
Abstract
OBJECTIVE
The observed heterogeneity of autism spectrum disorder (ASD)—and the diversity of rare germline mutations with which it has been associated—has been difficult to reconcile with knowledge of its pronounced heritability in the population.
Methods
This article reviews and synthesizes recent family and developmental studies incorporating behavioral indices of inherited risk for ASD.
Results
Autism may arise from critical combinations of early inherited neurobehavioral susceptibilities—some specific to autism, some not—each of which may be traceable to partially-independent sets of genetic variation. These susceptibilities and their respective genetic origins may not relate to the characterizing symptoms of autism (after it develops) in a straightforward way, and may account for “missing heritability” in molecular genetic studies.
Conclusions
Within-individual aggregations of a finite set of early inherited neurobehavioral susceptibilities—each individually common in the population—may account for a significant share of the heritability of ASD. Comprehensive identification of these underlying traits my help elucidate specific early intervention targets in individual patients, especially if autism represents a developmental consequence of earlier-interacting susceptibilities. Scientific understanding of the early ontogeny of autism will benefit from epidemiologically-rigorous, genetically-informative studies of robust endophenotypic candidates whose inter-relationships in infancy are mapped and normed.
Similar content being viewed by others

Introduction
More than a decade ago, in a seminal sibling study to refine understanding of the heritability of autism, Spiker, Risch, and colleagues1 explored the foundational question of whether the characterizing traits and features of autism spectrum disorder (ASD) bred true in autism-affected families. Their report documented the startling (and at the time perhaps under-appreciated) observation that what clustered in individual families affected by multiplex autism was not specific profiles or patterns of the three characterizing symptoms of the disorder (social deficits, language/communication deficits, and repetitive/stereotypic motor behavior), rather impairment along a single continuous severity dimension, the heritability of which roughly equated to the broad heritability of autism in the population. Stated another way, the severity of the syndrome as a whole bred truer than that of its ostensible component parts. This finding recapitulated the conclusions of other reports that sub clinical characteristics of the entire autistic syndrome (i.e., sub clinical elevations in all three DSM-IV criterion domains)—the so-called broader-autism phenotype2,3—were more commonly manifest in the unaffected relatives of individuals with categorically diagnosed autistic syndromes than in the general population. These two principal findings motivated a body of research which I reviewed in a previous article in this journal, entitled, The Quantitative Nature of Autistic Social Impairment.4
How could it be that the total severity of the syndrome itself bred truer than its own symptoms? Or that sub clinical versions of the entirety of the autistic syndrome were traceable in unaffected family members? Numerous explorations of the associations between genotype and phenotype in ASD have now converged upon the same fundamental principle of a single continuous severity dimension, the nature of which, until recently, has been enigmatic. In this article, I will describe how the “downward extension” of quantitative characterization of human behavior from childhood (i.e., after the time when autistic symptomatology usually first manifests) to infancy (i.e., before autism develops) is beginning to provide new clues to understanding the manner in which genetic variation relates to the neuropsychiatric impairments that define the condition. This review will cover recent advances which have suggested that ASD may arise from specific combinations of early inherited neurobehavioral susceptibilities—some specific to ASD, some not—and that generally do not bear direct 1:1 associations with specific ASD symptoms. A key implication for future pediatric research is that linking genetic variants to these earlier underlying traits rather than to the subsequent symptoms of ASD (once it develops) may be more productive in relating genes to brain to behavior and in devising effective approaches to preventive intervention.5,6
Behavioral manifestations of polygenic risk for autism
It is first important to contextualize the scientific relevance of elucidating tractable early developmental phenotypes, in light of contemporary understanding of the causal structure of autism, a principal aspect of which is that the heritability of the condition is pronounced, on the order of 85%.7 The past decade has seen remarkable advances in identifying de novo (germline) mutations that each represent rare monogenic causes of ASD,8,9 though, by definition, account for none of the syndrome’s substantial heritability. The number of genes implicated by such studies is over one hundred and growing by the month; each rare syndrome engendered by a deleterious mutation in a given gene presents critical scientific opportunity to explore: (a) how hemizygous loss- or gain-of-function of that gene can engender a phenocopy of ASD with or without co-occurring neurologic impairments such as epilepsy or intellectual disability; (b) how convergent pathogenic mechanisms triggered by mutations in disparate genes might be shared and therefore represent common therapeutic targets; and (c) how less disruptive variations in the same genes might contribute incremental causal influence on autistic syndromes.
