication.
Targeting the RAS/RAF/MAPK pathway for cancer therapy
Our understanding of the roles played by RAS/RAF/MAPK components in both normal physiology and pathological conditions has significantly progressed. In the realm of therapeutic interventions, RAS/RAF/MAPK inhibitors have emerged as promising targets for addressing BRAF-mutated cancers and other disorders. These inhibitors have gained approval from the FDA and are employed either as standalone treatments or in combination with two or more agents. These FDA-approved RAS/RAF/MAPK-targeted medications, including their most up-to-date information, are represented in Table 2 in the latest medication guide available at www.accessdata.fda.gov.
RAS/RAF/MAPK inhibitors
RAS/Raf/MAPK inhibitors represent a category of precision therapies employed in the management of diverse cancers, especially those marked by mutations in the RAS/RAF/MAPK pathway (Fig. 7a). These pathway constituents play a critical role in regulating cell proliferation, differentiation, and survival. However, when mutations disrupt their normal function, they can contribute to the initiation and progression of cancer.

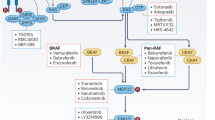
RAS/RAF/MAPK-targeted therapy in BRAF-mutated malignancies. a Functional classifications of BRAF mutations. In Class I, BRAF mutants (e.g., V600E) transmit signals via a monomeric form independent of RAS activation, leading to increased extracellular signal-related kinase (ERK) activation. In Class II, BRAF mutants (e.g., K601E) are RAS-independent, forming mutant–mutant BRAF dimers. RAF inhibitors (RAFi), such as Vemurafenib and Dabrafenib, block both Class I and II RAF kinases. In Class III, mutant BRAF (e.g., D287H, V459L) exhibits increased RAS binding and heterodimer formation between mutant BRAF and wild-type CRAF. MEK inhibitors (MEKi), such as Trametinib or Cobimetinib, show an additive effect when combined with RAFi for cancer treatment. b RAS/RAF/MAPK-targeted therapies. Specific Inhibitors targeting the RAS/RAF/MAPK pathway represent as each group of action: EGFR agonists (EGFRi), RAS inhibitors (RASi), RAF inhibitors (RAFi), MEK inhibitors (MEKi), and ERK inhibitors (ERKi). Arrows and bars indicate stimulating and inhibiting signals, respectively. This figure was created with BioRender.com
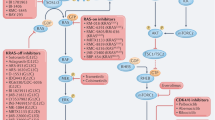
KRAS-targeted therapy
The proto-oncogene KRAS plays a crucial role in cell signaling pathways governing cell growth and differentiation. In various cancers, mutations in the KRAS gene have been linked to poor prognosis and a limited range of targeted treatment options. In Western nations, lung adenocarcinomas exhibit KRAS mutations in 20–40% of cases, while the prevalence is slightly lower, around, in Asian countries.303 KRAS mutations pose therapeutic challenges as they are often associated with resistance to specific target therapies, such as epidermal growth factor receptor (EGFR) inhibitors in non-small cell lung cancer (NSCLC).304 While the development of the direct RAS inhibitors has been proven challenging due to the nature of RAS protein, ongoing research and clinical trials are exploring strategies and therapies to effectively target KRAS-mutant tumors.
Cetuximab and Panitumumab
Cetuximab and Panitumumab are monoclonal antibody therapies that target the epidermal growth factor receptor (EGFR) and are employed in the treatment of various tumors, including head and neck and colorectal cancer.305 Importantly, these drugs are approved and effective exclusively in KRAS wild-type patients with advanced colorectal cancer.306 While they are not direct RAS-mutant targeted therapeutics, they may occasionally have an indirect impact on RAS signaling pathways.307 For instance, when combined with Cetuximab, LSN3074753 (a pan-RAFi) demonstrates synergistic anticancer efficacy in BRAF or KRAS-mutant CRC PDX models.308 It has been suggested that colorectal cancers with the KRASG13D mutation may respond more favorably to Cetuximab or Panitumumab treatment compared to other more prevalent KRAS mutations.309 In the realm of KRAS-mutant targeted therapeutics, Adagrasib (MRTX849) demonstrated favorable tolerability and exhibited anticancer activity in patients with advanced solid tumors harboring the KRASG12C mutation in a first-in-human phase I/IB clinical trial (KRYSTAL-1) (NCT03785249).310
Sotorasib
The U.S. FDA-approved Sotorasib (LumakrasTM, Amgen) has been employed in the treatment of advanced NSCLC patients with and KRASG12C mutation who have undergone at least one prior systemic therapy.311 The FDA’s rapid approval of Sotorasib serves as a remarkable example of recent expeditious approvals for clinically effective drugs. In phase I clinical trials, Sotorasib demonstrated promising anticancer activity in heavily pretreated patients with advanced solid tumors bearing the KRASG12C mutation (NCT03600883).312 Subsequently, in phase II clinical trials, Sotorasib therapy provided sustained clinical benefit without revealing new safety concerns in previously treated KRASG12C-mutated NSCLC patients (NCT03600883). It is noteworthy that Sotorasib’s oral availability is significantly restricted by CYP3A, while its brain accumulation is robustly constrained by ABCB1.313
Adagrasib
Adagrasib (KRAZATITM, Mirati Therapeutics) is an orally administered, and highly effective small molecule inhibitor, irreversibly covalent binding to KRAS. It have been developed for the treatment of solid tumors harboring the KRASG12C oncogenic driver mutation, including NSCLC and CRC.314 Tian et al. unequivocally demonstrates the promising effectiveness and acceptable safety profile of Adagrasib based on multiple registered interventional clinical trials (e.g., NCT05375994 and NCT05472623) in patients with KRASG12C-mutated NSCLC.315 They have also recommended further research to explore Adagrasib’s potential in various contexts and combination therapies. The US FDA has granted approval for expanded access to Adagrasib (MRTX849) in patients with advanced solid tumors carrying the KRASG12C mutation (NCT05162443), citing data from several interventional clinical trials (e.g., NCT04975256, NCT05853575, NCT03785249, NCT04330664, and NCT05609578). A recent phase I/II study showcased Adagrasib’s antitumor activity in heavily pretreated patients with metastatic CRC bearing mutant KRASG12C, both as oral monotherapy and in combination with Cetuximab (NCT03785249).316 The pharmacokinetics (PK) of Adagrasib is constrained by the CYP3A and ABCB1 activity, and it can be modified by mouse plasma carboxylesterase 1 c.317
BRAF-mutant targeted therapy
RAF inhibitors (RAFi)
Mutations of RAF are frequently found in melanoma and other cancers. The prevailing mutation of RAF is BRAFV600E. Abnormal activation of the RAS/RAF/MAPK pathway, a hallmark of cancer, often originates from genetic alterations in RAF-encoding genes or RAF upstream genes. Consequently, the RAF kinase family is a promising target for potential cancer therapies. The RAF kinase activity is frequently disrupted in human cancer due to RAF itself or mutations affecting the upstream regulators and downstream effectors proteins as depicted in Fig. 7b.
PLX4720, a potent and selective RAF inhibitor, has demonstrated robust antitumor activity against RAF-mutant melanoma in vitro and in vivo.318 Vemurafenib (PLX4032), an analog of PLX4720, exhibits improved pharmacokinetic properties compared to PLX4720, and has received approval for the treatment of advanced melanomas and other cancers harboring RAF mutations.319,320 Vemurafenib has shown exceptional efficacy with manageable side effects, both as a monotherapy and when combined with MEK inhibitors, for patients with RAF mutant cancer. Dabrafenib is another effective therapeutic option for melanomas bearing the V600E BRAF mutation, achieving tumor shrinkage in over 90% of patients with half experiencing partial to complete responses.321,322 Furthermore, clinical phase III studies have demonstrated that Encorafenib, a BRAF inhibitor, either standalone treatment or in combination with MEK inhibitors, significantly prolongs progression-free survival compared to Vemurafenib.323 In previous studies involving patients with BRAFV600E metastatic melanoma, treatment of Vemurafenib, Dabrafenib, and Encorafenib resulted in dose-dependent tumor suppression and improved overall survival rates relative to other therapeutic approaches.324,325 Notably, patients with BRAF-mutant melanoma have displayed significant responses to BRAF inhibitors such as vemurafenib (PLX4032, RG7204) and Dabrafenib (GSK2118436), while those with BRAF wild-type melanoma have not shown similar responses.326
Despite the encouraging beneficial observed with RAF inhibitors, approximately half of the patients treated with RAFi experience cancer recurrence due to the development of resistance to the RAF inhibitors within 6–7 months of initiating treatment.327
MEK inhibitors (MEKi)
While activated MEK mutations are relatively rare in human tumors, mutations in upstream genes like RAS or RAF are implicated in over 85% of malignancies, often resulting in elevated MEK activity.328 Targeting MEK has thus emerged as a promising and cutting-edge therapeutic approach. Combinations of MEK and BRAF inhibitors have received FDA approval and have proven effective in inhibiting tumor growth in both preclinical models and patients with RAS or RAF mutations.329 One notable MEK inhibitor, Trametinib, was identified progression-free and overall survival in patients with BRAFV600E-mutated metastatic melanoma.330 Furthermore, combinations of Dabrafenib and Trametinib led to improved progression-free survival in patients with metastatic BRAFV600E-mutated melanoma.331 Cobimetinib (GDC-0973) is another highly selective allosteric MEKi that has demonstrated effectiveness in BRAFV600E/K-mutant patients as well as in cell lines with BRAF- or KRAS-mutations.332 The FDA approved Cobimetinib in combination with Vemurafenib for the treatment of BRAFV600E/K-mutant and unresectable melanoma in 2015.333 Binimetinib, also known as MEK162, ARRY-162, or ARRY-438162, is an effective oral MEKi approved for patients with BRAFV600E/K-mutant and unresectable melanoma. The FDA approved the combination therapy of Binimetinib and Encorafenib in 2018334. In 2020, Selumetinib (KOSELUGO, AstraZeneca), a highly specific MEKi, is approved for the treatment of pediatric children with neurofibromatosis type 1 (NP1) who have symptomatic, inoperable plexiform neurofibromas (PN).335 Selumetinib is also under evaluation for use in other cancers such as melanoma, gliomas, and non-small cell lung cancers, where MEK1/2 is overexpressed.336
Ongoing clinical studies on the combination of BRAFi and MEKi
In patients with BRAF mutated cancer, a molecular mechanism analysis has revealed potential target for RAS/RAF/MAPK inhibitors. This analysis has led to a significant shift in the treatment approach for patients with BRAF mutated cancer. It is believed that inhibiting both BRAF and MEK greatly can improve clinical outcomes for melanoma patients by slowing or preventing the development of resistance to BRAFi alone.337 In Table 3, we have compiled an update overview of clinical studies related to combination of BRAFi and MEKi based on information from the ClinicalTrials.go database.
Vemurafenib and Cobimetinib
Hoffmann-La Roche/Genentech sponsored a Phase III clinical trial that employed a double-blind, placebo-controlled design to compare Vemurafenib alone with a combination of Vemurafenib and Cobimetinib (GDC-0973). This trial focused on patients who had not received prior treatment and were diagnosed with BRAFV600-mutattion-positive, unresectable locally advanced, or metastatic melanoma (NCT01689519). Although there was a slight increase in treatment-related side effects, the addition of Cobimetinib to Vemurafenib led to a significant improvement in progression-free survival (PSR) among patients with BRAFV600-mutated metastatic melanoma.337 Also, a recent clinical study demonstrated that the combination of Vemurafenib and Cobimetinib produced a partial response or better in 15 out of patients with papillary craniopharyngiomas, as part of a single-group study (NCT03224767). The average reduction in tumor size was 91%, and progression-free survival (PFS) was 87% at 12 months (95% CI, 57 to 98) and 58% at 24 months.338 Numerous ongoing studies are currently underway, with participants either receiving intervention or undergoing examination to assess the clinical and pathological responses to Vemurafenib and Cobimetinib in BRAF-positive cancers (NCT05768178, NCT02036086).
Encorafenib and Binimetinib
An observational study has been initiated to investigate the real-world effectiveness, impact on quality of life, safety, and tolerability of Encorafenib in combination with Binimetinib for the treatment of unresectable advanced or metastatic BRAF cancers in Germany, Austria and Switzerland (NCT04045691). Several clinical trials are currently underway, with participants either receiving an intervention or undergoing examination to assess the safety, tolerability and efficacy of combining Encorafenib with Binimetinib in patients with BRAFV600-mutant metastatic cancers, including pancreatic cancer (PC) (NCT04390243), NSCLC (NCT05195632, NCT03915951), hairy cell leukemia (HCL) (NCT04324112), multiple myeloma (MM) (NCT02834364) and melanoma (NCT05767879) Despite the significant improvement in survival seen with combination therapy involving of BRAFi and MEKi for BRAFV600-mutant melanomas, there remain limited options for targeted therapy in cases of BRAFnonV600-mutant melanomas. It has been observed that BRAFV600 mutations often co-occur with NF1 deletion, and that these mutations can signal as monomer and dimers when NF1 loss present.339 Consequently, in BRAFnonV600-mutant melanomas with co-occurring NF1 loss-of-function mutations, the combination of BRAFi targeting both monomeric and dimeric BRAF, along with MEK inhibition, has been shown to significantly reduce cell viability in vitro and tumor growth in vivo. Recently, two ongoing clinical studies are exploring whether patients with BRAFnonV600-mutant melanomas may benefit from the currently FDA-approved combination therapy of Encorafenib (BRAFi) and Bonimetinib (MEKi), which is reserved for BRAFnonV600-patients (NCT03839342, NCT03843775). These results are considered hypothesis-generating and will need further confirmation in larger clinical trials in the future.
Dabrafenib and Trametinib
Recent reports have shown that previously untreated individuals with metastatic melanoma and BRAFV600E/K mutations exhibited superior responses to the combination therapy of Dabrafenib and Trametinib compared to Dabrafenib alone.340 The randomized Phase III study funded by GlaxoSmithKline compared the efficacy of combination therapy involving Dabrafenib and Trametinib to that of administered alongside a placebo (Dabrafenib monotherapy) (NCT01584648). Moreover, the Novartis designed non-interventional study has initiated to evaluate the use of Dabrafenib in combination with Trametinib as adjuvant treatment for Chinese patients with stage III melanoma carrying the BRAFV600E mutation after complete resection (NCT04666272). Additionally, a managed access program (MAP) cohort clinical trial provides guidelines to physician for the treatment and monitoring of Trametinib/Dabrafenib, in eligible patients diagnosed with advanced NSCLC featuring the BRAFV600E/K-activating mutations (CTMT212X2002I, NCT04507919).
Assessment of a dual RAF/MEK inhibitor: VS-6766
A novel RAF/MEK inhibitor CH5126766/RO5126766 was discovered while screening for compounds that induce p27. It exhibits more prolonged inhibition of MAPK signaling compared to PD0325901, a well-known ATP-competitive MEK inhibitor.341 VS-6766, also known as CH5126766, is a first-in-class dual RAF-MEK inhibitor currently under investigation as a potential treatment for multiple myeloma and solid tumors harboring different RAS-RAF mutations. It targets two key nodes in the RAS/RAF/MAPK signaling pathway, demonstrating both safety and promising efficacy.342 Numerous interventional clinical trials are ongoing to evaluate the effectiveness of VS-6766, both as a standalone therapy and in combination regimens (Table 4). Notably, an intermittent dosage regimen of VS-6766 and Defactinib has shown clinical activity in patients with recurrent low-grade serous ovarian cancer (LGSOC). These findings have paved the way for an ongoing registration-directed study investigating combination of VS-6766 and Defactinib in patients with recurrent LGSOC (NCT04625270).
Targeted therapy in pediatric BRAF-mutational gliomas
The BRAFV600E mutations gas been associated with reduced responsiveness to conventional treatment in pediatric low-grade gliomas (pLGG).343 Recent findings from the phase II TADPOLE trial suggest that targeted therapy focusing on the RAS/RAF/MAPK signaling is beneficial for pediatric brain tumors with BRAF mutations. In earlier clinical studies, Dabrafenib demonstrated significant clinical efficacy and tolerability, both as monotherapy and in combination with trametinib, for the treatment of pLGG with BRAFV600E mutations (NCT01677741). These results have spurred further exploration of this combination as a first-line therapy (NCT02684058). Notably, Dabrafenib plus Trametinib, when used as a first-line therapy, achieved significantly higher response rates, longer progression-free survival, and improved safety profiles compared to standard monotherapy in pLGG with BRAFV600E mutations.344 In this randomized study, the combination therapy of Dabrafenib and Trametinib showed an overall response rate (ORR) of 47%, while monotherapy yielded an ORR of only 11%. Furthermore, the Dabrafenib and Trametinib combination significantly extended median progression-free survival compared to chemotherapy (20.1 months vs. 7.4 months) and led to clinical improvement in 86% of patients, as opposed to 46% in the monotherapy group. Additionally, the study with BRAFV600E -mutant pediatric high-grade gliomas (pHGG) demonstrated that combining Dabrafenib with Trametinib exhibits an enhanced ORR of 56 %, associated with long-lasting responses and improved duration of response (22.2 months), overall survival (32.8 months), and along with a favorable safety profile, in relapsed/refractory BRAFV600-mutant pHGG patients. In 2023, the FDA granted its approval for the utilization of the Dabrafenib and Trametinib combination as the primary treatment for pLGG in children aged 1 year or older. This milestone marks the successful outcomes of the TADPOLE trials in the pLGG population. Moreover, in 2022, the FDA also sanctioned the use of this combination therapy for patients aged six and above who were dealing with relapsed/refractory BRAFV600 solid tumors. This additional approval serves to reinforce the positive results observed in the pHGG cohort.
Combining BRAF and MEK inhibitors with other targeted therapy
Combining BRAF and MEK inhibitors with other targeted therapies is currently under investigation, aiming to prolong tumor responses.345 These therapies are also gaining traction in neoadjuvant and adjuvant settings (see Table 5).
Patients with BRAFV600-mutant malignancies are benefiting from RAF/MEKi inhibitor therapy. However, long-term efficacy is limited by disease progression in the brain due to inadequate pharmacokinetics (PK) and pharmacodynamics (PD).346,347 Researchers are working on enhancing the intrinsic properties of drugs, such as their PK and PD, to increase the effectiveness of RAS/RAF/MAPK inhibitors. One promising molecule is PF-07284890 (BRAFi), a potent brain-penetrant compound, currently undergoing evaluation in a first-in-human trial for patients (NCT05538130, NCT04543188).
Similar to BRAF/MEKi, combining adjuvant therapy together with specific checkpoint inhibitors, such as anti-PD-1 (Nivolumab and Pembrolizumab) or anti-CTLA-4 (Ipilimumab), has shown promising results. It leads to extended recurrence-free survival and a reduced occurrence of severe adverse events in patients who have undergone resection for stage IIIB, IIIC, or IV melanoma (NCT02388906, NCT04949113, NCT01972347). Additionally, the combination of these immune checkpoint inhibitors with BRAF/MEK-targeted medications appears to be a more viable treatment option due to their immunomodulatory action (NCT02967692, NCT04655157, NCT02858921). Furthermore, a phase I trial study has demonstrated the potential synergy between anti-PD-1 inhibitor and other BRAF/MEK inhibitors in treating patients with BRAF-mutant stage IIIC-IV melanoma (NCT03543969). Meanwhile, a phase II trial is evaluating the impact of Pembrolizumab, Dabrafenib, and Trametinib as a neoadjuvant therapy for patients with BRAFV600E-mutated anaplastic thyroid cancer (ATC) before surgical intervention (NCT04675710). An ongoing neoadjuvant phase II study of Dabrafenib, Trametinib and/or Pembrolizumab in BRAFV600-mutant respectable stage IIIB/C melanoma is investigating the effect of Pembrolizumab, Dabrafenib, and/or Trametinib in reducing tumor sizes before surgery and preventing melanoma recurrence after surgery in patients with BRAFV600-mutant respectable stage IIIB/C melanoma (NCT02858921).