Despite the demonstrated role of de novo variants, however, it is polygenic liability that accounts for the majority of the population-attributable risk for ASD,10,11 including that majority of cases in which ASD is not accompanied by intellectual disability. If common allelic variations are cumulatively responsible for (a) most cases of autism, and (b) the range of sub clinical autistic traits that encompass the general population distribution—these aggregate among the unaffected relatives of ASD probands and are minimally correlated with variation in intellectual functioning—inherited developmental phenotypes may represent far more parsimonious targets for understanding biology than can currently be identified linking molecular genetic data to diagnosis alone.
Notably, deleterious de novo mutations can combine with polygenic risk within an individual to give rise to an autistic syndrome.12,13 Tracing the respective effects of each type of genetic influence to a given behavioral phenotype—especially when such influences co-occur in an individual—has yielded remarkable new insights into understanding the manner in which polygenic risk influences brain and behavior. For example, Moreno DeLuca et al.14 showed that de novo 16p11.2 deletions incurred a predictable deleterious “shift” in the severity of autistic impairment in offspring (on the order of one to two standard deviations) in relation to what would be expected on the basis of the average quantitative trait burden (for each respective trait) of his/her parents. Thus, children with de novo deletions of 16p11.2 whose parents fell in the below-average range for social communication—yet still entirely within normal limits for adults—were significantly more likely to be affected by clinical-level ASD than those whose parents’ social communication was above average. Recognition that some mutations induce a predictable shift against a measurable genetic background rather than exerting an absolute effect represents an important advance in understanding clinical genomic variation (“Shift Happens”15). A corollary is that background variation within the range of normality can exert a range of quantitative effects on the phenotypic expression of a superimposed “deleterious” mutation.
Population-wide, the inherited behavioral endophenotype indexed by sub clinical variation in the characterizing traits and features of ASD among parents (as measured, for example, using the Social Responsiveness Scale) is now known to account for approximately 10 per cent of autistic trait variation among their offspring16 and to proportionately elevate risk for a clinical autistic syndrome.17,18 A question arises, then, to what extent the remaining inherited risk might be captured by measurements of one or more pre-diagnostic behavioral phenotypes representing causal liability prior to the onset of a diagnosable autistic syndrome.
Insights from a first generation of prospective studies of later-born infant siblings
A proliferation of studies of the early development of ASD among high-risk infant siblings of children with ASD has been particularly informative in this regard19,20,21,22,23,24 and reviewed by Szatmari et al.25 The conclusions of these studies have converged upon several important themes. First, the characterizing traits and features of ASD are usually absent prior to the age of 12 months—even when ascertaining very subtle versions of those traits among children who go on to develop severe autistic syndromes.26 Second, joint indices of (a) sensorimotor dysfunction; (b) nuanced components of early precursors of social communication (including social motivation, joint attention, infant vocalization, and response to name); and (c) global cognitive development may more robustly predict the development of ASD than any single index,24,26 but this is only beginning to be understood and will require a next generation of prospective recurrence studies with adequate sample sizes and much more complete phenotypic characterization. Third, when considering predictors for which epidemiologic prevalence in the general population is known (this is not common and a nagging scientific gap), the positive predictive value of each of these factors (or their combinations) for ASD is often more robust in the context of recurrence (i.e., the study of infant siblings of children affected by ASD) than in the general population.27 This suggests the possibility that some of the measurable early behavioral predictors of ASD may (a) arise independently from familial ASD risk and (b) subsequently interact with ASD-specific familial risk to amplify liability to the clinical syndrome. Examples of early developmental phenotypes that have been studied in an epidemiologic context and exhibit this pattern—i.e., that may constitute more robust predictors of within-family recurrence of autism than general population risk—include variation in (i) visual social engagement, (ii) motor coordination, and (iii) attention problems, each of which are described in greater detail below. A counter-example is accelerated head growth, which is highly familial but weakly associated with ASD in the general population, and a poor predictor of recurrence in ASD-affected families.28
Since many of the “first generation” high-risk infant sibling studies were designed exclusively to focus on “autism-specific” symptoms, the relative difficulty in identifying first year of life behavioral predictors homologous to the characterizing features of the diagnosis has appropriately motivated efforts to progressively widen the search space for early predictors of the condition, to phenotypes that are common among children with autism but not necessarily specific to ASD. The first major clues in this regard have come from observations of delayed sensorimotor development23,24 and/or the dissociation of typical correlations between motor and language development29 among high-risk infants who ultimately develop ASD. This complements a growing body of studies of school-aged children that have repeatedly documented motor coordination impairments occurring among children with ASD,30,31,32,33 at a frequency that rivals that of language impairment in ASD, though still not yet represented in the DSM5 diagnostic criteria for the disorder, and traditionally excluded because of its “non-specificity” for autism—this is a point to which I will return below. A major obstacle to research in establishing the role of these impairments in the developmental ontogeny of autism is that the methods for measuring meaningful variation in the first year of life are not yet as precise or reliable as those that apply later in development.
Specification of autism endophenotypes appreciable in infancy and early childhood
It is important to emphasize that the road to confirmation, that an early behavioral liability reflects an independent causal influence on autism, is long. First, since autism is largely heritable, a behavioral liability must be shown to be substantially heritable in a genetic epidemiologic context. Second, it must be shown that it aggregates among family members of clinically-affected individuals. Third, it must be shown to be at least partially independent from (minimally correlated with) other behavioral liabilities that are already established as causal influences on ASD.34 Fourth, it must predict ASD, either in the general population,35 or its recurrence in families affected by autism. These are the characteristics of endophenotypes. Notably, phenotypes which predict autism within a family recurrence context (i.e., later-born infant sibling studies) but do NOT predict autism as strongly in an epidemiologic context (i.e., general population studies) constitute behavioral liabilities that are not necessarily specific to autism. They may be common in the general population but require interactions with (or joint effects of) other influential factors to engender autism, in the absence of which their predictive influence is more modest.
For most candidate endophenotypes—whether behavioral, psychophysiologic,36 electrophysiologic,37,38 serologic39, or brain-imaging-based40,41,42,43 (these are outside of the scope of this review and their relation to most behavioral endophenotypes currently unknown)—what we know is generally limited to observed associations with autism in case control or recurrence studies, and it is therefore difficult to know the extent to which an observed association reflects causation of the condition (as opposed to a correlate or secondary consequence). Since a great deal hinges upon whether or not a candidate endophenotype reflects a causal influence, it should be made a scientific priority for candidate endophenotypes to be subjected to the systematic complement of studies—epidemiologic, family recurrence, prospective prediction, and association with other early predictors—that would “convert” a correlate or co-occuring trait to the status of endophenotype and to clarify its relative contribution to population-attributable risk for the condition. If the number of early-appreciable ASD endophenotypes that encompass the totality of polygenic liability to autism is relatively low, precise characterization of early neurobehavioral profiles that predict autism may be well within reach, if such systematic studies are pursued.
The notion of autism as an acquired “diversion” of typical development in the setting of a critical aggregation of early behavioral liabilities would be consistent with the heritability of autism (once it develops) as lying along a single continuous severity dimension,1 and carries with it the profound implication that the condition may not be “fixed” or inevitable, rather prevented before it arises (but perhaps not later) if there are ways to resolve the earlier deficits,6 a consideration supported by other studies described below.
Non-ASD-specific contributions to the cause of autism
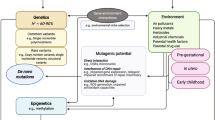
In recent analyses of the role of non-ASD-specific neurodevelopmental traits on ASD occurrence or recurrence in families, it was observed that Attention-Deficit Hyperactivity Disorder (ADHD) symptoms and motor coordination impairment jointly account for a very significant share of the variance (over 50%) in (a) categorical ASD recurrence; (b) quantitative trait severity;33 and (c) that siblings of children with ADHD have a four-fold increase in the incidence of autism.44 These findings are in keeping with earlier observations from general population studies45 of substantial genetic overlap between autistic traits and symptoms of either inattention/hyperactivity and developmental coordination disorder. Impairments in attention and motor coordination are strongly genetically-influenced but not specific to autism, and may relate to autism by amplifying the effect of “ASD-specific” factors early in development. Although such traits are sometimes referred to as “co-morbidities,” these and other research observations46,47 suggest that they may be actual causes of the autistic syndrome. Rather than being discarded (or mathematically regressed away in research studies48) it may be as important to include measurement of these traits as it is to measure traits that have traditionally been viewed as more “specific” to autism. A schematic depiction of the genetic epidemiologic relationship between ADHD and autism supported by these data is provided in Fig. 1.