However, a randomized phase II trial, advanced melanoma patients with BRAFV600E/K mutations who received a combination of Dabrafenib (BRAFi), Trametinib (MEKi), and Pembrolizumab (a PD-1-blocking antibody) displayed numerically longer progression-free survival (PFR) and duration of response but experienced with a higher rate of grade 3/4 adverse events compared to those treated with the doublet therapy of Dabrafenib and Trametinib along with a placebo (NCT02130466).
In addition to the established interventions using BRAF and MEK inhibitors, HSP90 inhibitors for patients with the BRAFV600-mutant melanoma are under investigation due to their manageable side-effect profiles when combined with BRAFi.348 HSP90 inhibitors, such as AT13387 or XL888, offer a promising strategy to combat drug resistance resulting from BRAF and MEK inhibition in melanomas.348 Clinical trials investigating the combination of BRAF/MEKi with XL888 or AT13387 are currently underway (NCT02721459, NCT02097225). Moreover, targeting the CDK4 pathway has emerged as a therapeutic approach for patients with BRAF-mutant metastatic melanomas.349 A patient-centered translational study has shown improved clinical outcomes for individuals with stage III or stage IV metastatic BRAF and NRAS wild-type melanoma who do not respond to standard therapy, typically immunotherapy. Lastly, there are ongoing clinical trials exploring various combinations with BRAF/MEKi, which information can be found in Table 5, provided by ClinicalTrials.gov.
RAFi resistance
The discovery of small-molecule BRAF inhibitors was the start of a revolution in the treatment of advanced melanoma.324,350 Some BRAF inhibitors have demonstrated promising results for the treatment of patients with melanoma featuring the BRAFV600E mutation. Despite the exceedingly positive effects of these inhibitors, the development of drug resistance is a widely occurring phenomenon in patients with cancer.16,351 RAFi resistance can be characterized as either intrinsic acquired or adaptive.14
Intrinsic RAFi resistance
Although cancers featuring activating BRAF mutations respond well to RAFi, a considerable fraction of melanoma cells harboring the BRAF V600E mutation show inherent resistance to RAFi.8 BRAFV600E–mutant tumors do not sensitize to either Vemurafenib or Trametinib due to high cyclin D1 and YAP1 expression.8,352,353,354 Activation of phosphatase and tensin homolog (PTEN), forkhead box O3, AKT, or insulin-like growth factor 1 (IGF-1) generally leads to the development of intrinsic RAFi resistance due to the suppression of apoptosis mediated by BCL2-interacting mediator of cell death.355,356,357,358 Intrinsic RAFi resistance represents an adaptation mediated by transcriptional and epigenetic rewiring.359 Relief from the human EGFR 3 feedback inhibition mediated by RAFis and MEK inhibitors reduces the anticancer effects of these treatments against BRAF-mutant thyroid carcinomas.360 Human growth factor (HGF) released by stromal cells activates the HGF receptor (MET), reactivating the MAPK and PI3K–AKT signaling pathways, resulting in the rapid development of RAFi resistance.361
Acquired RAFi resistance
Acquired RAFi resistance generally develops due to the competitive burden introduced by drug treatment, which results in diverse genetic abnormalities or the acquisition of therapy-induced de novo cellular epigenetic and transcriptional reprogramming events.362 In nearly all cases, acquired drug resistance is linked to secondary mutations in RAF, leading to the activation of parallel signaling pathways (such as RTKs) and the “on-target” accumulation of mutations in the RAF pathway.363,364,365,366,367,368 These secondary mutations often occur at “gatekeeper” residues within a RAF ATP-binding site, preventing inhibitors from binding to the existing hydrophobic pocket of the ATP-binding domain. A BRAF mutation at Thr259 confers resistance to some anticancer reagents (e.g., SB590885 and PLX4720), although intrinsic kinase activity is maintained.363 Acquired resistance to Vemurafenib is related to the increased expression of several RTKs, especially PDGF receptor. In addition, CRAF overexpression leads to acquired resistance to BRAF inhibition mediated by a RAF kinase switch in melanoma.368,369 MAP3K8 (COT) reactivates ERK in some melanoma samples that are resistant to RAFis and MEK inhibitors.370,371
Adaptive RAFi resistance
Adaptive resistance to RAFi arises from internal compensating mechanisms, often termed adaptive responses or feedback loops. These mechanisms enhance the survival and proliferation potential of a subset of the initial tumor population, ultimately promoting tumor growth. Such adaptive RAFi resistances can manifest in two main forms: non-transcriptional adaptive response, which regulate post-translational modifications of upstream kinases, and transcriptionally mediated adaptive responses. Many of these inherent resistance mechanisms are primarily underscored by the reactivation of the MAPK signaling pathway, consequently, reduces patient responses. Notably, the reactivation of MAPK via paradoxical signaling (as described in section 4.3) accounts for a significant portion of adaptive mechanisms, contributing to the diminished sensitivity to pathway-targeted drugs and overall patient responses.372
RAFi adaptation to RAF dimerization: feedback inhibition and paradoxical activation in RAF-mutant cancers
Aberrant ERK1/2 signaling can lead to oncogenicity, depending on the extent of ERK1/2 activation373. ERK dysregulation is associated with cell death and senescence,374,375 and the feedback mechanisms in ERK signaling are closely connected to cellular dysfunction.376 The negative feedback of ERK signaling is regulated directly via ERK pathway phosphorylation376 or indirectly through de novo gene expression, such as those encoding dual-specificity phosphatases (DUSPs) and Sprouty (SPRY).377,378 ERK-mediated feedback regulation directly targets RAF proteins, reducing dimerization.379 RAFi resistance reactivates ERK signaling and induces RAFi insensitivity via mechanisms that bypass RAF-dependent signaling.322 ERK reactivation is very subtlety regulated at multiple levels via “intra-pathway” feedback loops and mediated by phosphorylation, intracellular localization, and complex formation (Fig. 8a).380. Under normal conditions, RAS-activated RAF dimerizes and phosphorylates the downstream effectors MEK and ERK. Interestingly, ERK also activates other genes (e.g., DUSPs and SPRY) that inhibit the ERK pathway via a negative feedback loop, resulting in RAS dephosphorylation, which weakens the signal.14,15,378,381,382,383 The network of negative feedback loops limits ERK activation driven by the direct phosphorylation of components in the MAPK cascade.384,385,386 In BRAF-mutant cancers, the MEK–ERK axis is constitutively active, independent of RAS-mediated RAF dimerization, and low levels of basal RAS activation combined with direct phosphorylation result in the strong activation of negative feedback regulators (SPRY and DUSP), inhibiting signaling intermediates, such as EGFR and SOS. As a result, signaling is hampered downstream of activated receptors in BRAFV600E–mutant cells. Treatment with Vemurafenib, a first‐generation ATP-competitive RAFi, results in a temporary reduction in ERK activity,324 which rapidly recovers in BRAF-mutant cancer cells,387 possibly due to the inhibition of ERK-dependent negative feedback.15,388 The restoration of RTK signaling increases RAS-GTP levels, followed by an increase in the homo- or heterodimerization of RAF, which paradoxically reactivates ERK signaling in the presence of RAFis.389 This paradoxical activation of RAFi-insensitive BRAFV600E–mutant dimers result in the formation of a new steady-state signaling network, even in the presence of RAFis.389,390 Thus, RAFi are ineffective against these tumors due to the development of constitutive BRAF-mutant dimers or the transactivation of BRAF–CRAF heterodimers.389,391

Mechanistic basis of RAF inhibitor (RAFi) resistance. a RAFi resistance in RAF-mutant cancer. BRAFV600E–mutant cancers are characterized by hyperactive extracellular signal-related kinase (ERK) signaling during early disease, resulting in negative feedback inhibition of upstream signaling. RAS-GTP expression is minimal, but monomeric BRAFV600E responds to signals. Under these conditions, RAFi inhibits ERK signaling. Subsequent treatment with Vemurafenib suppresses ERK-dependent negative feedback, restoring receptor tyrosine kinase (RTK) signaling. Despite the presence of RAFi, RTK signal restoration elevates RAS-GTP levels, drives the formation of RAFi-insensitive RAF dimers, and reactivates ERK signaling. Over time, negative feedback pathways are partially restored, and a new steady-state condition featuring reactivated ERK signaling develops. b RAFi resistance mechanisms. RAFi resistance promotes significant RAF dimerization through growth factors or RTK activation, NRAS mutation (NRAS Q61), NF1 loss, expression of RAF splice variants (p61), or overexpression of BRAF or CRAF. Reactivation of ERK signaling and RAFi resistance can also develop in a RAF dimerization–independent manner involving MEK mutations or RAF bypass activation by COT (an ERK kinase kinase). This figure was created with BioRender.com
New RAFi families, such as pan-RAF dimer inhibitors and paradox breakers (dimer breakers), appear to be more effective therapeutic options than previous monomeric-targeting RAFis in tumors with RAF dimer–induced signals (Fig. 8b).392,393,394 pan-RAF dimer inhibitors (e.g., LXH254 and LY3009120) bind directly with RAF dimeric domains and inhibit RAF dimerization in BRAFV600E–mutant tumors. However, RAS mutation or amplification, BRAFV600E amplification, and intragenic deletion or BRAF splice variants can lead to RAFi resistance in patients with BRAFV600E–mutant tumors.390,395,396 Importantly, these inhibitors target both active RAF dimers and monomers to block ERK signaling in cancer cells.397 Most pan-RAF inhibitors (e.g., AZ628, Belvarafenib, CCT196969, CCT241161, LY3009120, and TAK-580 [MLN2480]) are successful when tested using in vitro cellular assays.398,399 However, the use of these inhibitors in patients has been limited by a lack of selectivity for mutant BRAF.400 Although LY3009120 exhibits antitumor effects in patients with mutant RAF dimers, persistent treatment with this drug causes severe resistance due to BRAFV600E dimerization.390 A limited dose escalation has been examined to determine the maximally tolerated dose in patients with RAS-mutant cancer, indicating the potential of this drug as an anticancer therapy.400 LXH254, an inhibitor of RAF dimerization, shows specific paralog selectivity, preventing dimeric formation of BRAF and CRAF but not ARAF.397 These drugs are unable to suppress MAPK signaling in normal cells because they inhibit mutant RAF dimers and monomers at great potency compared with their effects on wild-type RAF dimers. As a result, V600E–mutant BRAF that dimerizes with other RAF mutants or with wild-type BRAF or CRAF exhibit drug resistance due to the unintended paradoxical activation of ERK,322,389,393 which may also be responsible for the elevated toxicity and disruption in RAF signaling observed in non-cancer cells. As a result, these medications prove ineffectual when confronted with tumors carrying BRAFnonV600E-mutations. Additionally, an increase in the expression of BRAFV600E -dimers can lead to acquire resistance, primarily because of unintentional paradoxical activation of ERK.390 This paradoxical activation might also be accountable for heightened toxicity and disturbances in RAF signaling, which are observable in non-cancer cells.401
In addition to pan-RAF dimer inhibitors, paradox breakers (e.g., PLX8394 and PLX7904) disrupt homo- and heterodimers that contain BRAF, resulting in reduced paradoxical ERK activation in cells expressing both mutant and wild-type RAF.394,402 These inhibitors selectively bind BRAF to suppress ERK1/2 in BRAFV600E–mutant cells without inducing the paradoxical activation of ERK1/2 observed in RAS-mutant cells. PLX8394 specifically inhibits BRAF in colon adenocarcinoma cells, preventing the paradoxical activation of the MAPK pathway403 with greater potency than Encorafenib, a selective inhibitor of active BRAF monomers, in BRAF-mutant colorectal cancer.404 However, PLX8394 deteriorates ERK signaling via the specific dissociation of BRAF–containing dimers (BRAF homodimers and BRAF–CRAF heterodimers) but has no effect on CRAF homodimers or dimers containing ARAF due to differences in the amino acid residues within the N-terminal region of the kinase domain among RAF isoforms.402 In addition, PLX8394 activates RAF signaling via BRAF dimerization in several melanoma cells.405 RTK-induced mammalian target of rapamycin (mTOR) activity and other bypass signaling may be able to compensate for decreased p-ERK levels when using paradox breakers.406
The ability of RAF-mutant tumors to adapt to RAFi is primarily due to an increase in RAF dimerization (Fig. 8b, RAF dimerization–dependent).106 Mutant RAF dimerization is often associated with RAFi resistance. An inhibitory bypass mechanism can also result in resistance to RAFi, via RAF-independent ERK reactivation (Fig. 8b, RAF dimerization–independent). For example, mutations in the MEK kinase COT (MAP3K8) and MEK1 (encoded by MAP2K1) are closely related to inhibitory bypass resistance that develops in response to RAFi.371,407 RAFi-resistant tumor cells show the rapid recovery of MAPK pathway activation, allowing escape from RAFi therapy; therefore, the total blockade of the entire pathway is essential for stimulating apoptosis in RAF-mutant cancers.408 The combination of RAFi together with other downstream inhibitors (e.g., MEK inhibitors) can maximize MAPK pathway inhibition and minimize cancer resistance. Compared with typical single-agent chemotherapy, Trametinib, a MEK inhibitor, enhances progression-free and overall survival rates in patients with metastatic melanoma who harbor a BRAFV600E/V600K mutations.19 However, BRAFV600E–mutant tumors still can develop resistance to combination RAFi and MEK inhibitor therapy over the course of months to years.409 Most patients who are treated with RAFi and MEK inhibitor combination therapy develop mild (Grade 1–2) toxicity in response to these drugs.22,410,411 Therefore, a solution to the underlying toxicity induced by RAFi is necessary to develop effective therapies for RAF-mutant malignancies.
Strategies to overcome chemoresistance related to targeting the RAS/RAF/MAPK pathway
The hyperactivation of the RAS/RAF/MAPK signaling pathway plays a significant role in the development of numerous human malignancies. While current targeted therapies like BRAFi and MEKi show varying levels of effectiveness across different cancer types, s substantial number of patients develop resistance relatively quickly.412 Ongoing cancer research is now actively addressing the challenge of countering both intrinsic and acquired drug resistance to small-molecule RAF inhibitors. Strategies involving combination therapies are been exploring as potential approaches to address RAF inhibitor resistance.413
Utilizing existing combination therapeutic strategies against RAFi resistance
The RAS/RAF/MAPK inhibitors currently in use have been through clinical development as combination therapies. They can either vertically target specific proteins within the MAPK pathway or horizontally inhibit multiple signaling pathways.414
Vertical pathway inhibition
BRAF-targeted single-agent therapy frequently results in the development of drug resistance due to MAPK pathway reactivation. The vertical blockade of MAPK signaling mediated by RAFi may be supplemented by other agents targeting the intermittent pathway (RTK–RAF–MEK–ERK).415,416,417 Combination therapy has become an important therapeutic goal in class-specific polytherapy.417,418 A vertical blockade combining the RAFi Dabrafenib with the EGFR inhibitor Panitumumab and the MEK inhibitor Trametinib is able to greatly suppress MAPK signaling, demonstrating improved therapeutic efficacy in metastatic colon cancer compared with other therapies.419,420 Although several therapeutic approaches using various medication doses and regimens can extend therapeutic responses, these strategies can also accelerate the development of resistance mechanisms. Continuous drug-related toxicity is also a risk, and the intermittent targeting of mutant BRAF can lead to the development of new drug-resistant tumors.418,421,422,423 As Vemurafenib-resistant cancer cells are reliant on the presence of the drug, withdrawal of drug treatment could be sufficient to induce tumor regression.418,422 Although the development of resistance to multiple inhibitors targeting the MAPK pathway remains to be addressed, this drug combination approach improves patient outcomes.424,425
Horizontal pathway inhibition
Targeting multiple signaling pathways simultaneously, known as horizontal pathway inhibition, can be an effective strategy to counteract potential compensatory or escape mechanisms that selective pathway inhibition might trigger. Cancer cells develop heterogenous resistance mechanisms to evade various drugs, but most of these mechanisms involve the reactivation of MEK–ERK signaling, combined with enhanced signaling output via the PI3K–AKT–mTOR and other pathways.426 Therefore, understanding the MAPK-related mechanisms underlying the development of resistance to RAFi and combination therapies may improve their potential therapeutic effects.427,428 Reactivation of ERK signaling can also occur via MAPK-independent pathways, including the bypassing of the paradoxical MAPK arm through PI3KC mutations,429,430 activation of an AKT–ETS-1–mediated positive feedback loop,431 loss of PTEN activity432 overexpression of mTOR,433 or manipulation of autophagy proteins (ATGs)434,435 (Fig. 8b, MAPK-independent). The combined inhibition of both the RAF–MEK and PI3K–AKT–mTOR pathways has a synergistic pro-apoptotic effect. For example, the addition of an AKT or mTOR inhibitor to Vemurafenib therapy or MEK inhibitor therapy results in synergistic effects.436 SHP2 inhibitors efficiently bypass the paradoxical MAPK signaling arm in RAS-mutant tumors treated with RAFi.437,438
The sequential or intermittent dosing of BRAF/MEK inhibitors
Patients with metastatic melanoma harboring activating BRAF mutations have demonstrated improved survival when receiving combination therapy with both BRAFi and MEKi.439 Unfortunately, the therapeutic effectiveness of such treatments often proves transient, with resistance emerging within a few months.440 Intermittent therapy has been explored as a strategy to halt or delay resistance to BRAF inhibition.422 In pursuit of curation approach for melanoma patients with BRAF mutations, researchers have proposed that modifying dosing patterns could prevent the development of lethal drug resistance and extend the durability of BRAFi responses.441 The optional implementation of this strategy in treating advanced BRAFV600E-mutant melanoma patients is currently under investigation in randomized phase II and III clinical trials comparing intermittent and continuous dosing schedules of BRAFi in combination with MEKi (NCT02224781, NCT02196181). However, one completed trial did not need its primary objective of improving progression-free survival, indicating that the experimental intermittent schedule of both Vemurafenib and Cobimetinib did not demonstrate superior anticancer activity compared to the regular continuous schedule (NCT02583516).
Potential alternative strategies for further optimizing the use of RAF/MEKi
The potential of different combination therapies is being explored to delay the emergence of resistance to MAPKi or to precisely target BRAF/MEKi-resistance. Recent preclinical research has identified alternative strategies for combating resistance, including bolstering apoptosis, modulating autophagy, and directing attention on mitochondrial metabolism and phenotyping switch (Fig. 9a).

The possible therapeutic strategies for overcome resistance to RAS/RAF/MAPK inhibitors. a Alternative approaches to overcome MAPK inhibitor (RAFi) resistance. b Targeting autophagy. In cancer cells, autophagy-related genes are functionally and physically associated with mitogen-activated protein kinase (MAPK)-targeted therapy and cancer resistance induced by MAPK signaling inhibitors (e.g., RAFi, MEKi). The regulatory mechanisms involved in autophagy induction and the mediators that regulate autophagy are described in each rectangle. Arrows and bars indicate stimulating and inhibiting signals, respectively. This figure was created with BioRender.com
Targeting mitochondrial and energy metabolism
Metabolic disorders have long been associated with cancers, characterized by metabolic reprogramming, which involves alterations in metabolic pathways enabling cancer cells to proliferate rapidly, survive in hypoxia and nutrient-deprived conditions, and evade the immune system.442 Furthermore, metabolic reprogramming events play a crucial role in tumorigenesis, drug resistance, and metastases in melanomas.443 Research on chemotherapy-resistant, slow-cycling BRAFV600E melanomas has shown increased level of oxidative phosphorylation (OXPHOS) enzymes and blocking them resulted in cell death. Additionally, the pivotal role of the microphthalmia-associated transcription factor (MITF) and PGC1A in regulating phenotyping plasticity is indispensable for steering cellular metabolism towards OXPHOS. A two-tiered strategy involves combining anticancer drugs that target rapidly proliferating melanoma cells with drugs that suppress the slow-cycling, drug-resistant subpopulation. Several drugs targeting mitochondrial respiration have been investigated in preclinical settings with promising results.444 For instances, β-sitosterol, due to its favorable tolerability, excellent bioavailability, and ability to inhibit mitochondrial respiration, serves as a viable adjuvant to BRAFi therapy for individuals with or at risk for melanoma brain metastases. Combining Vemurafenib with either β-sitosterol or a functional knockdown of mitochondrial complex I completely eliminated BRAFi resistance.444 Also, uncouplers like SR4 and Niclosamide that disrupt mitochondrial OXPHOS may be effective as first-line adjuvant treatments for melanoma patients who are not responding to MAPK inhibitors.445
Cancer starvation therapy, which relies on glucose restriction to induce oxidative stress and slow tumor growth has proven effective in curbing the rapid multiplication of cancer cells.446 The glucose analog 2-Deoxy-D-glucose (2-DG), when used in combination with Cisplatin or Erlotinib, enhances the cytotoxicity of head and neck squamous cell carcinoma (HNSCC) cells through metabolic oxidative stress. While 2-DG treatment alone may not cause significant cell death in most cancer cells, it does sensitize them to oxidative stress induced by radiotherapy or chemotherapy. Using 2-DG as an alternative therapeutic approach for the treatment of radioresistant and highly glycolytic cervical malignancies involves inhibiting intracellular redox metabolism and glycolysis.447 Also, some transcriptional factors, including hypoxia-inducible factor-1 (HIF1), MYC, and MONDOA (MLXIP), involve the regulation of glycolysis by inhibiting BRAF in melanomas, and further the combined inhibition of BRAF and glycolysis triggers cell death in BRAFi-resistant melanoma cells.448 Although there is currently no clinical evidence of this, targeting glucose metabolism could potentially be adapted therapeutically to overcome BRAFi resistance, particularly in pancreatic cancer driven by KRAS-dependent resistance to MAPK inhibition.449
Therapeutic modulation of metabolic difference in lipid metabolism can be a promising strategy to overcome BRAFi resistance. AMP-activated protein kinase (AMPK), a nutritional sensor present in both healthy and malignant cells, regulates lipid breakdown, phosphorylated, and inactivates acetyl-CoA carboxylase, preventing fatty acid production, and promotes lipid utilization through beta-oxidation in mitochondria.450 AMPK-dependent phosphorylation of BRAF at serine 729 reduces MAPK signal in BRAF wild-type cells by Inhibiting BRAF interaction with CRAF and KSR1.451 A study elucidated how 14-3-3 controls dimerization-driven RAF activation and mitigate the negative side effects of RAF inhibitors in cancer therapy.452 Additionally, the combination of Phenformin and PLX4720 resulted in tumor regression in some mice models. This study suggests that combining AMPK activators, such as Phenformin, with BRAF inhibitors may offer significant therapeutic advantages in melanoma treatment.
Targeting phenotyping switching
Phenotypic switching and metabolic reprogramming play distinct roles in the development of resistance in therapy-resistant clones of BRAFV600-mutated melanoma cells.453 The significance of MITF, a primary transcription factor in melanocyte differentiation/dedifferentiation, has been emphasized in relation to phenotypic switching. It has been observed that drug-sensitive cell lines and patient biopsies typically exhibit high MITF levels, whereas inherently resistant cell lines and patient biopsies demonstrated low MITF expression but elevated levels of NF-kB signaling and the receptor tyrosine kinase AXL.454 These MITF-low/NF-kB-high melanomas display resistance to ERK, RAF, and MEK inhibition in vitro. Remarkably, MITF expression show an inverse correlation with AXL expression, suggesting that cell lines can be categorized based on their metastatic potential regardless of whether they carry the BRAF or NRAS mutations.455 Additionally, Wnt5a signaling directly influences the motility and invasion of metastatic melanoma cells with low MITF levels.456 The phenomenon of EMT observed in cancer cells bears a striking resemblance to phenotypic switching.457 Exploring phenotypic switching may be essential in developing novel therapeutics capable of restoring sensitivity to RAFi or MEKi, providing a critical avenue for overcoming resistance.
Targeting the PI3K/AKT, tumor microenvironment and inflammatory responses
Resistance commonly develops in the majority of advancing melanomas through the reactivation of MAPK signaling, often driven by alterations in BRAF, NRAS, and MEK.458 While the activation of the PI3K/AKT pathway does not limit patient responses to BRAF/MEK inhibition, a small subset of resistant melanoma relies on the compensatory PI3K/AKT signaling cascade.459 Initial clinical trials with PI3K inhibitors in combination with Vemurafenib have shown promising results.460 In challenging-to-treat BRAF-mutant mCRC patients, a combination therapy comprising Alpelisib (a PI3K inhibitor), Encorafenib, and Cetuximab (an anti-EGFR) has demonstrated effectiveness.461 Notably, in BRAFV600E-mutated CRC cancers, activation of the PI3K/AKT pathway serves as a mechanism for both innate and acquired resistance to BRAF inhibitors, with proposed combination approaches to improve outcomes in this challenging patient population.462 PTEN, a major antagonist PI3K, is frequently mutated in various cancer tissues and is implicated in 17% of melanoma cases, 10% of colorectal cancer cases, and 4% of lung adenocarcinoma cases.463 Mutations in PTEN, resulting loss of function, are associated with a higher proportion of BRAF-mutant alleles, and are linked to lower progression-free survival (PFS), overall survival (OS), and response rates in melanoma patients. PTEN loss induces BRAFi-resistance in melanoma cells by inhibiting BIM expression.355 The involvement of AKT3 and the activation of FOXO3a play a role in enhancing apoptosis when PLX4720 and a PI3K inhibitor are combined to treat PTEN-negative cells.355 Importantly, HSP90 inhibition appears to be more effective in restoring BIM expression and downregulating Mcl-1 expression compared to the combined MEK/PI3K inhibitor therapy, making it a potentially highly effective strategy for managing the diverse array of resistance mechanism observed in BRAF resistance.464 AEBP1 (adipocyte enhancer-binding protein 1) is significantly upregulated in melanoma cells resistant to PLX4032 due to over-activation of the PI3K/AKT/CREB signal pathway. Blockage of this signaling pathway effectively restores the PLX4032-resistant phenotype of melanoma cells.
The development of resistance to BRAFi resistance in melanoma is a well-recognized challenge, stemming not only from genomic or epigenetic aberrations but also from the crucial role played by the tumor microenvironment. In response to BRAF inhibition, melanoma cells and fibroblasts undergo microenvironmental changes that enhance PI3K/AKT survival signaling, allowing tumor cells to evade treatment.465 When treated with Emurafenib, melanoma cells release TGF-β, triggering fibroblasts to increase expression levels of α-smooth muscle actin (α-SMA), neuregulin (NRG), and fibronectin. Paradoxically, Vemurafenib’s off-target effects lead to the secretion of hepatocyte growth factor (HGF) by fibroblasts. Furthermore, tumor-associated macrophages (TAMs) contribute to the development of a pro-tumorigenic microenvironment that fosters resistance to MAPKi therapy by providing an abundance of oxygen, nutrients (resulting in hypoxia and metabolic stress), and extracellular matrix proteins.466 Subsequently, TAMs secrete angiotensin, COX-2, IFN, and IL-1, further promoting the growth and metastasis of melanoma.467 Thus, to enhance the effectiveness of combination therapy for melanoma patients, targeting spatiotemporal interactions within tumor microenvironments holds significant promise.
Inflammatory signals establish a novel epigenetic program that silences a specific set of genes contributing to inflammation-induced cellular transformation in tumor cells.468 Tumor-induced inflammatory responses have adverse effects on the adaptive immune system and open up possibilities for new therapeutic strategies address the immunological dysfunctions caused by tumors.469 A novel independent predictor of Anthracycline/Taxane neoadjuvant chemotherapy response in breast cancer is the presence of tumor-associated lymphocytes, aiding healthcare professionals in identifying patients who will benefit most from this treatment.470 Pharmacological targeting of key factors derived from tumor-associated inflammation presents a unique strategy to eliminate therapy-resistant tumors. Celecoxib, a COX-2 inhibitor, significantly reduced tumor burden by 90% and notably delay tumor growth induced by the BRAFi inhibitor PLX7420.471 Copper chelation emerges as a potential treatment strategy for a specific subset of tumors characterized by activating BRAFV600E mutations. Copper chelators, commonly used to treat Wilson’s disease, inhibit tumor formation in human or mouse cells harboring BRAFV600E mutations or engineered to be resistant to BRAFi.472
Targeting apoptosis
Tumor cells often develop resistance to chemotherapy and radiation by disrupting the regulation of apoptosis-related mediators, especially by increasing the levels of anti-apoptotic BCL-2 family proteins.473 BH3 mimics have gained popularity as a means to induce cell death effectively by targeting the primary anti-apoptotic proteins without necessitating significant involvement of pro-apoptotic proteins.474 ABT-263, a BH3 mimic, when used in combination with selective BRAFi (PLX4720), enhances both the extent and speed of responses in treatment-naïve patients with BRAFV600E-mutated melanoma but it is less effective in individuals who have developed resistance to these drugs.475 Surprisingly, ABT-737 significantly increases the sensitivity of melanoma cell lines to conventional chemotherapeutics, leading to BIM-mediated apoptosis.476 Inhibiting anti-apoptotic BCL-2 proteins can improve primary PLX-4032 responses and reduces the development of resistance to both targeted and standard therapies.477
In addition to BCL-2 family proteins, STAT3 is a promising target for anti-apoptotic drug development against BRAFi resistance. Inhibiting the STAT3 pathway demonstrates significantly higher cytotoxicity compared to the currently used therapeutic drug PLX-4032. This approach targets both BRAF-mutant and WT melanoma cells without selectivity.478 Combing Vemurafenib with STAT3 silencing or miR-579-3p overexpression proves effective in overcoming Vemurafenib resistance in cancer cells.479 Furthermore, activation of the RAS-RAF-MAPK pathway is associated with the prevention of Caspase-3 activation, cell protection against apoptosis, and direct phosphorylation of caspase-9.213 Caspase-3 inhibits MEK1 through proteolytic means, leading to reduced pro-survival ERK signaling and increased susceptibility of cell to apoptosis.480
Endoplasmic reticulum (ER) stress can potentially activate various apoptotic signaling pathways in melanoma cells in a context-dependent manner. The MEK/ERK signaling pathway plays a pivotal role in preventing caspase-4 activation induced by ER stress. Additionally, the apoptosis repressor with caspase recruitment domain (ARC) proteins appears essential in preventing Caspase-8 activation in melanoma cells under ER stress.481 Inhibiting the MEK/ERK pathway makes melanoma cells more susceptible to apoptosis induced by ER stress, partially through caspase-4 activation, and is also associated with the inhibition of ER chaperon glucose-regulated protein 78 (GRP78) production.482
Expanding current strategies using high-throughput technology
High-throughput techniques for profiling tumor-associated gene expression, including miRNAs, have a great prospect for clinical applications. A recent advancement involves a high-throughput quantitative method based on RCA, enhancing the sensitivity of detecting miRNAs associated with Triple-negative breast cancer (TNBC) using fluorescence-encoded microspheres.483
Genome editing
The translation of gene editing from theory to clinical practice has been accelerated by recent advancements in programmable nucleases, including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and CRISPR/Cas9-associated nucleases.484 One of the pioneering pooled CRISPR screens involved a genome-scale knockout library of melanoma cells treated with the BRAF inhibitor, Vemurafenib, revealing genes responsible for treatment resistance.485 Another study found that CRISPR/Cas9 editing of RAS and MEK mutant cells led to resistance to BRAF and MEK inhibitors, but MEK1 Q56P restored sensitivity to the MEK/BRAF inhibitor combination, and KRAS G13D increased sensitivity to immunotherapy.486
Cancer vaccines
Cancer vaccines aim to activate latent or unresponsive tumor-specific T cells, bolstering the innate immune defense against cancer.487 Various cancer immunotherapies have been developed to trigger tumor-specific immune response in tumor patients. Clinical trials have demonstrated the effectiveness of cytosine-phosphorothioate-guanine oligodeoxynucleotides (CpG ODN) as a cancer vaccine adjuvant. The GO-PEI-OVA-PEG-CpG nano-vaccine exhibited favorable safety profiles and significant anticancer efficacy, extending mouse survival, and limiting tumor growth in vivo. When combined with NLG918 (antitumor immunotherapy), the vaccine displayed even greater therapeutic.488 Peptide-based drug delivery system, like cancer vaccines, offer an additional promising avenue to enhance existing BRAFi and MEKi strategies. An early-phase I clinical trial is therapy with the administration of six melanoma helper peptides (6MHP). The trial also assesses the impact of peptide vaccination, BRAF inhibitors, and MEK inhibitors on the immune system using participant blood and tumor samples. Initial results indicate that 77% of patients exhibited antibody responses to 6MHP by week 7, peaking 6 weeks after the last vaccine and persisting for 6 months (NCT02382549).
Leveraging online resources for compound screening
Resources for studying the RAF-centered signaling network are expanding, facilitating the discovery of protein sets that physically interact with MAPK network through high-throughput techniques, such as protein-interaction based screening approaches.489 The development of the NanoBRET screening platform employs live-cell bioluminescence resonance energy transfer (BRET) to identify substances that modify the binding between activated KRAS and CRAF kinase.489 Additionally, a subtractive forward two-hybrid method is employed to identify small molecule that disrupt the interaction between RAS and RAF.490 Furthermore, numerous online databases, including Cell Circuits, NCBI GEO, DIP, BIND, KEGG, NetPath, LINCS, BioGrid, and expert systems, enable individual researcher to construct and query protein interaction maps tailored to their gene of interest.
Targeting autophagy: a new therapeutic avenue for RAS/RAF/MAPK inhibitor-resistance
Autophagy is generally affected by a wide range of anticancer medications, including DNA-damaging agents, microtubule-targeted therapies, antimetabolites, death receptor agonists, hormonal agents, antiangiogenic agents, proteasome inhibitors, histone deacetylase inhibitors, and kinase inhibitors.262 Both chemotherapeutic agents and ionizing radiation stimulate autophagy by inducing autophagosome formation.262,491,492 Some agents promote autophagosome formation but prevent lysosome fusion, consequently blocking autophagic flux.493 For example, mTOR inhibitors, such as Rapamycin and Temsirolimus, are well-known anticancer drugs that directly promote autophagy by disrupting the activity of the negative autophagy regulator mTOR complex 1 (mTORC1).494 ABT-737, a Bcl-2 inhibitor, directly targets the core autophagy machinery.495,496
Some autophagy inhibitors can attenuate tumor growth. Autophagy inhibition increases Icariin-induced cytotoxicity in colorectal cancer cells.497 In addition, several PI3K inhibitors, including 3-Methyladenine, Wortmannin, and LY294002, used as anticancer therapeutics, directly inhibit autophagy.498,499 Anticancer therapies that activate autophagy frequently induce a pro-survival response that may contribute to the development of drug resistance and refractory cancer.500,501,502,503 Therefore, drugs that target autophagy should be considered for combination therapy strategies together with other anticancer drugs. The antimalarial drugs Chloroquine (CQ) and Hydroxychloroquine (HCQ) are currently being studied as potential components of combination therapies together with standard treatments in multiple tumor types.504,505,506
Autophagy-associated RAFi resistance
Two recent studies have delved into the potential therapeutic benefits of combining autophagy inhibitors with targeted therapy.507,508 Based on a wealth of preclinical research, clinical trials, and recent literature, we have compiled a summary of autophagy-related RAFi-resistance and potential therapeutic interventions. MAPK inhibitors are used as conventional therapy in cancer patients with activated RAF mutations. However, their antitumorigenic effects and clinical benefits are temporary due to the eventual development of drug resistance and relapse. Drug resistance can occur when upstream factors activate these signaling pathways, bypassing RAF inhibition (Fig. 9b). The MAPK pathway is often aberrantly stimulated by BCR-ABL oncoproteins through direct binding with RAS activators, and aberrant MAPK activation plays significant roles in the onset and progression of leukemia.509 In addition, MAPK phosphorylation is frequently associated with autophagic structures, suggesting that microtubule-associated protein 1 A/1B-light chain 3 (LC3)-II–positive membranes and ATG5- and ATG12-positive pre-autophagosomes may serve as scaffolds or cellular signaling platforms for the RAF–MEK–ERK cascade, facilitating ERK pathway activation510. The ATG7–ATG5–ATG12–LC3-II cascade modulates MAPK phosphorylation in an unconventional manner (Fig. 9b). Atg4b, an LC3-specific protease, catalyzes the conversion of the LC3 precursor into its active forms and deconjugates modifiers from phospholipids.511,512
AMPK plays a crucial in promoting resistance to RAS-RAF-MAPK pathway inhibitors triggering autophagy.513 The oncogenic BRAF negatively regulates the tumor suppressor LKB1, fostering the proliferation of melanoma cells. Consequently, the RAS/RAF/MAPK pathway initiates autophagy activation through the LKB1-AMPK-ULK1 signaling axis. To induce mTORC1 inhibition and cell cycle arrest in response to energy stress, AMPK must phosphorylate Raptor, a part of mTORC1.514 The oncogenic BRAF leads to heightened basal autophagy and increased resistance to apoptosis in cutaneous melanomas, also causing chronic ER stress.515 ER stress-induced autophagy is diminished in cells lacking IRE1 or treated with a JNK inhibitor, highlighting the essential role of the IRE1-JNK pathway in autophagy activation following ER stress.516 Activation of the IRE1/ASK1/JNK and TRB3 pathways is induced by p38 activation, which is driven by BRAFV600E.515
BCL-2 family members (BCL-2, BCL-xL, and MCL-1) inhibit autophagy, whereas BNIP3 stimulates autophagy by releasing Beclin1 from the BCL2/Beclin1 complex.496 p53 family isoforms contribute to acquired resistance to targeted MAPK inhibitors in melanoma cells,517 and p53-dependent activation of damage-regulated autophagy modulator-1 (DRAM-1) triggers autophagy.518 RAF-mutant–induced chronic ER stress stimulates basal autophagy, and RAF-mediated p38 activation augments the IRE1–ASK1–JNK and Tribbles 3 (TRB3) pathways.519 DMP1 also acts as a critical molecule connecting oncogenic RAS/RAF/MEK/ERK signaling with the tumor-suppressive ARF–MDM2–p53 pathway.520 In conclusion, RAFi resistance in cancer is often associated with increased autophagy, driven by multiple interconnected pathways and molecular events, suggesting that understanding these mechanism may have implications for developing more effective treatment strategies in cancer.