Mechanisms by which genetic influences that are non-specific to autism may compound autistic severity and incur “comorbid” affectation with non-ASD-specific traits (ADHD as an example here). 1. Specific influences on ASD 2. Amplification of ASD severity by a non-ASD-specific causal influence. ADHD attention-deficit/hyperactivity disorder, ASD autism spectrum disorder. Note that in this model, inherited liabilities that are initially independent become correlated (a) by definition when contributing to the development of autism, and (b) by reciprocal interaction over the course of later development, after the respective syndromes of disability are manifest
In general, behavioral comorbidity is the rule rather than the exception in ASD. An ASD “comorbidity” can have several fundamentally-distinct causal origins: (i) it can arise from chance co-occurrence of ASD with a causally independent liability (e.g., ASD and diabetes); (ii) it can arise as a secondary symptom of ASD (e.g., ASD and aggression); or (iii) it can be the late manifestation of a cause of ASD. Recently, Hawks et al.49 traced the respective origins of autistic and general psychopathologic traits to infancy in an epidemiologic study of 314 twins. Standardized ratings of variation in autistic traits and other non-ASD-related behavioral traits, which are substantially correlated throughout childhood (after the time when autism develops), were found to be independent from one another and traceable to distinct genetic structures. The authors concluded that the commonly-observed co-occurence of specific psychiatric syndromes with autism may arise from interactions between autistic symptoms (after they arise) and genetically independent neuropsychiatric liabilities, suggesting opportunities for preventive amelioration of outcomes of these interactions over the course of development.
It is well known that rare, highly deleterious genetic variants associated with autism are also associated with intellectual disability, ADHD, epilepsy, or schizophrenia across different families. A reconceptualization of autism in which some of its contributing components are specific to autism but others are non-specific provides a direct way of accounting for pleiotropy in studies of genotype-phenotype association in developmental disabilities research. Lahey and colleagues have invoked the concept of “behavioral pleiotropy” on the basis of discovery of highly heritable symptom clusters accounting for a major share of the attributable causal variance across a wide diversity of neuropsychiatric syndromes, including mood and anxiety disorders, disruptive behavior disorders, and substance use disorders.50 One of the core symptom domains derived from factor analysis of psychiatric interview data, is the capacity for emotion regulation. Other lines of investigation have identified additional independent behavioral “building blocks” of psychopathology51 that can be tested for alignment with domains of mental functioning that comprise the NIH matrix known as RDoC (Research Domain Criteria), which attempts to parse psychopathology into its developmental causal structure, rather than on the basis of manifestations of symptoms after a syndrome of impairment or decompensation emerges.
In the future, interventions targeted to early developmental liabilities that may contribute risk for ASD (whether specific or not specific to ASD) can be directly tested for their ability to reduce long-term disability in ASD, particularly among infants known to be at elevated risk. Given the high prevalence of motor impairments and attention/hyperactivity problems in ASD, for example, either or both might serve as important early targets for intervention, both with respect to reducing non-specific risk and to reducing so-called “comorbidity” in affected children. For example, studies have shown that both exercise and developmental opportunities to improve visuomotor proficiency (through balance exercises and martial arts) may have surprisingly broad positive effects on executive functioning, inattention/hyperactivity, and social behavior in young school-aged children.52,53
A final critical corollary of causal influence of non-ASD-specific factors is that they will (to the extent that they are operative) seriously confound biomarker studies—including studies of the association between phenotype and (i) genotype; (ii) brain imaging; and (iii) serologic markers—in any study in which they are not uniformly ascertained among both cases and controls. In other words, if control subjects carry quantitative aggregations of the same traits that are contributing to autism susceptibility (in cases), the only signal that can be derived is from the difference. If the difference between most patients with ASD and most controls is that trait liabilities are more diverse (and cumulative) than individually severe, any signal that would be otherwise generated by a contributory trait would be significantly diminished or lost. This has major consequences for study design and raises the possibility that inclusion of non-ASD-specific risk factors in genetic research (again, quantified in both cases and controls) might actually account for elements of “missing heritability” in autism, and may help resolve apparent discrepancies between genetic epidemiologic and molecular genetic research in estimating the extent of genetic overlap between autism and other neuropsychiatric impairments (such as ADHD). For example, case-control genetic studies of ASD rarely if ever ascertain or control for sub clinical ADHD traits among the controls, and case control genetic studies of ADHD rarely if ever ascertain or control for sub clinical ASD traits among the controls. This holds true and may confound not only genetic research but all biomarker studies, including investigations in neuroimaging, electrophysiology, and proteomics. Future studies that consider the presence of sub clinical traits of contributing non-specific behavioral impairments in both cases and controls will increase precision in capturing biological signatures that are not necessarily specific to ASD but nevertheless (paradoxically) contributing to its severity.