Targeting autophagy to overcome RAFi resistance as a therapeutic strategy
BRAFV600E-mutant MAPK activation in cancer highlights the potential therapeutic benefits of using MAPK inhibitors, such as Vemurafenib, which have demonstrated beneficial outcomes when used to treat patients with late-stage melanoma.324 In addition, pharmacological inhibitors of BRAFV600E-mutant and MEK have been used to treat metastatic carcinoma.331,337,521 However, the rapid development of drug resistance has limited the use of these drugs in cancer therapy, resulting in temporary benefits lasting from several months to less than 2 years.522 When combined with immune checkpoint inhibitors targeting programmed death protein 1 or ipilimumab targeting cytotoxic T lymphocyte–associated protein 4, these medications greatly enhance life expectancy up to 4 years in 50% of patients with metastatic melanoma.523,524 However, combination therapy using RAFis or MEK inhibitors together with immune checkpoint inhibitors remains restricted to patients with certain types of cancer.525,526,527,528 To enhance therapeutic efficacy, a wide range of adaptive responses must be studied, including therapy resistance mechanisms and autophagy, which enable drug sequestration and cell survival.
Inhibition of adaptive protective autophagy induced by MAPK re-sensitization in RAFi resistance
Numerous avenues that lead to acquired resistance to RAFis generally utilize multiple methods to target the same or parallel pathways.366 Vertical inhibition of the MAPK signaling pathway using combinations of RAFi together with MEK inhibitors has received FDA approval as a first-line treatment strategy for patients with advanced RAF-mutant melanoma, NSCLC, and thyroid cancer. Suppression of RAF-mutant signaling promotes autophagy in cancer cells,25,26 suggesting that targeting autophagy in cancer cells may be desirable when using pathway-targeted inhibitors in RAF-mutant cancer.529 Many tumors harboring RAS and RAF mutations develop an “addiction” to autophagy, which is required to maintain cellular homeostasis; thus, the inhibition of protective autophagy induced by MAPK pathway activation may represent a therapeutic option in RAS- and RAF-mutant malignancies.435,530,531,532,533 The genetic suppression of autophagy genes may also represent a novel approach for the treatment of lung cancers harboring mutant RAS534 or BRAFV600E.535 Pharmacologic RAF inhibition dramatically increases autophagy in RAFi-resistant melanoma cells,26 and the genetic or pharmacological inhibition of autophagy counters RAFi resistance in brain tumors.434 Additionally, inhibiting autophagy in RAFi-resistant tumor cells in vitro and in patients reverses drug resistance.434 Combining autophagy inhibition with MEK inhibition increases apoptotic cell death in a RAF-mutant colorectal cancer cell line.536 The combined inhibition of autophagy and MEK significantly accelerates regression in RAF-mutant patient-derived colorectal cancer cells xenografted onto an animal model.537 Similarly, RAF-mutant thyroid cancer cells are sensitive to autophagy targeting in the presence of vemurafenib.320,538
Therapeutics success beyond autophagy targeted RAFi therapy
In cancer, autophagy plays adaptive and protective roles when MAPK signaling is hampered.539 Accordingly, combined treatment using both MAPK and autophagy inhibitors represents one of the best options for treating various carcinomas.26,531 However, tumor responses to autophagy inhibitors are not clinically consistent due to non-uniform permeability across tumor tissues and the potential toxicity of combination treatments using multiple chemotherapeutics.540
Because autophagy can act as a double-edged sword during tumorigenesis, autophagy-targeted treatment outcomes are highly dependent on cancer types, contexts, and stages.541,542 In addition, the inhibition of autophagy initiation and late fusion steps results in differential responses. For example, early-stage autophagy inhibition using ULK1/2 inhibitors (e.g., MRT 68921) decreases tumor cell death, whereas inhibitors (HCQ) that target the later fusion stage greatly increase cell death in mesothelioma cells.543 CQ and its variants, such as HCQ, are currently the most frequently tested autophagy inhibitors in cancer clinical trials. These chemicals impede the late fusion stage between autophagosomes and lysosomes, causing deacidification and impairing enzymatic function.540,544,545 Interestingly, these inhibitors lack selectivity, affecting total lysosomal function.546 However, CQ largely improved the sensitivity of Vemurafenib in an ex vivo primary cell culture derived from patients harboring the BRAFV600E mutation435. Autophagy-independent vascular normalization of CQ inhibits tumor invasion and metastasis by enhancing chemotherapeutic effects.547,548 The autophagy inhibitor HCQ has been more widely used in clinical trials over CQ because HCQ is less toxic at optimum doses.504,549,550,551 HCQ shows beneficial effects and synergistic effects when combined with the mTOR inhibitor, Temsirolimus, in melanoma552 and breast cancer.553,554 Importantly, HCQ affects chemotherapy and radiation sensitivity in non-selective cancers.555 Lys05, a novel lysosomal autophagy inhibitor, has also been suggested as a potential therapeutic agent for cancer.556,557
CQ and HCQ are non-selective autophagy inhibitors, and specific small-molecule inhibitors that target earlier stages of autophagy are preferred for cancer therapy.547,558,559,560 ULK1, a key autophagy regulator, can be specifically targeted by cellular energy status regulators, such as mTORC1 and AMPK.561 AMPK, a low-energy sensor, activates ULK1 to induce autophagy.562 ATG13, an autophagy protein in the ULK1 complex, is a static autophagy marker that corresponds closely with autophagic flux in mesothelioma.563 In addition, multiple ATP-competitive ULK1 kinase inhibitors have demonstrated compelling evidence to support clinical use,564,565 such as the successful suppression of NSCLC growth by SBI-0206965.565,566 Vacuolar protein sorting 34 (VPS34), a component of the Beclin1 complex, also plays an important early role in autophagosome formation and is considered a potential therapeutic target.567,568 Several VPS34 inhibitors (e.g., SAR405) have demonstrated anticancer effects in kidney carcinoma cells,569 including VPS34-IN1, a particularly powerful and selective VPS34 inhibitor with cancer therapeutic potential.570 Furthermore, transitional small molecules tend to form prolonged associations with target proteins, leading to the inhibition of their enzymatic activity. This extended binding duration poses challenges in devising small molecule structures and countering drug resistance resulting from target mutations. In contrast, the proteolysis-targeting chimera (PROTAC) exhibits the ability to specifically eliminate target proteins, thereby intensifying the cytotoxic impact on mutant tumors. In the realm of both fundamental research and pharmaceutical advancement, the autophagy-targeting chimera (AUTOTAC) provides a versatile platform for precise proteolysis.571 Also, high-throughput technology presents a promising approach for identifying selective autophagy inhibitors that are both effective and less cytotoxic when used in combination therapy. Arzonol, for instance, emerged as a distinct chemotherapeutic candidate through high-throughput screening of natural compounds aimed at modulating autophagy.572 Overall, targeting autophagy initiation, together with improved investigations of the functional mechanisms underlying existing chemotherapies, could be crucial for developing novel cancer therapies.
Conclusion and future directions
RAF inhibitors exhibit exceptional clinical efficacy in patients with RAF-mutant carcinoma, although their therapeutic effects are restricted due to the development of drug resistance. Recent in vitro and in vivo translational studies have elucidated the molecular basis underlying the development of RAFi resistance in cancer. Obtaining additional biological insights into the process of drug resistance represents a necessary, long-term goal for the development of new RAFi and combination treatments able to slow or prevent the development of drug resistance in cancer. The combined regulation of MAPK signaling and RAFi-induced autophagy may represent a potential strategy for overcoming drug resistance and inform the development of novel cancer therapies. Because autophagy can act as either a pro-death or a pro-survival factor in tumorigenesis, targeting autophagy is highly dependent on cancer type and stage. Autophagy activation can promote tumorigenesis, and autophagy inhibition can improve the therapeutic efficacy of RAFi. A better understanding of the molecular mechanisms and precise targets underlying the complex process of autophagy modulation could lead to the development of agonists or blockers that could serve as potential therapeutic agents for future use in RAF-mutant cancers.
Pharmacological autophagy inhibitors hold significant potential in combating resistance to RAF inhibitors in cancer patients with RAF mutations. Recent research highlights the efficacy of compounds like 3-Methyladenine and hydroxychloroquine (HCQ) in suppressing autophagy when combined with anti-EGFR monoclonal antibodies (mAbs) and checkpoint inhibitors. The BAMM trial (BRAF Autophagy and MEK Inhibition in Melanoma), which combined Dabrafenib, Trametinib, and HCQ, demonstrated these combinations are effective and safe in patients with BRAFV600-mutant melanoma. Beyond classical autophagy inhibitors, emerging indirect inhibitors like ABT-737, natural products such as Resveratrol, and synthetic compounds like Quinacrine may warrant clinical investigation. The AUTOTAC chemical biology platform has garnered significant attention within the area of targeted protein degradation. It employs bifunctional molecules that consist of ligand binding to specific targets linked with ligands targeting the autophagy processes. Furthermore, recent advancements in autophagy-based degraders and molecular adhesives, such as autophagosome-tethering compounds (AATEC) or lysosome-targeting chimera (LYTAC), have enabled the successful degradation of numerous target proteins by routing them to the lysosome.
Also, the genetic inhibition of autophagy genes via RNA interference and CRISPR/Cas9 targeting holds promising. Evaluating the safety of combing BRAF and MEK inhibitor therapy with melanoma helper peptides (6MHP) administration assesses the impact on the immune system. In addition, cancer vaccines aim to activate tumor-specific T cells, strengthening the immune defenses. Future research emphasizes high-throughput technology for preclinical evaluations of cytotoxicity and combination therapy between RAS/RAF/MAPK inhibitors and other compounds.
References
Zhang, W. & Liu, H. T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18 (2002).
Gysin, S., Salt, M., Young, A. & McCormick, F. Therapeutic strategies for targeting ras proteins. Genes Cancer 2, 359–372 (2011).
Karnoub, A. E. & Weinberg, R. A. Ras oncogenes: split personalities. Nat. Rev. Mol. Cell Biol. 9, 517–531 (2008).
Wang, W., Fang, G. & Rudolph, J. Ras inhibition via direct Ras binding–is there a path forward? Bioorg. Med. Chem. Lett. 22, 5766–5776 (2012).
Spiegel, J. et al. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 10, 613–622 (2014).
Sayyed-Ahmad, A. & Gorfe, A. A. How to make an undruggable enzyme druggable: lessons from ras proteins. Adv. Protein Chem. Struct. Biol. 122, 181–202 (2020).
Cuadrado, A. et al. H-ras and raf-1 cooperate in transformation of NIH3T3 fibroblasts. Oncogene. 8, 2443–2448 (1993).
Fedorenko, I. V., Paraiso, K. H. & Smalley, K. S. Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem. Pharmacol. 82, 201–209 (2011).
Downward, J. Targeting RAS signaling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22 (2003).
Leicht, D. T. et al. Raf kinases: function, regulation and role in human cancer. Biochim. Biophys. Acta 1773, 1196–1212 (2007).
Dillon, M. et al. Progress on Ras/MAPK signaling research and targeting in blood and solid cancers. Cancers 13, 5059 (2021).
Hobbs, G. A., Der, C. J. & Rossman, K. L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129, 1287–1292 (2016).
Li, W., Chong, H. & Guan, K. L. Function of the Rho family GTPases in Ras-stimulated Raf activation. J. Biol. Chem. 276, 34728–34737 (2001).
Holderfield, M., Deuker, M. M., McCormick, F., McMahon, M. & Targeting, R. A. F. kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 14, 455–467 (2014).
Yaeger, R. & Corcoran, R. B. Targeting alterations in the RAF-MEK pathway. Cancer Discov. 9, 329–341 (2019).
Haarberg, H. E. & Smalley, K. S. Resistance to Raf inhibition in cancer. Drug Discov. Today Technol. 11, 27–32 (2014).
Oddo, D. et al. Molecular landscape of acquired resistance to targeted therapy combinations in BRAF-mutant colorectal cancer. Cancer Res. 76, 4504–4515 (2016).
Hatzivassiliou, G. et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 501, 232–236 (2013).
Flaherty, K. T. et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 367, 107–114 (2012).
Michielin, O. et al. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-updagger. Ann. Oncol. 30, 1884–1901 (2019).
Adamopoulos, C. et al. Exploiting allosteric properties of RAF and MEK inhibitors to target therapy-resistant tumors driven by oncogenic BRAF signaling. Cancer Discov. 11, 1716–1735 (2021).
Welsh, S. J. & Corrie, P. G. Management of BRAF and MEK inhibitor toxicities in patients with metastatic melanoma. Ther. Adv. Med. Oncol. 7, 122–136 (2015).
Sinha, R. et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, prevention and management of the main treatment-related skin toxicities. Br. J. Dermatol. 167, 987–994 (2012).
Yun, C. W. & Lee, S. H. The roles of autophagy in cancer. Int. J. Mol. Sci. 19, 3466 (2018).
Goulielmaki, M. et al. BRAF associated autophagy exploitation: BRAF and autophagy inhibitors synergise to efficiently overcome resistance of BRAF mutant colorectal cancer cells. Oncotarget. 7, 9188–9221 (2016).
Ma, X. H. et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Invest. 124, 1406–1417 (2014).
Lee, C. S. et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc. Natl Acad. Sci. USA 116, 4508–4517 (2019).
Molina, J. R. & Adjei, A. A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 1, 7–9 (2006).
Harvey, J. J. An unidentified virus which causes the rapid production of tumours in mice. Nature 204, 1104–1105 (1964).
Tur’ianov, M. et al. [Present-day characteristics of etiology, epidemiology and clinical aspects of diphtheria in adults]. Sov. Med. 1, 75–7 (1991).
Scolnick, E. M., Rands, E., Williams, D. & Parks, W. P. Studies on the nucleic acid sequences of Kirsten sarcoma virus: a model for formation of a mammalian RNA-containing sarcoma virus. J. Virol. 12, 458–463 (1973).
Rapp, U. R. et al. Structure and biological activity of v-raf, a unique oncogene transduced by a retrovirus. Proc. Natl Acad. Sci. USA 80, 4218–4222 (1983).
Moelling, K. et al. Serine- and threonine-specific protein kinase activities of purified gag-mil and gag-raf proteins. Nature 312, 558–561 (1984).
Bonner, T. I. et al. Structure and biological activity of human homologs of the raf/mil oncogene. Mol. Cell Biol. 5, 1400–1407 (1985).
Huleihel, M. et al. Characterization of murine A-raf, a new oncogene related to the v-raf oncogene. Mol. Cell Biol. 6, 2655–2662 (1986).
Sturgill, T. W., Ray, L. B., Erikson, E. & Maller, J. L. Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 334, 715–718 (1988).
Courchesne, W. E., Kunisawa, R. & Thorner, J. A putative protein kinase overcomes pheromone-induced arrest of cell cycling in S. cerevisiae. Cell 58, 1107–1119 (1989).
Ahn, N. G., Weiel, J. E., Chan, C. P. & Krebs, E. G. Identification of multiple epidermal growth factor-stimulated protein serine/threonine kinases from Swiss 3T3 cells. J. Biol. Chem. 265, 11487–11494 (1990).
Kyriakis, J. M. et al. Raf-1 activates MAP kinase-kinase. Nature 358, 417–421 (1992).
Zhang, X. F. et al. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 364, 308–313 (1993).
Troppmair, J., Hartkamp, J. & Rapp, U. R. Activation of NF-kappa B by oncogenic Raf in HEK 293 cells occurs through autocrine recruitment of the stress kinase cascade. Oncogene. 17, 685–690 (1998).
Kane, R. C. et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 12, 7271–7278 (2006).
Ambrosini, G. et al. Sorafenib inhibits growth and mitogen-activated protein kinase signaling in malignant peripheral nerve sheath cells. Mol. Cancer Ther. 7, 890–896 (2008).
Bollag, G. et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 11, 873–886 (2012).
Wright, C. J. & McCormack, P. L. Trametinib: first global approval. Drugs. 73, 1245–1254 (2013).
Menzies, A. M. & Long, G. V. Dabrafenib and trametinib, alone and in combination for BRAF-mutant metastatic melanoma. Clin. Cancer Res. 20, 2035–2043 (2014).
New Medical Devices. P&T. 40, 792–806 (2015).
Braicu, C. et al. A comprehensive review on MAPK: a promising therapeutic target in cancer. Cancers 11, 1618 (2019).
Krall, E. B. et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife 6, e18970 (2017).
Kim, E. K. & Choi, E. J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 1802, 396–405 (2010).
Karin, M. Mitogen activated protein kinases as targets for development of novel anti-inflammatory drugs. Ann. Rheum. Dis. 63(Suppl. 2), ii62–ii64 (2004).
Coskun, M., Olsen, J., Seidelin, J. B. & Nielsen, O. H. MAP kinases in inflammatory bowel disease. Clin. Chim. Acta 412, 513–520 (2011).
Yu, W., Song, X. & Liu, Y. TRB3 regulates pulmonary interstitial fibrosis through the MAPK signaling pathway. Int. J. Clin. Exp. Pathol. 12, 3247–3257 (2019).
Pearson, G. et al. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 22, 153–183 (2001).
Johnson, G. L. & Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 298, 1911–1912 (2002).
Qi, M. & Elion, E. A. MAP kinase pathways. J. Cell Sci. 118, 3569–3572 (2005).
Chen, Z. et al. MAP kinases. Chem. Rev. 101, 2449–2476 (2001).
Coulombe, P. & Meloche, S. Atypical mitogen-activated protein kinases: structure, regulation and functions. Biochim. Biophys. Acta 1773, 1376–1387 (2007).
Raman, M., Chen, W. & Cobb, M. H. Differential regulation and properties of MAPKs. Oncogene. 26, 3100–3112 (2007).
Cargnello, M. & Roux, P. P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83 (2011).
Ramos, J. W. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 40, 2707–2719 (2008).
Levitzki, A. Protein kinase inhibitors as a therapeutic modality. Acc. Chem. Res. 36, 462–469 (2003).
Faivre, S., Djelloul, S. & Raymond, E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Semin. Oncol. 33, 407–420 (2006).
Du, Z. & Lovly, C. M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 17, 58 (2018).
Edin, M. L. & Juliano, R. L. Raf-1 serine 338 phosphorylation plays a key role in adhesion-dependent activation of extracellular signal-regulated kinase by epidermal growth factor. Mol. Cell Biol. 25, 4466–4475 (2005).
Hamden, K. E. et al. Raf and VEGF: emerging therapeutic targets in Kaposi’s sarcoma-associated herpesvirus infection and angiogenesis in hematopoietic and nonhematopoietic tumors. Leukemia. 19, 18–26 (2005).
Dibb, N. J., Dilworth, S. M. & Mol, C. D. Switching on kinases: oncogenic activation of BRAF and the PDGFR family. Nat. Rev. Cancer 4, 718–727 (2004).
Sebolt-Leopold, J. S. et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 5, 810–816 (1999).
Roberts, P. J. & Der, C. J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 26, 3291–3310 (2007).
Wennerberg, K., Rossman, K. L. & Der, C. J. The Ras superfamily at a glance. J. Cell Sci. 118, 843–846 (2005).
Downward, J. Control of ras activation. Cancer Surv. 27, 87–100 (1996).
Herrero, A. & Crespo, P. RAS dimers: the novice couple at the RAS-ERK pathway ball. Genes 12, 1556 (2021).
Scolnick, E. M., Papageorge, A. G. & Shih, T. Y. Guanine nucleotide-binding activity as an assay for src protein of rat-derived murine sarcoma viruses. Proc. Natl Acad. Sci. USA 76, 5355–5359 (1979).
Hennig, A. et al. Ras activation revisited: role of GEF and GAP systems. Biol. Chem. 396, 831–848 (2015).
Hall, B. E., Bar-Sagi, D. & Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl Acad. Sci. USA 99, 12138–12142 (2002).
Chen, K., Zhang, Y., Qian, L. & Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 14, 116 (2021).
Vetter, I. R. & Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science. 294, 1299–1304 (2001).