Specifying measurable autism endophenotypes in infancy
Sub clinical aggregations of the characterizing traits and features of autism, exhibit a unitary factor structure, whether ascertained among close relatives of individuals with ASD or in the general population, and carry the qualifying features of an endophenotype, one which exhibits robust trait-like stability over the life course from age 3 years through adulthood.54 The identification of other candidate endophenotypes that are appreciable/measurable in infancy, prior to the timing of the onset of autistic syndromes may prove critical to understanding the early origins of autism and to account for a larger share of the total polygenic risk than may be traceable to behavior variation later in childhood. One of the more promising early predictors of recurrence in autism is the measurement of social visual attention/engagement using eye-tracking methods, invoked by the observation that one of the most pathognomonic features of the autistic syndrome is abnormality in eye contact. Using methodologies that had identified predictors of autism recurrence in the first year of life,55 we recently observed that variation in the allocation of attention to social aspects of the visual environment is under stringent genetic control among infants in the general population, with effects traceable to the active seeking of social information.56 The indices that were most highly heritable—preferential attention to eye and mouth regions of the face—were those that distinguish typically-developing children from those with autism.
These results implicated deficits in social visual engagement as a neurodevelopmental endophenotype—for both population-wide variation in social-information-seeking and for autism. The replicated observation that most young children who develop autism manifest relative deficiencies in this early developmental competency offers a potential bridge for understanding specific relationships between genes, brain, and behavior in the development of autism. Social visual engagement relates to the active construction and maintenance of an ecological niche that mediates social attachment and buffers risk and adversity in early childhood, whether genetic or environmental in origin.55,56 Johnson and colleagues57 hypothesized that key features of autism may be the end result of relative deficiencies in such adaptive mechanisms, rather than a direct consequence of neural pathology. In this view autism is not “inevitable,” but a developmental adaptation to genetic perturbation, engendered on rare occasions by highly deleterious mutations and perhaps more commonly by permutations and combinations of discrete “sets” of polygenic variation respectively indexed by the behavioral traits described herein. If the particular “adaptation” (or maladaptation when severe) comprised by the autistic syndrome is itself relatively parsimonious, it would reconcile the causal heterogeneity observed in ASD and explain the original observation by Spiker that it is the syndrome (not its symptoms) that breeds true, as well as the subsequent observations of a unitary factor structure manifest across the entire range of severity of autistic impairment that occurs in nature.
In any given child, a critical accumulation of developmental disruptions may need to co-aggregate (additively) to engender the autistic syndrome. In the example of social visual engagement, while nearly all children affected by autism exhibit significant atypicalities, a small proportion of unaffected infants in the general population exhibited the same atypicalities,56 suggesting that such deficits may be necessary but not sufficient to cause clinical autistic syndromes, and may be genetically independent from other components of inherited risk. A pilot eye-tracking study of individual children representing a diverse set of loss-of-function mutations in NF1—a recently-recognized quantitative trait locus for autistic impairment58,59—made precisely this observation: that decreased attention to faces was associated with elevated autism traits in the affected children, even though many controls had comparably low attention to faces.
Specific polygenic liabilities that are contributory but not independently sufficient to cause ASD would be predicted to be more powerful predictors of autism in multiplex families than in the general population, because of a higher burden of ASD risk within those families in general. Other key candidates for independent endophenotypic contributions to the development of autism (which can be subjected to rigorous analysis across study designs in the manner described above) include more fine-grained contributors to variation in social attention,60,61,62 nuanced elements of visual search,63 impairment in the developmental capacity for error-based learning or predictive modeling,64,65 primary deficits in social motivation;66 abnormalities in speech perception;67,68 and deficits in cerebellar learning that might exert joint abnormality in the domains of social and motor functioning.69,70 A next generation of early developmental studies will need to discern their relative contributions to the development of autism and the extent to which their respective biological origins are independent or shared.