Schopel, M. et al. The small GTPases Ras and Rheb studied by multidimensional NMR spectroscopy: structure and function. Biol. Chem. 398, 577–588 (2017).
Heidecker, G. et al. Mutational activation of c-raf-1 and definition of the minimal transforming sequence. Mol. Cell Biol. 10, 2503–2512 (1990).
Marx, M. et al. A novel oncogene related to c-mil is transduced in chicken neuroretina cells induced to proliferate by infection with an avian lymphomatosis virus. EMBO J. 7, 3369–3373 (1988).
Wellbrock, C., Karasarides, M. & Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 5, 875–885 (2004).
Niault, T. S. & Baccarini, M. Targets of Raf in tumorigenesis. Carcinogenesis. 31, 1165–1174 (2010).
Kolch, W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J. 351(Pt 2), 289–305 (2000).
Trakul, N. & Rosner, M. R. Modulation of the MAP kinase signaling cascade by Raf kinase inhibitory protein. Cell Res. 15, 19–23 (2005).
Carey, K. D., Watson, R. T., Pessin, J. E. & Stork, P. J. The requirement of specific membrane domains for Raf-1 phosphorylation and activation. J. Biol. Chem. 278, 3185–3196 (2003).
Goitre, L., Trapani, E., Trabalzini, L. & Retta, S. F. The Ras superfamily of small GTPases: the unlocked secrets. Methods Mol. Biol. 1120, 1–18 (2014).
Hancock, J. F. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell Biol. 4, 373–384 (2003).
Cox, A. D. & Der, C. J. Ras history: the saga continues. Small GTPases 1, 2–27 (2010).
Dharmawardhane, S. et al. Regulation of macropinocytosis by p21-activated kinase-1. Mol. Biol. Cell 11, 3341–3352 (2000).
Stokoe, D. & McCormick, F. Activation of c-Raf-1 by Ras and Src through different mechanisms: activation in vivo and in vitro. EMBO J. 16, 2384–2396 (1997).
Winston, L. A. & Hunter, T. JAK2, Ras, and Raf are required for activation of extracellular signal-regulated kinase/mitogen-activated protein kinase by growth hormone. J. Biol. Chem. 270, 30837–30840 (1995).
Firnau, M. B. & Brieger, A. CK2 and the hallmarks of cancer. Biomedicines 10, 1987 (2022).
Nan, X. et al. Single-molecule superresolution imaging allows quantitative analysis of RAF multimer formation and signaling. Proc. Natl Acad. Sci. USA 110, 18519–18524 (2013).
Maurer, G., Tarkowski, B. & Baccarini, M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 30, 3477–3488 (2011).
Tran, T. H. et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat. Commun. 12, 1176 (2021).
Chao, F. A. et al. Insights into the cross talk between effector and allosteric lobes of KRAS from methyl conformational dynamics. J. Am. Chem. Soc. 144, 4196–4205 (2022).
Stokoe, D. et al. Activation of Raf as a result of recruitment to the plasma membrane. Science 264, 1463–1467 (1994).
Leevers, S. J., Paterson, H. F. & Marshall, C. J. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 369, 411–414 (1994).
Herrmann, C., Martin, G. A. & Wittinghofer, A. Quantitative analysis of the complex between p21ras and the Ras-binding domain of the human Raf-1 protein kinase. J. Biol. Chem. 270, 2901–2905 (1995).
Nassar, N. et al. The 2.2 A crystal structure of the Ras-binding domain of the serine/threonine kinase c-Raf1 in complex with Rap1A and a GTP analogue. Nature 375, 554–560 (1995).
Nguyen, K. et al. Exploring CRD mobility during RAS/RAF engagement at the membrane. Biophys. J. 121, 3630–3650 (2022).
Williams, J. G. et al. Elucidation of binding determinants and functional consequences of Ras/Raf-cysteine-rich domain interactions. J. Biol. Chem. 275, 22172–22179 (2000).
Mott, H. R. et al. The solution structure of the Raf-1 cysteine-rich domain: a novel ras and phospholipid binding site. Proc Natl Acad Sci USA 93, 8312–8317 (1996).
Hekman, M. et al. Associations of B- and C-Raf with cholesterol, phosphatidylserine, and lipid second messengers: preferential binding of Raf to artificial lipid rafts. J. Biol. Chem. 277, 24090–24102 (2002).
Freeman, A. K., Ritt, D. A. & Morrison, D. K. The importance of Raf dimerization in cell signaling. Small GTPases 4, 180–185 (2013).
Rajakulendran, T. et al. A dimerization-dependent mechanism drives RAF catalytic activation. Nature 461, 542–545 (2009).
Matallanas, D. et al. Raf family kinases: old dogs have learned new tricks. Genes Cancer 2, 232–260 (2011).
Ritt, D. A., Monson, D. M., Specht, S. I. & Morrison, D. K. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol 30, 806–819 (2010).
Dougherty, M. K. et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17, 215–224 (2005).
Brummer, T., Naegele, H., Reth, M. & Misawa, Y. Identification of novel ERK-mediated feedback phosphorylation sites at the C-terminus of B-Raf. Oncogene 22, 8823–8834 (2003).
Yang, S. & Liu, G. Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinoma. Oncol. Lett. 13, 1041–1047 (2017).
Shin, S. Y. et al. Positive- and negative-feedback regulations coordinate the dynamic behavior of the Ras-Raf-MEK-ERK signal transduction pathway. J. Cell Sci. 122, 425–435 (2009).
Sturm, O. E. et al. The mammalian MAPK/ERK pathway exhibits properties of a negative feedback amplifier. Sci. Signal. 3, ra90 (2010).
Kamioka, Y. et al. Multiple decisive phosphorylation sites for the negative feedback regulation of SOS1 via ERK. J. Biol. Chem. 285, 33540–33548 (2010).
Waters, S. B. et al. Desensitization of Ras activation by a feedback disassociation of the SOS-Grb2 complex. J. Biol. Chem. 270, 20883–20886 (1995).
Trujillo, J. I. MEK inhibitors: a patent review 2008–2010. Expert Opin. Ther. Pat. 21, 1045–1069 (2011).
Chung, E. & Kondo, M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol. Res. 49, 248–268 (2011).
Schulze, A. et al. The transcriptional response to Raf activation is almost completely dependent on Mitogen-activated Protein Kinase Kinase activity and shows a major autocrine component. Mol. Biol. Cell 15, 3450–3463 (2004).
Mansour, S. J. et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966–970 (1994).
Cowley, S., Paterson, H., Kemp, P. & Marshall, C. J. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 77, 841–852 (1994).
Avruch, J. et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog. Horm. Res. 56, 127–155 (2001).
Cuadrado, A. & Nebreda, A. R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417 (2010).
Kim, C. et al. The kinase p38 alpha serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat. Immunol. 9, 1019–1027 (2008).
Nebreda, A. R. & Porras, A. p38 MAP kinases: beyond the stress response. Trends Biochem. Sci. 25, 257–260 (2000).
Regan, J. et al. Pyrazole urea-based inhibitors of p38 MAP kinase: from lead compound to clinical candidate. J. Med. Chem. 45, 2994–3008 (2002).
Li, M. et al. TRAF6-p38/JNK-ATF2 axis promotes microglial inflammatory activation. Exp. Cell Res. 376, 133–148 (2019).
Bennett, B. L. et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl Acad. Sci. USA 98, 13681–13686 (2001).
Carboni, S. et al. AS601245 (1, 3-benzothiazol-2-yl (2-[[2-(3-pyridinyl) ethyl] amino]-4 pyrimidinyl) acetonitrile): a c-Jun NH2-terminal protein kinase inhibitor with neuroprotective properties. J. Pharmacol. Exp. Ther. 310, 25–32 (2004).
Bogoyevitch, M. A. et al. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim. Biophys. Acta 1804, 463–475 (2010).
Sabapathy, K. et al. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell 15, 713–725 (2004).
Tournier, C. et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874 (2000).
Hoang, V. T. et al. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 392, 51–59 (2017).
Wang, X. et al. Activation of extracellular signal-regulated protein kinase 5 downregulates FasL upon osmotic stress. Cell Death Differ. 13, 2099–2108 (2006).
Zehorai, E., Yao, Z., Plotnikov, A. & Seger, R. The subcellular localization of MEK and ERK–a novel nuclear translocation signal (NTS) paves a way to the nucleus. Mol. Cell Endocrinol. 314, 213–220 (2010).
Mulloy, R., Salinas, S., Philips, A. & Hipskind, R. A. Activation of cyclin D1 expression by the ERK5 cascade. Oncogene. 22, 5387–5398 (2003).
Roberts, O. L. et al. ERK5 is required for VEGF-mediated survival and tubular morphogenesis of primary human microvascular endothelial cells. J. Cell Sci. 123, 3189–3200 (2010).
Deleris, P. et al. Activation loop phosphorylation of the atypical MAP kinases ERK3 and ERK4 is required for binding, activation and cytoplasmic relocalization of MK5. J. Cell Physiol. 217, 778–788 (2008).
Abe, M. K. et al. ERK8, a new member of the mitogen-activated protein kinase family. J. Biol. Chem. 277, 16733–16743 (2002).
Klevernic, I. V. et al. Characterization of the reversible phosphorylation and activation of ERK8. Biochem. J. 394, 365–373 (2006).
Kant, S. et al. Characterization of the atypical MAPK ERK4 and its activation of the MAPK-activated protein kinase MK5. J. Biol. Chem. 281, 35511–35519 (2006).
Aberg, E. et al. Docking of PRAK/MK5 to the atypical MAPKs ERK3 and ERK4 defines a novel MAPK interaction motif. J. Biol. Chem. 284, 19392–19401 (2009).
Schumacher, S. et al. Scaffolding by ERK3 regulates MK5 in development. EMBO J. 23, 4770–4779 (2004).
Aberg, E. et al. Regulation of MAPK-activated protein kinase 5 activity and subcellular localization by the atypical MAPK ERK4/MAPK4. J. Biol. Chem. 281, 35499–35510 (2006).
Seternes, O. M. et al. Activation of MK5/PRAK by the atypical MAP kinase ERK3 defines a novel signal transduction pathway. EMBO J. 23, 4780–4791 (2004).
Abe, M. K., Kuo, W. L., Hershenson, M. B. & Rosner, M. R. Extracellular signal-regulated kinase 7 (ERK7), a novel ERK with a C-terminal domain that regulates its activity, its cellular localization, and cell growth. Mol. Cell Biol. 19, 1301–1312 (1999).
Saelzler, M. P. et al. ERK8 down-regulates transactivation of the glucocorticoid receptor through Hic-5. J. Biol. Chem. 281, 16821–16832 (2006).
Henrich, L. M. et al. Extracellular signal-regulated kinase 7, a regulator of hormone-dependent estrogen receptor destruction. Mol. Cell Biol. 23, 5979–5988 (2003).
Kojima, H. et al. STAT3 regulates Nemo-like kinase by mediating its interaction with IL-6-stimulated TGFbeta-activated kinase 1 for STAT3 Ser-727 phosphorylation. Proc. Natl Acad. Sci. USA 102, 4524–4529 (2005).
Nakhaei-Rad, S. et al. Structural fingerprints, interactions, and signaling networks of RAS family proteins beyond RAS isoforms. Crit. Rev. Biochem. Mol. Biol. 53, 130–156 (2018).
Kolch, W. Coordinating ERK/MAPK signaling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 6, 827–837 (2005).
Smith, F. D. et al. AKAP-Lbc enhances cyclic AMP control of the ERK1/2 cascade. Nat. Cell Biol. 12, 1242–1249 (2010).
Dumaz, N. & Marais, R. Integrating signals between cAMP and the RAS/RAF/MEK/ERK signalling pathways. Based on the anniversary prize of the Gesellschaft fur Biochemie und Molekularbiologie Lecture delivered on 5 July 2003 at the Special FEBS Meeting in Brussels. FEBS J. 272, 3491–3504 (2005).
Fischer, A., Muhlhauser, W. W. D., Warscheid, B. & Radziwill, G. Membrane localization of acetylated CNK1 mediates a positive feedback on RAF/ERK signaling. Sci. Adv. 3, e1700475 (2017).
Amaddii, M. et al. Flotillin-1/reggie-2 protein plays dual role in activation of receptor-tyrosine kinase/mitogen-activated protein kinase signaling. J. Biol. Chem. 287, 7265–7278 (2012).
Harder, T. & Kuhn, M. Selective accumulation of raft-associated membrane protein LAT in T cell receptor signaling assemblies. J. Cell Biol. 151, 199–208 (2000).
Thome, C. H. et al. NTAL is associated with treatment outcome, cell proliferation and differentiation in acute promyelocytic leukemia. Sci. Rep. 10, 10315 (2020).
Jin, T. et al. PAQR10 and PAQR11 mediate Ras signaling in the Golgi apparatus. Cell Res. 22, 661–676 (2012).
Chen, C. & Noble, S. M. Post-transcriptional regulation of the Sef1 transcription factor controls the virulence of Candida albicans in its mammalian host. PLoS Pathog. 8, e1002956 (2012).
Lemmon, M. A. & Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell. 141, 1117–1134 (2010).
Kouhara, H. et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell 89, 693–702 (1997).
Hadari, Y. R. et al. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc. Natl Acad. Sci. USA 98, 8578–8583 (2001).
Lamothe, B. et al. The docking protein Gab1 is an essential component of an indirect mechanism for fibroblast growth factor stimulation of the phosphatidylinositol 3-kinase/Akt antiapoptotic pathway. Mol. Cell Biol. 24, 5657–5666 (2004).
Xiao, B. et al. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature 376, 188–191 (1995).
Levy-Apter, E. et al. Adaptor protein GRB2 promotes Src tyrosine kinase activation and podosomal organization by protein-tyrosine phosphatase ϵ in osteoclasts. J. Biol. Chem. 289, 36048–36058 (2014).
Marie-Cardine, A. et al. SHP2-interacting transmembrane adaptor protein (SIT), a novel disulfide-linked dimer regulating human T cell activation. J. Exp. Med. 189, 1181–1194 (1999).
Morrison, D. K. & Davis, R. J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 19, 91–118 (2003).
Ritt, D. A. et al. CK2 Is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr. Biol. 17, 179–184 (2007).
Schaeffer, H. J. et al. MP1: a MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science 281, 1668–1671 (1998).
Pudewell, S. & Ahmadian, M. R. Spotlight on accessory proteins: RTK-RAS-MAPK modulators as new therapeutic targets. Biomolecules 11, 895 (2021).
McNulty, D. E. et al. MAPK scaffold IQGAP1 binds the EGF receptor and modulates its activation. J. Biol. Chem. 286, 15010–15021 (2011).
Ishibe, S., Joly, D., Liu, Z. X. & Cantley, L. G. Paxillin serves as an ERK-regulated scaffold for coordinating FAK and Rac activation in epithelial morphogenesis. Mol. Cell 16, 257–267 (2004).
DeFea, K. A. et al. beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 (2000).
Posada, I. M. et al. ASPP2 is a novel pan-ras nanocluster scaffold. PLoS One 11, e0159677 (2016).
Li, M. et al. Mechanistic insights into the long-range allosteric regulation of KRAS Via neurofibromatosis Type 1 (NF1) scaffold upon SPRED1 loading. J. Mol. Biol. 434, 167730 (2022).
Mason, J. M., Morrison, D. J., Basson, M. A. & Licht, J. D. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 16, 45–54 (2006).
Zhou, L. & Hanemann, C. O. Merlin, a multi-suppressor from cell membrane to the nucleus. FEBS Lett. 586, 1403–1408 (2012).
Inder, K. L., Hill, M. M. & Hancock, J. F. Nucleophosmin and nucleolin regulate K-Ras signaling. Commun. Integr Biol. 3, 188–190 (2010).
Inder, K. L. et al. Nucleophosmin and nucleolin regulate K-Ras plasma membrane interactions and MAPK signal transduction. J. Biol. Chem. 284, 28410–28419 (2009).
Ikedife, J., He, J. & Wei, Y. PEA-15 engages in allosteric interactions using a common scaffold in a phosphorylation-dependent manner. Sci. Rep. 12, 116 (2022).
Lee, S. E. et al. RGS14 is a natural suppressor of both synaptic plasticity in CA2 neurons and hippocampal-based learning and memory. Proc. Natl Acad. Sci. USA 107, 16994–16998 (2010).
Vomastek, T. et al. Modular construction of a signaling scaffold: MORG1 interacts with components of the ERK cascade and links ERK signaling to specific agonists. Proc. Natl Acad. Sci. USA 101, 6981–6986 (2004).
Blazevits, O. et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci. Rep. 6, 24165 (2016).
Yin, G., Haendeler, J., Yan, C. & Berk, B. C. GIT1 functions as a scaffold for MEK1-extracellular signal-regulated kinase 1 and 2 activation by angiotensin II and epidermal growth factor. Mol. Cell Biol. 24, 875–885 (2004).
Villalobo, A., Ishida, H., Vogel, H. J. & Berchtold, M. W. Calmodulin as a protein linker and a regulator of adaptor/scaffold proteins. Biochim. Biophys. Acta Mol. Cell Res. 1865, 507–521 (2018).
Jang, H., Stevens, P., Gao, T. & Galperin, E. The leucine-rich repeat signaling scaffolds Shoc2 and Erbin: cellular mechanism and role in disease. FEBS J. 288, 721–739 (2021).
Park, J. et al. Deficiency in the LIM-only protein FHL2 impairs assembly of extracellular matrix proteins. FASEB J. 22, 2508–2520 (2008).
Chivers, S. B., Brackley, A. D. & Jeske, N. A. Raf kinase inhibitory protein reduces bradykinin receptor desensitization. J. Neurochem. 162, 156–165 (2022).
Dautel, G. & Merle, M. Pronator quadratus free muscle flap for treatment of palmar defects. J. Hand. Surg. Br. 18, 576–578 (1993).
Rath, O. et al. The RKIP (Raf-1 Kinase Inhibitor Protein) conserved pocket binds to the phosphorylated N-region of Raf-1 and inhibits the Raf-1-mediated activated phosphorylation of MEK. Cell Signal. 20, 935–941 (2008).
Granovsky, A. E. & Rosner, M. R. Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res. 18, 452–457 (2008).
Escara-Wilke, J., Yeung, K. & Keller, E. T. Raf kinase inhibitor protein (RKIP) in cancer. Cancer Metastasis Rev. 31, 615–620 (2012).
Zou, Q. et al. RKIP suppresses the proliferation and metastasis of breast cancer cell lines through up-regulation of miR-185 targeting HMGA2. Arch Biochem. Biophys. 610, 25–32 (2016).
Du, Y. et al. MiR-543 promotes proliferation and epithelial-mesenchymal transition in prostate cancer via targeting RKIP. Cell Physiol. Biochem. 41, 1135–1146 (2017).
Li, Y. et al. Bmi-1-induced miR-27a and miR-155 promote tumor metastasis and chemoresistance by targeting RKIP in gastric cancer. Mol. Cancer 19, 109 (2020).
Raquel-Cunha, A., Cardoso-Carneiro, D., Reis, R. M. & Martinho, O. Current status of Raf Kinase Inhibitor Protein (RKIP) in lung cancer: Behind RTK Signal. Cells 8, 442 (2019).
Kim, H. S. et al. Reduced expression of Raf-1 kinase inhibitory protein predicts regional lymph node metastasis and shorter survival in esophageal squamous cell carcinoma. Pathol. Res. Pract. 208, 292–299 (2012).
Koelzer, V. H. et al. Geographic analysis of RKIP expression and its clinical relevance in colorectal cancer. Br. J. Cancer 108, 2088–2096 (2013).
Minoo, P. et al. Loss of raf-1 kinase inhibitor protein expression is associated with tumor progression and metastasis in colorectal cancer. Am. J. Clin. Pathol. 127, 820–827 (2007).
Huang, W. et al. Downregulation of RKIP promotes radioresistance of nasopharyngeal carcinoma by activating NRF2/NQO1 axis via downregulating miR-450b-5p. Cell Death Dis. 11, 504 (2020).
Jing, S. H., Gao, X., Yu, B. & Qiao, H. Raf Kinase Inhibitor Protein (RKIP) Inhibits Tumor Necrosis Factor-alpha (TNF-alpha) Induced Adhesion Molecules Expression in Vascular Smooth Muscle Bells by Suppressing (Nuclear Transcription Factor-kappaB (NF-kappaB) Pathway. Med. Sci. Monit. 23, 4789–4797 (2017)..
Wottrich, S. et al. Inverse correlation between the metastasis suppressor RKIP and the metastasis inducer YY1: Contrasting roles in the regulation of chemo/immuno-resistance in cancer. Drug Resist. Updat. 30, 28–38 (2017).
Yang, K. et al. KRAS promotes tumor metastasis and chemoresistance by repressing RKIP via the MAPK-ERK pathway in pancreatic cancer. Int. J. Cancer 142, 2323–2334 (2018).
Yuan, L. et al. Reduced RKIP enhances nasopharyngeal carcinoma radioresistance by increasing ERK and AKT activity. Oncotarget 7, 11463–11477 (2016).
Wang, A. et al. Reduced RKIP expression levels are associated with frequent non-small cell lung cancer metastasis and STAT3 phosphorylation and activation. Oncol. Lett. 13, 3039–3045 (2017).
Al-Mulla, F. et al. A new model for raf kinase inhibitory protein induced chemotherapeutic resistance. PLoS One 7, e29532 (2012).
He, Q. Y. et al. Reduction of RKIP expression promotes nasopharyngeal carcinoma invasion and metastasis by activating Stat3 signaling. Oncotarget 6, 16422–16436 (2015).
Baccarini, M. Second nature: biological functions of the Raf-1 “kinase”. FEBS Lett. 579, 3271–3277 (2005).
Hong, L., Wang, Y., Chen, W. & Yang, S. MicroRNA-508 suppresses epithelial-mesenchymal transition, migration, and invasion of ovarian cancer cells through the MAPK1/ERK signaling pathway. J. Cell Biochem. 119, 7431–7440 (2018).
Tang, Q., Wu, J., Zheng, F., Hann, S.S. & Chen, Y. Emodin Increases Expression of Insulin-Like Growth Factor Binding Protein 1 through Activation of MEK/ERK/AMPKα and Interaction of PPARγ and Sp1 in Lung Cancer. Cell Physiol. Biochem. 41, 339–357 (2017).
Baek, J. H. et al. Hypoxia-induced VEGF enhances tumor survivability via suppression of serum deprivation-induced apoptosis. Oncogene 19, 4621–4631 (2000).
Huang, Y. et al. miR‑101 regulates the cell proliferation and apoptosis in diffuse large B‑cell lymphoma by targeting MEK1 via regulation of the ERK/MAPK signaling pathway. Oncol. Rep. 41, 377–386 (2019).
Lefloch, R., Pouyssegur, J. & Lenormand, P. Total ERK1/2 activity regulates cell proliferation.Cell Cycle 8, 705–711 (2009).
Allan, L. A. et al. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat. Cell Biol. 5, 647–654 (2003).
Malumbres, M. et al. Cellular response to oncogenic ras involves induction of the Cdk4 and Cdk6 inhibitor p15(INK4b). Mol. Cell Biol. 20, 2915–2925 (2000).
Chang, M. C. et al. Mesothelin enhances invasion of ovarian cancer by inducing MMP-7 through MAPK/ERK and JNK pathways. Biochem. J. 442, 293–302 (2012).
Bian, C. X. et al. P70S6K 1 regulation of angiogenesis through VEGF and HIF-1alpha expression. Biochem. Biophys. Res. Commun. 398, 395–399 (2010).
Chen, J. et al. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc. Natl Acad. Sci. USA 98, 7783–7788 (2001).
Nolan, A. A., Aboud, N. K., Kolch, W. & Matallanas, D. Hidden targets in RAF signaling pathways to block oncogenic RAS signaling. Genes 12, 553 (2021).
Huser, M. et al. MEK kinase activity is not necessary for Raf-1 function. EMBO J. 20, 1940–1951 (2001).
Galabova-Kovacs, G. et al. ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle 5, 1514–1518 (2006).
Bui, N. L. et al. Bad phosphorylation as a target of inhibition in oncology. Cancer Lett. 415, 177–186 (2018).
Wang, H. G., Rapp, U. R. & Reed, J. C. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 87, 629–638 (1996).
Marliss, E. B., Caron, D., Albisser, A. M. & Zinman, B. Present and future expectations regarding insulin infusion systems. Diabetes Care 4, 325–327 (1981).
Matsuzawa, A. et al. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: advanced findings from ASK1 knockout mice. Antioxid. Redox. Signal. 4, 415–425 (2002).
Alavi, A. S., Acevedo, L., Min, W. & Cheresh, D. A. Chemoresistance of endothelial cells induced by basic fibroblast growth factor depends on Raf-1-mediated inhibition of the proapoptotic kinase, ASK1. Cancer Res. 67, 2766–2772 (2007).
Du, J., Cai, S. H., Shi, Z. & Nagase, F. Binding activity of H-Ras is necessary for in vivo inhibition of ASK1 activity. Cell Res. 14, 148–154 (2004).
Yamaguchi, O. et al. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J. Clin. Invest. 114, 937–943 (2004).
O’Neill, E., Rushworth, L., Baccarini, M. & Kolch, W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 306, 2267–2270 (2004).
O’Neill, E. & Kolch, W. Taming the Hippo: Raf-1 controls apoptosis by suppressing MST2/Hippo. Cell Cycle 4, 365–367 (2005).
Kilili, G. K. & Kyriakis, J. M. Mammalian Ste20-like kinase (Mst2) indirectly supports Raf-1/ERK pathway activity via maintenance of protein phosphatase-2A catalytic subunit levels and consequent suppression of inhibitory Raf-1 phosphorylation. J. Biol. Chem. 285, 15076–15087 (2010).
Rauch, J. et al. Heterogeneous nuclear ribonucleoprotein H blocks MST2-mediated apoptosis in cancer cells by regulating A-Raf transcription. Cancer Res. 70, 1679–1688 (2010).
Galan, J. A. & Avruch, J. MST1/MST2 protein kinases: regulation and physiologic roles. Biochemistry. 55, 5507–5519 (2016).
Mielgo, A. et al. A MEK-independent role for CRAF in mitosis and tumor progression. Nat. Med. 17, 1641–1645 (2011).
Joukov, V. & De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 11, eaar4195 (2018).
Advani, S. J. et al. Kinase-independent role for CRAF-driving tumour radioresistance via CHK2. Nat. Commun. 6, 8154 (2015).
Khazak, V., Astsaturov, I., Serebriiskii, I. G. & Golemis, E. A. Selective Raf inhibition in cancer therapy. Expert Opin. Ther. Targets 11, 1587–1609 (2007).
Galaktionov, K., Jessus, C. & Beach, D. Raf1 interaction with Cdc25 phosphatase ties mitogenic signal transduction to cell cycle activation. Genes Dev. 9, 1046–1058 (1995).
Dhawan, P. & Richmond, A. A novel NF-kappa B-inducing kinase-MAPK signaling pathway up-regulates NF-kappa B activity in melanoma cells. J. Biol. Chem. 277, 7920–7928 (2002).
Cheung, M., Sharma, A., Madhunapantula, S. V. & Robertson, G. P. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 68, 3429–3439 (2008).
Palmieri, G. et al. Main roads to melanoma. J. Transl. Med. 7, 86 (2009).
Amiri, K. I. & Richmond, A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 24, 301–313 (2005).
Templer, D. I. et al. Exploration of head injury without medical attention. Percept. Mot. Skills 75, 195–202 (1992).
Choux, M. et al. Hydatid cyst of the fourth ventricle in a six-year-old child. Mod. Probl. Paediatr. 18, 273–275 (1976).
Baumann, B. et al. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc. Natl Acad. Sci. USA 97, 4615–4620 (2000).
Jette, C. & Thorburn, A. A Raf-induced, MEK-independent signaling pathway regulates atrial natriuretic factor gene expression in cardiac muscle cells. FEBS Lett. 467, 1–6 (2000).
Ehrenreiter, K. et al. Raf-1 regulates Rho signaling and cell migration. J. Cell Biol. 168, 955–964 (2005).
Ehrenreiter, K. et al. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell 16, 149–160 (2009).
Piazzolla, D. et al. Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J. Cell Biol. 171, 1013–1022 (2005).
Niault, T. et al. From autoinhibition to inhibition in trans: the Raf-1 regulatory domain inhibits Rok-alpha kinase activity. J. Cell Biol. 187, 335–342 (2009).
Kern, F. et al. Essential, non-redundant roles of B-Raf and Raf-1 in Ras-driven skin tumorigenesis. Oncogene 32, 2483–2492 (2013).
Janosch, P. et al. The Raf-1 kinase associates with vimentin kinases and regulates the structure of vimentin filaments. FASEB J. 14, 2008–2021 (2000).
Ku, N. O., Fu, H. & Omary, M. B. Raf-1 activation disrupts its binding to keratins during cell stress. J. Cell Biol. 166, 479–485 (2004).
De Luca, A. et al. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets. 16(Suppl 2), S17–S27 (2012).
Moelling, K. et al. Regulation of Raf-Akt Cross-talk. J. Biol. Chem. 277, 31099–31106 (2002).
Nguyen, L. K. et al. Competing to coordinate cell fate decisions: the MST2-Raf-1 signaling device. Cell Cycle 14, 189–199 (2015).
Romano, D. et al. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer Res. 70, 1195–1203 (2010).
Matallanas, D. et al. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 27, 962–975 (2007).
Mizushima, N. Autophagy: process and function. Genes Dev. 21, 2861–2873 (2007).
Ravikumar, B. et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 90, 1383–1435 (2010).
Khandia, R. et al. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells 8, 674 (2019).
Mulcahy Levy, J. M. & Thorburn, A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 27, 843–857 (2020).
Thorburn, A., Thamm, D. H. & Gustafson, D. L. Autophagy and cancer therapy. Mol. Pharmacol. 85, 830–838 (2014).
Kroemer, G., Marino, G. & Levine, B. Autophagy and the integrated stress response. Mol. Cell 40, 280–293 (2010).
Rubinsztein, D. C., Marino, G. & Kroemer, G. Autophagy and aging. Cell 146, 682–695 (2011).
Onorati, A. V., Dyczynski, M., Ojha, R. & Amaravadi, R. K. Targeting autophagy in cancer. Cancer. 124, 3307–3318 (2018).
Kim, J. H. et al. Raf/MEK/ERK can regulate cellular levels of LC3B and SQSTM1/p62 at expression levels. Exp. Cell Res. 327, 340–352 (2014).
Xie, X. et al. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 5, 410–423 (2015).
Liu, H., He, Z. & Simon, H. U. Autophagy suppresses melanoma tumorigenesis by inducing senescence. Autophagy 10, 372–373 (2014).
Wu, Y., Zhang, T., Zhang, X. & Gao, Q. Decoding the complexity of metastasis. Cancer Biol. Med. 19, 284–288 (2022).
Massague, J. & Obenauf, A. C. Metastatic colonization by circulating tumour cells. Nature. 529, 298–306 (2016).
Bergers, G. & Fendt, S. M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 21, 162–180 (2021).
Faubert, B., Solmonson, A. & DeBerardinis, R. J. Metabolic reprogramming and cancer progression. Science 368, eaaw5473 (2020).
Kelley, D. E. & Mandarino, L. J. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes. 49, 677–683 (2000).
Randle, P. J., Garland, P. B., Hales, C. N. & Newsholme, E. A. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1, 785–789 (1963).
Vriens, K. et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature 566, 403–406 (2019).
Triki, M. et al. mTOR Signaling and SREBP activity increase FADS2 expression and can activate sapienate biosynthesis. Cell Rep. 31, 107806 (2020).
Elia, I., Doglioni, G. & Fendt, S. M. Metabolic hallmarks of metastasis formation. Trends Cell Biol. 28, 673–684 (2018).
Krebs, A. M. et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529 (2017).
Pastushenko, I. et al. Identification of the tumour transition states occurring during EMT. Nature 556, 463–468 (2018).
Hao, Y., Baker, D. & Ten Dijke, P. TGF-beta-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 20, 2767 (2019).
Lee, M. K. et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 26, 3957–3967 (2007).
Mulder, K. M. & Morris, S. L. Activation of p21ras by transforming growth factor beta in epithelial cells. J. Biol Chem. 267, 5029–5031 (1992).
Wong, A. et al. FRS2 alpha attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc. Natl Acad. Sci. USA 99, 6684–6689 (2002).
Yuan, X. et al. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J. Hematol. Oncol. 7, 87 (2014).
Cai, J. et al. Simultaneous overactivation of Wnt/beta-catenin and TGFbeta signalling by miR-128-3p confers chemoresistance-associated metastasis in NSCLC. Nat Commun 8, 15870 (2017).
Zhang, N. et al. The interaction of the senescent and adjacent breast cancer cells promotes the metastasis of heterogeneous breast cancer cells through notch signaling. Int. J. Mol. Sci. 22, 849 (2021).
Kolbe, N. et al. Data-based stochastic modeling reveals sources of activity bursts in single-cell TGF-beta signaling. PLoS Comput. Biol. 18, e1010266 (2022).
Moustakas, A. & Heldin, C. H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 98, 1512–1520 (2007).
Kudo-Saito, C., Shirako, H., Takeuchi, T. & Kawakami, Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell 15, 195–206 (2009).
Ko, C. C., Hsieh, Y. Y. & Yang, P. M. Long non-coding RNA MIR31HG promotes the transforming growth factor beta-induced epithelial-mesenchymal transition in pancreatic ductal adenocarcinoma cells. Int. J. Mol. Sci. 23, 6559 (2022).
Janda, E. et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J. Cell Biol. 156, 299–313 (2002).
Xie, L. et al. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 6, 603–610 (2004).
Wang, Y. et al. Autophagy inhibition specifically promotes epithelial-mesenchymal transition and invasion in RAS-mutated cancer cells. Autophagy 15, 886–899 (2019).
Qin, W. et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget 6, 39839–39854 (2015).
Qiang, L. & He, Y. Y. Autophagy deficiency stabilizes TWIST1 to promote epithelial-mesenchymal transition. Autophagy. 10, 1864–1865 (2014).
Alizadeh, J. et al. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 1865, 749–768 (2018).
Liang, C. et al. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy 16, 486–500 (2020).
Peng, Y. F. et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy 9, 2056–2068 (2013).
Lv, Q., Hua, F. & Hu, Z. W. DEDD, a novel tumor repressor, reverses epithelial-mesenchymal transition by activating selective autophagy. Autophagy. 8, 1675–1676 (2012).
Chen, S. & Guan, J. L. Tumor-promoting and -suppressive roles of autophagy in the same mouse model of BrafV600E-driven lung cancer. Cancer Discov. 3, 1225–1227 (2013).
Karsli-Uzunbas, G. et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927 (2014).
Strohecker, A. M. & White, E. Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy. 10, 384–385 (2014).
Yuan, M. et al. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal. Transduct. Target Ther. 4, 61 (2019).
Massarelli, E. et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin. Cancer Res. 13, 2890–2896 (2007).
Bou-Assaly, W. & Mukherji, S. Cetuximab (erbitux). AJNR Am. J. Neuroradiol. 31, 626–627 (2010).
Petrelli, F. et al. Cetuximab and panitumumab in KRAS wild-type colorectal cancer: a meta-analysis. Int. J. Colorectal. Dis. 26, 823–833 (2011).
Tejpar, S. et al. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol. 30, 3570–3577 (2012).
Yao, Y. M. et al. Mouse PDX trial suggests synergy of concurrent inhibition of RAF and EGFR in colorectal cancer with BRAF or KRAS mutations. Clin Cancer Res 23, 5547–5560 (2017).
Schirripa, M. et al. Phase II study of single-agent cetuximab in KRAS G13D mutant metastatic colorectal cancer. Ann. Oncol. 26, 2503 (2015).
Ou, S. I. et al. First-in-human phase I/IB dose-finding study of adagrasib (MRTX849) in patients with advanced KRAS(G12C) solid tumors (KRYSTAL-1). J. Clin. Oncol. 40, 2530–2538 (2022).
Nakajima, E. C. et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 28, 1482–1486 (2022).
Skoulidis, F. et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384, 2371–2381 (2021).
Loos, N. H. C. et al. ABCB1 limits brain exposure of the KRAS(G12C) inhibitor sotorasib, whereas ABCB1, CYP3A, and possibly OATP1a/1b restrict its oral availability. Pharmacol. Res. 178, 106137 (2022).
De, S. K. First approval of adagrasib for the treatment of non-small cell lung cancer harboring a KRASG12C mutation. Curr. Med. Chem. 31, 266–272 (2024).
Tian, H., Yang, Z. & He, J. Adagrasib: a landmark in the KRAS(G12C)-mutated NSCLC. MedComm (2020) 3, e190 (2022).
Yaeger, R. et al. Adagrasib with or without cetuximab in colorectal cancer with mutated KRAS G12C. N. Engl. J. Med. 388, 44–54 (2023).
Loos, N. H. C. et al. Pharmacokinetics of the KRAS(G12C) inhibitor adagrasib is limited by CYP3A and ABCB1, and influenced by binding to mouse plasma carboxylesterase 1c. Biomed. Pharmacother. 166, 115304 (2023).
Tsai, J. et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl Acad. Sci. USA 105, 3041–3046 (2008).
Bollag, G. et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599 (2010).
Hyman, D. M. et al. Vemurafenib in Multiple Nonmelanoma Cancers with BRAF V600 Mutations. N. Engl. J. Med. 373, 726–736 (2015).
Ballantyne, A. D. & Garnock-Jones, K. P. Dabrafenib: first global approval. Drugs 73, 1367–1376 (2013).
Lito, P., Rosen, N. & Solit, D. B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 19, 1401–1409 (2013).
Koelblinger, P., Thuerigen, O. & Dummer, R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr. Opin. Oncol. 30, 125–133 (2018).
Chapman, P. B. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 (2011).
Delord, J. P. et al. Phase I dose-escalation and -expansion study of the BRAF inhibitor encorafenib (LGX818) in metastatic BRAF-mutant melanoma. Clin. Cancer Res. 23, 5339–5348 (2017).
Sosman, J. A. et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 366, 707–714 (2012).
Johnson, D. B. et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur. J. Cancer 51, 2792–2799 (2015).
Song, Y. et al. Targeting RAS-RAF-MEK-ERK signaling pathway in human cancer: current status in clinical trials. Genes Dis. 10, 76–88 (2023).
Kirchberger, M. C. et al. MEK inhibition may increase survival of NRAS-mutated melanoma patients treated with checkpoint blockade: Results of a retrospective multicentre analysis of 364 patients. Eur. J. Cancer 98, 10–16 (2018).
Yamaguchi, T. et al. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. Int. J. Oncol. 39, 23–31 (2011).
Flaherty, K. T. et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694–1703 (2012).
Sarkisian, S. & Davar, D. MEK inhibitors for the treatment of NRAS mutant melanoma. Drug Des. Dev. Ther. 12, 2553–2565 (2018).
Andrlova, H., Zeiser, R. & Meiss, F. Cobimetinib (GDC-0973, XL518). Recent Results Cancer Res. 211, 177–186 (2018).