Sex: To date, one of the most robust predictors of all
Since the male-to-female ASD recurrence ratio stands at 3:1 (manifest from the time of early childhood when autism is first diagnosed), and since the vast majority of all molecular genetic risk tagged to autism lie on autosomes rather than sex chromosomes, sex-specific reduction of the phenotypic expression of genetic susceptibility is believed to occur in the form of a categorical reduction in ASD risk among females, with the absence of a “Carter Effect”.71,72,73 This suggests that most (not all) females are relatively “protected” from most (not all)—inherited forms of autism,28,74,75 and that sex represents a resiliency factor that operates against numerous (heterogeneous) genetic liabilities to ASD. A recent infant sibling recurrence study76 is very notable in this regard: it identified several first year of life predictors of autism (early ASD symptomatology, speed of attentional disengagement, and variation in gaze following), none of which were found to be inherently sexually dimorphic, but all of which more strongly predicted ASD in male than in female siblings of ASD probands, suggesting an interaction between these early life variables and sex in their influence on ASD.
Just as sex may mitigate a diversity of genetic susceptibilities to ASD, targeted biological therapies may someday prove capable of reducing impairment across a multitude of causes, as has been the case for ADHD (i.e., treatment with stimulant medication) which is as genetically diverse as autism. The identification of a comprehensive array of contributing elements—both to liability and resilience—can contribute to a reconceptualization of autism as a developmental divergence engendered by a critical accumulation of inherited developmental liabilities, each of which constitutes a potential target for innovative preventive intervention efforts.
Conclusions and implications for future research
The latest generation of developmental studies of autism has built upon the seminal observation of Spiker and colleagues, and revealed that the “unitary” outcome of autism may represent a final common pathway predicted from an array of neurobehavioral susceptibilities that are appreciable before the syndrome is diagnosed, and that each may be engendered by partially independent sets of genetic variation that do not relate to symptoms in a straightforward way. Given that the autistic syndrome (deficits in social communication, restricted interests, and repetitive motor behavior) is recognizable across a remarkably wide range of severity from sub clinical to profoundly-impairing,77 it remains a very high scientific priority to understand how or why the characterizing traits of the condition “travel together” (e.g., shared neural circuitry, or a specifically recruited adaptation to early developmental stress) and how their severity might be jointly intensified by early co-aggregation with other neuropsychiatric liabilities. Thus, evidence is emerging that autism may be “fractionable” but not after it develops, rather before it develops. Some of the contributing early developmental liabilities appear not to be specific to ASD—these could account for a significant share of the “missing heritability” of autism, contribute to variable expression of monogenic syndromes, and relate to so-called “co-morbidities”—these are inappropriately named if they actually contribute to the causation of autism itself.
Linking biological processes in the early development of autism78 to a set of precursor behavioral traits rather than to a diagnosis of “autism” may advance personalized approaches to developmental intervention, especially if autism represents an epiphenomenon of earlier-interacting susceptibilities. Future research on the neurobiology of autism should focus on understanding why the co-aggregation of non-specific liabilities (e.g., inattention and motor coordination)—at levels near the pathologic ends of their respective distributions—might jointly amplify risk for a syndrome of profound social disability, with timing of onset in the second year of life.
A path forward for early behavioral phenotyping will involve the identification of highly heritable intermediate phenotypes that may more directly link inherited risk to brain and behavioral development than is possible by molecular genetic associations to symptomatology after autism develops. To elucidate an early endophenotypic structure for the development of autism will require testing of a next generation of viable early phenotypic candidates (both behavioral and neural-circuitry-based) in a manner that is epidemiologically rigorous, in which the distinction between (a) prediction of ASD in the general population and (b) prediction of ASD recurrence in families affected by ASD, is closely attended to. Particularly important are studies of the normative relationships between early endophenotypic candidates, to determine the extent to which such candidates are genetically independent, the extent to which they influence one another over the course of early development (even if they begin independently), and the extent to which they individually and/or jointly predict autism. Such studies will help generate norms for the relationships between key quantitative “axes” of early development—including sensorimotor function, general cognition, language, and the respective developmental capacities for reciprocial social behavior, attention, and emotion regulation—that may someday function in the same way that height-versus-weight charts inform pediatric practice, i.e., by the interpretation of a given variable score (e.g., weight) not in absolute terms but in relation to other core factors that influence it (e.g., height). For example, in monitoring the developmental progress of an infant, it would be extremely helpful to know just how much erosion in the capacity for emotion regulation to expect from a one standard deviation reduction in the capacity for reciprocal social behavior, and whether that relationship is linear or curvilinear. No such relational norms exist for early behavioral development.