Shirley, M. Encorafenib and Binimetinib: first global approvals. Drugs. 78, 1277–1284 (2018).
Casey, D. et al. FDA approval summary: selumetinib for plexiform neurofibroma. Clin. Cancer Res. 27, 4142–4146 (2021).
Khan, S. et al. Intermittent MEK inhibition for the treatment of metastatic uveal melanoma. Front. Oncol. 12, 975643 (2022).
Larkin, J. et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 371, 1867–1876 (2014).
Brastianos, P. K. et al. BRAF-MEK inhibition in newly diagnosed papillary craniopharyngiomas. N. Engl. J. Med. 389, 118–126 (2023).
Rajkumar, S. et al. Melanomas with concurrent BRAF non-p.V600 and NF1 loss-of-function mutations are targetable by BRAF/MEK inhibitor combination therapy. Cell Rep. 39, 110634 (2022).
Long, G. V. et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 371, 1877–1888 (2014).
Ishii, N. et al. Enhanced inhibition of ERK signaling by a novel allosteric MEK inhibitor, CH5126766, that suppresses feedback reactivation of RAF activity. Cancer Res. 73, 4050–4060 (2013).
Guo, C. et al. Intermittent schedules of the oral RAF-MEK inhibitor CH5126766/VS-6766 in patients with RAS/RAF-mutant solid tumours and multiple myeloma: a single-centre, open-label, phase 1 dose-escalation and basket dose-expansion study. Lancet Oncol. 21, 1478–1488 (2020).
Nobre, L. et al. Outcomes of BRAF V600E pediatric gliomas treated with targeted BRAF inhibition. JCO Precis. Oncol. 4, PO.19.00298 (2020).
Bouffet, E. et al. Dabrafenib plus Trametinib in Pediatric Glioma with BRAF V600 Mutations. N. Engl. J. Med. 389, 1108–1120 (2023).
Eroglu, Z. & Ozgun, A. Updates and challenges on treatment with BRAF/MEK-inhibitors in melanoma. Expert Opin. Orphan Drugs. 6, 545–551 (2018).
Selvasaravanan, K. D. et al. The limitations of targeting MEK signalling in Glioblastoma therapy. Sci. Rep. 10, 7401 (2020).
Sakji-Dupre, L. et al. Cerebrospinal fluid concentrations of vemurafenib in patients treated for brain metastatic BRAF-V600 mutated melanoma. Melanoma Res. 25, 302–305 (2015).
Eroglu, Z. et al. Combined BRAF and HSP90 Inhibition in Patients with Unresectable BRAF (V600E)-Mutant Melanoma. Clin. Cancer Res. 24, 5516–5524 (2018).
Nassar, K. W. et al. Targeting CDK4/6 represents a therapeutic vulnerability in acquired BRAF/MEK inhibitor-resistant melanoma. Mol. Cancer Ther. 20, 2049–2060 (2021).
Hauschild, A. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomized controlled trial. Lancet 380, 358–365 (2012).
Spagnolo, F. et al. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Onco. Targets Ther. 8, 157–168 (2015).
Smalley, K. S. et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 7, 2876–2883 (2008).
Lin, L. & Bivona, T. G. The Hippo effector YAP regulates the response of cancer cells to MAPK pathway inhibitors. Mol. Cell Oncol. 3, e1021441 (2016).
Lin, L. et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 47, 250–256 (2015).
Paraiso, K. H. et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 71, 2750–2760 (2011).
Yang, J. Y. et al. Activation of FOXO3a is sufficient to reverse mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor chemoresistance in human cancer. Cancer Res. 70, 4709–4718 (2010).
Shao, Y. & Aplin, A. E. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 70, 6670–6681 (2010).
De Bruyne, E. et al. IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood 115, 2430–2440 (2010).
Neel, D. S. & Bivona, T. G. Resistance is futile: overcoming resistance to targeted therapies in lung adenocarcinoma. NPJ Precis. Oncol. 1, 3 (2017).
Montero-Conde, C. et al. Relief of feedback inhibition of HER3 transcription by RAF and MEK inhibitors attenuates their antitumor effects in BRAF-mutant thyroid carcinomas. Cancer Discov. 3, 520–533 (2013).
Straussman, R. et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 487, 500–504 (2012).
Zaman, A., Wu, W. & Bivona, T. G. Targeting oncogenic BRAF: past, present, and future. Cancers 11, 1197 (2019).
Whittaker, S. et al. Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci. Transl. Med. 2, 35ra41 (2010).
Nazarian, R. et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010).
Lin, L. et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc. Natl Acad. Sci. USA 111, E748–E757 (2014).
Rizos, H. et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: spectrum and clinical impact. Clin. Cancer Res. 20, 1965–1977 (2014).
Garraway, L. A. & Hahn, W. C. On or off target: mutations, models, and predictions. Sci. Transl. Med. 2, 35ps28 (2010).
Montagut, C. et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 68, 4853–4861 (2008).
Villanueva, J. et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18, 683–695 (2010).
Sharma, V. et al. Registered report: COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Elife 5, e11414 (2016).
Johannessen, C. M. et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 (2010).
Saei, A. & Eichhorn, P. J. A. Adaptive responses as mechanisms of resistance to BRAF inhibitors in melanoma. Cancers 11, 1176 (2019).
Dhillon, A. S., Hagan, S., Rath, O. & Kolch, W. MAP kinase signalling pathways in cancer. Oncogene. 26, 3279–3290 (2007).
Unni, A. M. et al. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. Elife 7, e33718 (2018).
Leung, G. P. et al. Hyperactivation of MAPK signaling is deleterious to RAS/RAF-mutant melanoma. Mol. Cancer Res. 17, 199–211 (2019).
Lake, D., Correa, S. A. & Muller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell Mol. Life Sci. 73, 4397–4413 (2016).
Kidger, A. M. & Keyse, S. M. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin. Cell Dev. Biol. 50, 125–132 (2016).
Hanafusa, H., Torii, S., Yasunaga, T. & Nishida, E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signaling pathway. Nat. Cell Biol. 4, 850–858 (2002).
Ueki, K. et al. Feedback regulation of mitogen-activated protein kinase kinase kinase activity of c-Raf-1 by insulin and phorbol ester stimulation. J. Biol. Chem. 269, 15756–15761 (1994).
Friday, B. B. et al. BRAF V600E disrupts AZD6244-induced abrogation of negative feedback pathways between extracellular signal-regulated kinase and Raf proteins. Cancer Res. 68, 6145–6153 (2008).
Brondello, J. M., Brunet, A., Pouyssegur, J. & McKenzie, F. R. The dual specificity mitogen-activated protein kinase phosphatase-1 and -2 are induced by the p42/p44MAPK cascade. J. Biol. Chem. 272, 1368–1376 (1997).
Amit, I. et al. A module of negative feedback regulators defines growth factor signaling. Nat. Genet. 39, 503–512 (2007).
Kim, H. J. & Bar-Sagi, D. Modulation of signalling by Sprouty: a developing story. Nat. Rev. Mol. Cell Biol. 5, 441–450 (2004).
Avraham, R. & Yarden, Y. Feedback regulation of EGFR signalling: decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 12, 104–117 (2011).
Li, X., Huang, Y., Jiang, J. & Frank, S. J. ERK-dependent threonine phosphorylation of EGF receptor modulates receptor downregulation and signaling. Cell Signal. 20, 2145–2155 (2008).
Pratilas, C. A. et al. (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc. Natl Acad. Sci. USA 106, 4519–4524 (2009).
Joseph, E. W. et al. The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc. Natl Acad. Sci. USA 107, 14903–14908 (2010).
Wan, P. T. et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116, 855–867 (2004).
Poulikakos, P. I. et al. RAF inhibitors transactivate RAF dimers and ERK signaling in cells with wild-type BRAF. Nature 464, 427–430 (2010).
Yao, Z. et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 28, 370–383 (2015).
Poulikakos, P. I. et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390 (2011).
Karoulia, Z., Gavathiotis, E. & Poulikakos, P. I. New perspectives for targeting RAF kinase in human cancer. Nat. Rev. Cancer 17, 676–691 (2017).
Hatzivassiliou, G. et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 (2010).
Zhang, C. et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 526, 583–586 (2015).
Koumaki, K. et al. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Epsilon CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim. Biophys. Acta Mol. Basis Dis. 1867, 166061 (2021).
Karoulia, Z. et al. An integrated model of RAF inhibitor action predicts inhibitor activity against oncogenic BRAF signaling. Cancer Cell 30, 485–498 (2016).
Monaco, K. A. et al. LXH254, a Potent and Selective ARAF-Sparing Inhibitor of BRAF and CRAF for the Treatment of MAPK-Driven Tumors. Clin. Cancer Res. 27, 2061–2073 (2021).
Sun, Y. et al. A brain-penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low-grade astrocytomas. Neuro. Oncol. 19, 774–785 (2017).
Girotti, M. R. et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell 27, 85–96 (2015).
Sullivan, R. J. et al. A phase I study of LY3009120, a Pan-RAF inhibitor, in patients with advanced or metastatic cancer. Mol. Cancer Ther. 19, 460–467 (2020).
Noeparast, A. et al. Type II RAF inhibitor causes superior ERK pathway suppression compared to type I RAF inhibitor in cells expressing different BRAF mutant types recurrently found in lung cancer. Oncotarget 9, 16110–16123 (2018).
Yao, Z. et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nat. Med. 25, 284–291 (2019).
Tutuka, C. S. A. et al. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 16, 112 (2017).
Pickles, O. J. et al. Paradox breaker BRAF inhibitors have comparable potency and MAPK pathway reactivation to encorafenib in BRAF mutant colorectal cancer. Oncotarget 11, 3188–3197 (2020).
Botton, T. et al. Genetic heterogeneity of BRAF fusion kinases in melanoma affects drug responses. Cell Rep. 29, 573–588.e577 (2019).
Jain, P. et al. Overcoming resistance to single-agent therapy for oncogenic BRAF gene fusions via combinatorial targeting of MAPK and PI3K/mTOR signaling pathways. Oncotarget 8, 84697–84713 (2017).
Shi, H. et al. Preexisting MEK1 exon 3 mutations in V600E/KBRAF melanomas do not confer resistance to BRAF inhibitors. Cancer Discov. 2, 414–424 (2012).
Paraiso, K. H. et al. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 102, 1724–1730 (2010).
Frohlich, F., Gerosa, L., Muhlich, J. & Sorger, P. K. Mechanistic model of MAPK signaling reveals how allostery and rewiring contribute to drug resistance. Mol. Syst. Biol. 19, e10988 (2023).
Schreck, K. C. et al. RAF and MEK inhibitor therapy in adult patients with brain tumors: a case-based overview and practical management of adverse events. Neurooncol. Pract. 7, 369–375 (2020).
Daud, A. & Tsai, K. Management of treatment-related adverse events with agents targeting the MAPK pathway in patients with metastatic melanoma. Oncologist. 22, 823–833 (2017).
Carlino, M. S. et al. Antiproliferative effects of continued mitogen-activated protein kinase pathway inhibition following acquired resistance to BRAF and/or MEK inhibition in melanoma. Mol. Cancer Ther. 12, 1332–1342 (2013).
Smalley, K. S. & Flaherty, K. T. Integrating BRAF/MEK inhibitors into combination therapy for melanoma. Br. J. Cancer 100, 431–435 (2009).
Hendrikse, C. S. E. et al. The potential of RAS/RAF/MEK/ERK (MAPK) signaling pathway inhibitors in ovarian cancer: a systematic review and meta-analysis. Gynecol. Oncol. 171, 83–94 (2023).
Whittaker, S. R. et al. Combined Pan-RAF and MEK inhibition overcomes multiple resistance mechanisms to selective RAF inhibitors. Mol. Cancer Ther. 14, 2700–2711 (2015).
Del Curatolo, A. et al. Therapeutic potential of combined BRAF/MEK blockade in BRAF-wild type preclinical tumor models. J. Exp. Clin. Cancer Res. 37, 140 (2018).
Gao, Y. et al. Allele-specific mechanisms of activation of MEK1 mutants determine their properties. Cancer Discov. 8, 648–661 (2018).
Abdel-Wahab, O. et al. Efficacy of intermittent combined RAF and MEK inhibition in a patient with concurrent BRAF- and NRAS-mutant malignancies. Cancer Discov. 4, 538–545 (2014).
Corcoran, R. B. et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov. 8, 428–443 (2018).
Corcoran, R. B. et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2, 227–235 (2012).
Marshall, C. J. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185 (1995).
Das Thakur, M. et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 494, 251–255 (2013).
Kong, X. et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature 550, 270–274 (2017).
Welsh, S. J., Rizos, H., Scolyer, R. A. & Long, G. V. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur. J. Cancer 62, 76–85 (2016).
Xue, Y. et al. An approach to suppress the evolution of resistance in BRAF(V600E)-mutant cancer. Nat. Med. 23, 929–937 (2017).
Rittler, D. et al. Horizontal combination of MEK and PI3K/mTOR inhibition in BRAF mutant tumor cells with or without concomitant PI3K pathway mutations. Int. J. Mol. Sci. 21, 7649 (2020).
Corcoran, R. B. New therapeutic strategies for BRAF mutant colorectal cancers. J. Gastrointest. Oncol. 6, 650–659 (2015).
Dankner, M. et al. Classifying BRAF alterations in cancer: new rational therapeutic strategies for actionable mutations. Oncogene. 37, 3183–3199 (2018).
Candido, S. et al. The PIK3CA H1047R mutation confers resistance to BRAF and MEK inhibitors in A375 melanoma cells through the cross-activation of MAPK and PI3K-Akt pathways. Pharmaceutics 14, (2022).
Bonazzoli, E. et al. PI3K oncogenic mutations mediate resistance to afatinib in HER2/neu overexpressing gynecological cancers. Gynecol. Oncol. 153, 158–164 (2019).
Phuchareon, J., McCormick, F., Eisele, D. W. & Tetsu, O. EGFR inhibition evokes innate drug resistance in lung cancer cells by preventing Akt activity and thus inactivating Ets-1 function. Proc. Natl Acad. Sci. USA 112, E3855–E3863 (2015).
Schreck, K. C. et al. Deconvoluting mechanisms of acquired resistance to RAF inhibitors in BRAF(V600E)-Mutant Human Glioma. Clin. Cancer Res. 27, 6197–6208 (2021).
Menon, S. & Manning, B. D. Common corruption of the mTOR signaling network in human tumors. Oncogene. 27(Suppl 2), S43–S51 (2008).
Mulcahy Levy, J. M. et al. Autophagy inhibition overcomes multiple mechanisms of resistance to BRAF inhibition in brain tumors. Elife 6, e19671 (2017).
Levy, J. M. et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov. 4, 773–780 (2014).
Atefi, M. et al. Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One 6, e28973 (2011).
Nichols, R. J. et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol. 20, 1064–1073 (2018).
Liu, C. et al. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin Cancer Res 27, 342–354 (2021).
Eroglu, Z. & Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther. Adv. Med. Oncol. 8, 48–56 (2016).
Das Thakur, M. & Stuart, D. D. The evolution of melanoma resistance reveals therapeutic opportunities. Cancer Res. 73, 6106–6110 (2013).
Schreuer, M. et al. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAF(V600)-mutant melanoma: an open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 18, 464–472 (2017).
Fernandez, L. P., Gomez de Cedron, M. & Ramirez de Molina, A. Alterations of lipid metabolism in cancer: implications in prognosis and treatment. Front. Oncol. 10, 577420 (2020).
Abildgaard, C. & Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 21, 164–171 (2015).
Sundstrom, T. et al. Inhibition of mitochondrial respiration prevents BRAF-mutant melanoma brain metastasis. Acta Neuropathol. Commun. 7, 55 (2019).
Figarola, J. L. et al. Bioenergetic modulation with the mitochondria uncouplers SR4 and niclosamide prevents proliferation and growth of treatment-naive and vemurafenib-resistant melanomas. Oncotarget 9, 36945–36965 (2018).
Yu, J. et al. Advanced cancer starvation therapy by simultaneous deprivation of lactate and glucose using a MOF nanoplatform. Adv. Sci. 8, e2101467 (2021).
Simons, A. L. et al. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 67, 3364–3370 (2007).
Parmenter, T. J. et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 4, 423–433 (2014).
Rashmi, R. et al. Radioresistant cervical cancers are sensitive to inhibition of glycolysis and redox metabolism. Cancer Res. 78, 1392–1403 (2018).
Zhang, M. et al. Adipocyte-derived lipids mediate melanoma progression via FATP proteins. Cancer Discov. 8, 1006–1025 (2018).
Yuan, J. et al. The AMPK inhibitor overcomes the paradoxical effect of RAF inhibitors through blocking phospho-Ser-621 in the C terminus of CRAF. J Biol. Chem. 293, 14276–14284 (2018).
Yuan, P. et al. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc. Natl Acad. Sci. USA 110, 18226–18231 (2013).
Han, J. et al. Elucidating molecular mechanisms of acquired resistance to BRAF inhibitors in melanoma using a microfluidic device and deep sequencing. Genom. Inform. 19, e2 (2021).
Konieczkowski, D. J. et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov 4, 816–827 (2014).
Hoek, K. S. et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 19, 290–302 (2006).
Weeraratna, A. T. et al. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell 1, 279–288 (2002).
Kemper, K., de Goeje, P. L., Peeper, D. S. & van Amerongen, R. Phenotype switching: tumor cell plasticity as a resistance mechanism and target for therapy. Cancer Res. 74, 5937–5941 (2014).
Long, G. V. et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 5, 5694 (2014).
Irvine, M. et al. Oncogenic PI3K/AKT promotes the step-wise evolution of combination BRAF/MEK inhibitor resistance in melanoma. Oncogenesis 7, 72 (2018).
van Geel, R. et al. A phase Ib dose-escalation study of encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF-mutant colorectal cancer. Cancer Discov. 7, 610–619 (2017).
Mao, M. et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin. Cancer Res. 19, 657–667 (2013).
Forbes, S. A. et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017).
Catalanotti, F. et al. PTEN Loss-of-function alterations are associated with intrinsic resistance to BRAF inhibitors in metastatic melanoma. JCO Precis Oncol. 1, (2017).
Paraiso, K. H. et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin. Cancer Res. 18, 2502–2514 (2012).
Fedorenko, I. V. et al. BRAF inhibition generates a host-tumor niche that mediates therapeutic escape. J. Invest. Dermatol. 135, 3115–3124 (2015).
Almeida, F. V., Douglass, S. M., Fane, M. E. & Weeraratna, A. T. Bad company: microenvironmentally mediated resistance to targeted therapy in melanoma. Pigment Cell Melanoma Res. 32, 237–247 (2019).
Wang, H. et al. Pro-tumor activities of macrophages in the progression of melanoma. Hum. Vaccine Immunother. 13, 1556–1562 (2017).
Abu-Remaileh, M. et al. Chronic inflammation induces a novel epigenetic program that is conserved in intestinal adenomas and in colorectal cancer. Cancer Res. 75, 2120–2130 (2015).
Gallina, G. et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Invest. 116, 2777–2790 (2006).
Denkert, C. et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 28, 105–113 (2010).
Escuin-Ordinas, H. et al. COX-2 inhibition prevents the appearance of cutaneous squamous cell carcinomas accelerated by BRAF inhibitors. Mol. Oncol. 8, 250–260 (2014).
Brady, D. C. et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509, 492–496 (2014).
Dogan, E., Kara, H. G., Kosova, B. & Cetintas, V. B. in Metastasis (ed C. M. Sergi) (2022).
Mukherjee, N. et al. BH3 mimetics induce apoptosis independent of DRP-1 in melanoma. Cell Death Dis. 9, 907 (2018).
Wroblewski, D. et al. The BH3-mimetic ABT-737 sensitizes human melanoma cells to apoptosis induced by selective BRAF inhibitors but does not reverse acquired resistance. Carcinogenesis 34, 237–247 (2013).
Anvekar, R. A. et al. Sensitization to the mitochondrial pathway of apoptosis augments melanoma tumor cell responses to conventional chemotherapeutic regimens. Cell Death Dis. 3, e420 (2012).
Serasinghe, M. N. et al. Anti-apoptotic BCL-2 proteins govern cellular outcome following B-RAF(V600E) inhibition and can be targeted to reduce resistance. Oncogene 34, 857–867 (2015).
Alcolea, V. et al. Identification of a novel quinoxaline-isoselenourea targeting the STAT3 pathway as a potential melanoma therapeutic. Int. J. Mol. Sci. 20, 512 (2019).
Su, Y. et al. Targeting STAT3 restores BRAF inhibitor sensitivity through miR-759-3p in human cutaneous melanoma cells. Int. J. Clin. Exp. Pathol. 11, 2550–2560 (2018).
Moriizumi, H. et al. Caspase 3-specific cleavage of MEK1 suppresses ERK signaling and sensitizes cells to stress-induced apoptosis. FEBS Open Bio. 13, 684–700 (2023).
Chen, L. H. et al. Inhibition of endoplasmic reticulum stress-induced apoptosis of melanoma cells by the ARC protein. Cancer Res. 68, 834–842 (2008).
Jiang, C. C. et al. Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 67, 9750–9761 (2007).
Liu, J. et al. High-throughput quantitative detection of triple-negative breast cancer-associated expressed miRNAs by rolling circle amplification on fluorescence-encoded microspheres. Chin. Chem. Lett. 34, 108141 (2023).
Li, H. et al. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target Ther. 5, 1 (2020).
Olivieri, I., Vitali, C., Gemignani, G. & Pasero, G. Concomitant ankylosing spondylitis and DISH. J. Rheumatol. 16, 1170–1172 (1989).
Turner, E. et al. CRISPR/Cas9 Edited RAS & MEK Mutant Cells Acquire BRAF and MEK Inhibitor Resistance with MEK1 Q56P Restoring Sensitivity to MEK/BRAF Inhibitor Combo and KRAS G13D Gaining Sensitivity to Immunotherapy. Cancers 14, 5449 (2022).
Yao, M. et al. Research progress of nanovaccine in anti-tumor immunotherapy. Front. Oncol. 13, 1211262 (2023).
Zhang, L. et al. A novel therapeutic vaccine based on graphene oxide nanocomposite for tumor immunotherapy. Chin. Chem. Lett. 33, 4089–4095 (2022).
Durrant, D. E. et al. Development of a high-throughput NanoBRET screening platform to identify modulators of the RAS/RAF interaction. Mol. Cancer Ther. 20, 1743–1754 (2021).
Khazak, V. et al. A two-hybrid approach to identify inhibitors of the RAS-RAF interaction. Enzymes 33(Pt A), 213–248 (2013).
Levy, J. M. & Thorburn, A. Targeting autophagy during cancer therapy to improve clinical outcomes. Pharmacol. Ther. 131, 130–141 (2011).
Shen, S. et al. Association and dissociation of autophagy, apoptosis and necrosis by systematic chemical study. Oncogene 30, 4544–4556 (2011).
Kochl, R., Hu, X. W., Chan, E. Y. & Tooze, S. A. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 7, 129–145 (2006).
Hua, H. et al. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 12, 71 (2019).
Oltersdorf, T. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681 (2005).
Pattingre, S. et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 (2005).
Zhou, C. et al. AMPK-autophagy inhibition sensitizes icaritin-induced anti-colorectal cancer cell activity. Oncotarget 8, 14736–14747 (2017).
Blommaart, E. F. et al. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 243, 240–246 (1997).
Wu, Y. T. et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 285, 10850–10861 (2010).
Buchser, W. J. et al. Cell-mediated autophagy promotes cancer cell survival. Cancer Res. 72, 2970–2979 (2012).
Bahar, E., Han, S. Y., Kim, J. Y. & Yoon, H. Chemotherapy resistance: role of mitochondrial and autophagic components. Cancers 14, 1462 (2022).
Al-Ejeh, F. et al. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 29, 6085–6098 (2010).
John, S. et al. Regulation of estrogenic effects by beclin 1 in breast cancer cells. Cancer Res. 68, 7855–7863 (2008).
Manic, G. et al. Chloroquine and hydroxychloroquine for cancer therapy. Mol. Cell Oncol. 1, e29911 (2014).
Chen, N. & Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 11, 157–168 (2011).
Amaravadi, R. K. et al. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 17, 654–666 (2011).
Fratta, E. et al. Autophagy in BRAF-mutant cutaneous melanoma: recent advances and therapeutic perspective. Cell Death Discov. 9, 202 (2023).
Foth, M. & McMahon, M. Autophagy inhibition in BRAF-driven cancers. Cancers 13, 3498 (2021).
Ma, L. et al. Matrine inhibits BCR/ABL mediated ERK/MAPK pathway in human leukemia cells. Oncotarget 8, 108880–108889 (2017).
Martinez-Lopez, N. et al. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 4, 2799 (2013).
Kabeya, Y. et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J. Cell Sci. 117, 2805–2812 (2004).
Tanida, I. et al. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J. Biol. Chem. 279, 36268–36276 (2004).
Sanduja, S. et al. AMPK promotes tolerance to Ras pathway inhibition by activating autophagy. Oncogene 35, 5295–5303 (2016).
Gwinn, D. M. et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008).
Corazzari, M. et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 22, 946–958 (2015).
Ogata, M. et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 26, 9220–9231 (2006).
Vlasic, I., Horvat, A., Tadijan, A. & Slade, N. p53 family in resistance to targeted therapy of melanoma. Int. J. Mol. Sci. 24, 65 (2022).
Crighton, D. et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134 (2006).
Sreeramaneni, R. et al. Ras-Raf-Arf signaling critically depends on the Dmp1 transcription factor. Mol. Cell Biol. 25, 220–232 (2005).
Inbal, B. et al. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 157, 455–468 (2002).
Ascierto, P. A. et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. Lancet Oncol. 14, 249–256 (2013).
Gibney, G. T. et al. Paradoxical oncogenesis–the long-term effects of BRAF inhibition in melanoma. Nat. Rev. Clin. Oncol. 10, 390–399 (2013).
Yun, S. et al. Targeting immune checkpoints in unresectable metastatic cutaneous melanoma: a systematic review and meta-analysis of anti-CTLA-4 and anti-PD-1 agents trials. Cancer Med. 5, 1481–1491 (2016).
Weiss, S. A., Wolchok, J. D. & Sznol, M. Immunotherapy of melanoma: facts and hopes. Clin. Cancer Res. 25, 5191–5201 (2019).
Huynh, S. et al. Combined therapy with Anti-PD1 and BRAF and/or MEK inhibitor for advanced melanoma: a multicenter cohort study. Cancers 12, 1666 (2020).
Hu-Lieskovan, S. et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 7, 279ra241 (2015).
Dummer, R. et al. Rationale for immune checkpoint inhibitors plus targeted therapy in metastatic melanoma: a review. JAMA Oncol. 6, 1957–1966 (2020).
Sanlorenzo, M. et al. BRAF and MEK Inhibitors increase PD-1-positive melanoma cells leading to a potential lymphocyte-independent synergism with anti-PD-1 antibody. Clin. Cancer Res. 24, 3377–3385 (2018).
Li, S. et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 10, 1693 (2019).
Kinsey, C. G. et al. Protective autophagy elicited by RAF–>MEK–>ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 25, 620–627 (2019).
Bryant, K. L. et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 25, 628–640 (2019).
Goodall, M. L. et al. Development of potent autophagy inhibitors that sensitize oncogenic BRAF V600E mutant melanoma tumor cells to vemurafenib. Autophagy 10, 1120–1136 (2014).
Seton-Rogers, S. Eliminating protective autophagy in KRAS-mutant cancers. Nat. Rev. Cancer 19, 247 (2019).
Guo, J. Y. et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev. 27, 1447–1461 (2013).
Strohecker, A. M. et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 3, 1272–1285 (2013).
Koustas, E., Papavassiliou, A. G. & Karamouzis, M. V. The role of autophagy in the treatment of BRAF mutant colorectal carcinomas differs based on microsatellite instability status. PLoS One 13, e0207227 (2018).
Zhao, H. & Zheng, B. Dual Targeting of Autophagy and MEK in KRAS Mutant Cancer. Trends Cancer. 5, 327–329 (2019).
Wang, W. et al. Targeting Autophagy Sensitizes BRAF-Mutant Thyroid Cancer to Vemurafenib. J. Clin. Endocrinol. Metab. 102, 634–643 (2017).
Levy, J. M. M., Towers, C. G. & Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 (2017).
Chude, C. I. & Amaravadi, R. K. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 18, 1279 (2017).
Li, C. et al. Impact of autophagy inhibition at different stages on cytotoxic effect of autophagy inducer in glioblastoma cells. Cell Physiol. Biochem. 35, 1303–1316 (2015).
Palmeira dos Santos, C. et al. Comparative study of autophagy inhibition by 3MA and CQ on Cytarabine‑induced death of leukaemia cells. J. Cancer Res. Clin. Oncol. 140, 909–920 (2014).
Follo, C. et al. Inhibition of autophagy initiation potentiates chemosensitivity in mesothelioma. Mol. Carcinog. 57, 319–332 (2018).
Jain, V., Singh, M. P. & Amaravadi, R. K. Recent advances in targeting autophagy in cancer. Trends Pharmacol. Sci. 44, 290–302 (2023).
Shi, T. T., Yu, X. X., Yan, L. J. & Xiao, H. T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharmacol. 79, 287–294 (2017).
Piao, S. & Amaravadi, R. K. Targeting the lysosome in cancer. Ann. N. Y. Acad. Sci. 1371, 45–54 (2016).
Maes, H., Kuchnio, A., Carmeliet, P. & Agostinis, P. How to teach an old dog new tricks: autophagy-independent action of chloroquine on the tumor vasculature. Autophagy. 10, 2082–2084 (2014).
Maes, H. et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 26, 190–206 (2014).
Barnard, R. A. et al. Phase I clinical trial and pharmacodynamic evaluation of combination hydroxychloroquine and doxorubicin treatment in pet dogs treated for spontaneously occurring lymphoma. Autophagy 10, 1415–1425 (2014).
Warhurst, D. C. et al. Hydroxychloroquine is much less active than chloroquine against chloroquine-resistant Plasmodium falciparum, in agreement with its physicochemical properties. J. Antimicrob. Chemother. 52, 188–193 (2003).
Finbloom, D. S., Silver, K., Newsome, D. A. & Gunkel, R. Comparison of hydroxychloroquine and chloroquine use and the development of retinal toxicity. J. Rheumatol. 12, 692–694 (1985).
Xie, X., White, E. P. & Mehnert, J. M. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One 8, e55096 (2013).
Dragowska, W. H. et al. Induction of autophagy is an early response to gefitinib and a potential therapeutic target in breast cancer. PLoS One 8, e76503 (2013).
Cook, K. L. et al. Hydroxychloroquine inhibits autophagy to potentiate antiestrogen responsiveness in ER+ breast cancer. Clin. Cancer Res. 20, 3222–3232 (2014).
McAfee, Q. et al. Autophagy inhibitor Lys05 has single-agent antitumor activity and reproduces the phenotype of a genetic autophagy deficiency. Proc. Natl Acad. Sci. USA 109, 8253–8258 (2012).
Zhou, W., Guo, Y., Zhang, X. & Jiang, Z. Lys05 induces lysosomal membrane permeabilization and increases radiosensitivity in glioblastoma. J. Cell Biochem. 121, 2027–2037 (2020).
Amaravadi, R. K. & Winkler, J. D. Lys05: a new lysosomal autophagy inhibitor. Autophagy. 8, 1383–1384 (2012).
Maycotte, P. et al. Chloroquine sensitizes breast cancer cells to chemotherapy independent of autophagy. Autophagy 8, 200–212 (2012).
Eng, C. H. et al. Macroautophagy is dispensable for growth of KRAS mutant tumors and chloroquine efficacy. Proc. Natl Acad. Sci. USA 113, 182–187 (2016).
Zahedi, S. et al. Effect of early-stage autophagy inhibition in BRAF(V600E) autophagy-dependent brain tumor cells. Cell Death Dis. 10, 679 (2019).
Roach, P. J. AMPK -> ULK1 -> autophagy. Mol. Cell Biol. 31, 3082–3084 (2011).
Alers, S., Loffler, A. S., Wesselborg, S. & Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 32, 2–11 (2012).
Follo, C. et al. Autophagy initiation correlates with the autophagic flux in 3D models of mesothelioma and with patient outcome. Autophagy 12, 1180–1194 (2016).
Petherick, K. J. et al. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 290, 11376–11383 (2015).
Egan, D. F. et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol. Cell 59, 285–297 (2015).
Tang, F. et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 37, 3449–3458 (2017).
Dowdle, W. E. et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 16, 1069–1079 (2014).
Bilanges, B. & Vanhaesebroeck, B. Cinderella finds her shoe: the first Vps34 inhibitor uncovers a new PI3K-AGC protein kinase connection. Biochem. J. 464, e7–e10 (2014).
Ronan, B. et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 10, 1013–1019 (2014).
Bago, R. et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 463, 413–427 (2014).
Ji, C. H. et al. The AUTOTAC chemical biology platform for targeted protein degradation via the autophagy-lysosome system. Nat. Commun. 13, 904 (2022).
Deitersen, J. et al. High-throughput screening for natural compound-based autophagy modulators reveals novel chemotherapeutic mode of action for arzanol. Cell Death Dis. 12, 560 (2021).
Qiu, R. G., Chen, J., McCormick, F. & Symons, M. A role for Rho in Ras transformation. Proc. Natl Acad. Sci. USA 92, 11781–11785 (1995).
Qiu, R. G., Abo, A., McCormick, F. & Symons, M. Cdc42 regulates anchorage-independent growth and is necessary for Ras transformation. Mol. Cell Biol. 17, 3449–3458 (1997).
Qiu, R. G. et al. An essential role for Rac in Ras transformation. Nature 374, 457–459 (1995).
Soriano, O., Alcon-Perez, M., Vicente-Manzanares, M. & Castellano, E. The crossroads between RAS and RHO signaling pathways in cellular transformation, motility and contraction. Genes 12, 819 (2021).
Tang, Y., Yu, J. & Field, J. Signals from the Ras, Rac, and Rho GTPases converge on the Pak protein kinase in Rat-1 fibroblasts. Mol. Cell Biol. 19, 1881–1891 (1999).
Wang, Y., Senoo, H., Sesaki, H. & Iijima, M. Rho GTPases orient directional sensing in chemotaxis. Proc. Natl Acad. Sci. USA 110, E4723–E4732 (2013).
Zang, M., Hayne, C. & Luo, Z. Interaction between active Pak1 and Raf-1 is necessary for phosphorylation and activation of Raf-1. J. Biol. Chem. 277, 4395–4405 (2002).
Tran, N. H. & Frost, J. A. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J. Biol. Chem. 278, 11221–11226 (2003).
Zang, M. et al. Microtubule integrity regulates Pak leading to Ras-independent activation of Raf-1. insights into mechanisms of Raf-1 activation. J. Biol. Chem. 276, 25157–25165 (2001).
King, A. J. et al. The protein kinase Pak3 positively regulates Raf-1 activity through phosphorylation of serine 338. Nature 396, 180–183 (1998).
Cammarano, M. S., Nekrasova, T., Noel, B. & Minden, A. Pak4 induces premature senescence via a pathway requiring p16INK4/p19ARF and mitogen-activated protein kinase signaling. Mol. Cell Biol. 25, 9532–9542 (2005).
Cotteret, S., Jaffer, Z. M., Beeser, A. & Chernoff, J. p21-Activated kinase 5 (Pak5) localizes to mitochondria and inhibits apoptosis by phosphorylating BAD. Mol. Cell Biol. 23, 5526–5539 (2003).
McKay, M. M. & Morrison, D. K. Integrating signals from RTKs to ERK/MAPK. Oncogene. 26, 3113–3121 (2007).
Sun, H., King, A. J., Diaz, H. B. & Marshall, M. S. Regulation of the protein kinase Raf-1 by oncogenic Ras through phosphatidylinositol 3-kinase, Cdc42/Rac and Pak. Curr. Biol. 10, 281–284 (2000).
Chao, T. S. et al. Src tyrosine kinase mediates stimulation of Raf-1 and mitogen-activated protein kinase by the tumor promoter thapsigargin. Cancer Res. 57, 3168–3173 (1997).
Ziogas, A., Moelling, K. & Radziwill, G. CNK1 is a scaffold protein that regulates Src-mediated Raf-1 activation. J. Biol. Chem. 280, 24205–24211 (2005).
Grammatikakis, N. et al. p50(cdc37) acting in concert with Hsp90 is required for Raf-1 function. Mol. Cell Biol. 19, 1661–1672 (1999).
Wallace, A. G. & von Witt, R. Modifast and weight reduction. Med. J. Aust. 140, 598 (1984).
Xia, K. et al. Tyrosine phosphorylation of the proto-oncoprotein Raf-1 is regulated by Raf-1 itself and the phosphatase Cdc25A. Mol. Cell Biol. 19, 4819–4824 (1999).
Cheng, J. J., Wung, B. S., Chao, Y. J. & Wang, D. L. Sequential activation of protein kinase C (PKC)-alpha and PKC-epsilon contributes to sustained Raf/ERK1/2 activation in endothelial cells under mechanical strain. J. Biol. Chem. 276, 31368–31375 (2001).
Reusch, H. P. et al. Regulation of Raf by Akt controls growth and differentiation in vascular smooth muscle cells. J. Biol. Chem. 276, 33630–33637 (2001).
Luo, H., Rose, P. E., Roberts, T. M. & Dearolf, C. R. The Hopscotch Jak kinase requires the Raf pathway to promote blood cell activation and differentiation in Drosophila. Mol. Genet. Genom.267, 57–63 (2002).
Abraham, D. et al. Raf-1-associated protein phosphatase 2A as a positive regulator of kinase activation. J. Biol. Chem. 275, 22300–22304 (2000).
Raabe, T. & Rapp, U. R. Ras signaling: PP2A puts Ksr and Raf in the right place. Curr. Biol. 13, R635–R637 (2003).
Acknowledgements
We thank all the lab members for their thoughtful feedback on this study. All figures were created with BioRender.com.
Funding
This study was supported by grants from the Basic Science Research Program through the National Research Foundation of Korea (RS-2023-00219399, RS-2023-00238051) and by the Commercializations Promotion Agency for R&D Outcomes (COMPA) grant funded by the Korea government (MSIT) (1711173796).
Author information
Authors and Affiliations
Contributions
E.M.B. conceptualized, wrote the manuscript, and created all original figures. H.J.K. collected data for clinical trials, and edited the manuscript during revision. D.R.K. conceptualized, provided financial support, edited all figures, and contributed to writing the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bahar, M.E., Kim, H.J. & Kim, D.R. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Sig Transduct Target Ther 8, 455 (2023). https://doi.org/10.1038/s41392-023-01705-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01705-z
This article is cited by
-
Long-term response to MEK inhibitor monotherapy in a patient with papillary thyroid carcinoma harboring BRAF V600E mutation
International Cancer Conference Journal (2024)