In addition, studies of the influence (and amelioration) of any given behavioral liability on the development of ASD should be conducted whenever possible within study designs that incorporate control for any/all specifiable inherited risk for ASD, including genotype whenever possible, family genetic background liability for ASD (including affectation status of first degree relatives and endophenotypic characterization of parents), and phenotyping of subjects for a comprehensive array of known behavioral liabilities related to ASD (both specific and non-specific i.e., attention and motor coordination). Genetic and neurobiologic studies to discover new biomarkers must recognize that sub clinical variations of endophenotypic contributors to autism abound in “control” subjects, and will confound such studies if left unmeasured among the unaffected individuals in case-control research designs. Finally, it is possible that the discovery of polygenic risk signals for autism may be substantially refined if (a) linked to endophenotypes rather than the diagnosis of autism and (b) derived from populations representing the entire range of quantitative variation in the respective phenotype, and including measurement of all endophenotypic contributors to autism within both affected and unaffected subjects.
References
Spiker, D., Lotspeich, L. J., Dimiceli, S., Myers, R. M. & Risch, N. Behavioral phenotypic variation in autism multiplex families: evidence for a continuous severity gradient. Am. J. Med Genet 114, 129–136 (2002).
Piven, J., Palmer, P., Jacobi, D., Childress, D. & Arndt, S. Broader autism phenotype: evidence from a family history study of multiple-incidence autism families. Am. J. Psychiatry 154, 185–190 (1997).
Constantino, J. N. et al. Autistic social impairment in the siblings of children with pervasive developmental disorders. Am. J. Psychiatry 163, 294–296 (2006).
Constantino, J. N. The quantitative nature of autistic social impairment. Pediatr. Res. 69(5 Pt 2), 55R–62R (2011).
Constantino, J. N. & Charman, T. Diagnosis of autism spectrum disorder: reconciling the syndrome, its diverse origins, and variation in expression. Lancet 15, 279–291 (2016).
Shultz, S., Klin, A. & Jones, W. Neonatal transitions in social behavior and their implications for autism. Trends Cogn. Sci. 22, 452–469 (2018).
Sandin, S. et al. The heritability of autism spectrum disorder. JAMA 318, 1182–1184 (2017).
Sanders, S. J. He et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015).
Ramaswami, G. & Geschwind, D. H. Genetics of autism spectrum disorder. Handb. Clin. Neurol. 147, 321–329 (2018).
Gaugler, T. et al. Most genetic risk for autism resides with common variation. Nat. Genet. 46, 881–885 (2014).
Boyle, E. A., Li, Y. I. & Pritchard, J. K. An expanded view of complex traits: from polygenic to omnigenic. Cell 169, 1177–1186 (2017).
Yuen, R. K. et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med 21, 185–191 (2015).
Weiner et al 2017
Moreno-De-Luca, A. et al. The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry 72, 119–126 (2015).
Finucane, B., Challman, T. D., Martin, C. L. & Ledbetter, D. H. Shift happens: family background influences clinical variability in genetic neurodevelopmental disorders. Genet. Med. 18, 302–304 (2016).
Constantino, J. N. & Todd, R. D. Intergenerational transmission of subthreshold autistic traits in the general population. Biol. Psychiatry 57, 655–660 (2005).
Lyall, K. et al. Parental social responsiveness and risk of autism spectrum disorder in offspring. JAMA Psychiatry 71, 936–942 (2014).
Page, J. et al. Quantitative autistic trait measurements index background genetic risk for ASD in Hispanic families. Mol. Autism 7, 39 (2016).
Paul, R., Fuerst, Y., Ramsay, G., Chawarska, K. & Klin, A. Out of the mouths of babes: vocal production in infant siblings of children with ASD. J. Child Psychol. Psychiatry 52, 588–598 (2011).
Messinger, D. et al. Beyond autism: a baby siblings research consortium study of high-risk children at three years of age. J. Am. Acad. Child Adolesc. Psychiatry 52, 300–308 (2013).
Miller, M. et al. Response to name in infants developing autism spectrum disorder: a prospective study. J. Pediatr. 183, 141–146 (2017).
Ozonoff, S. et al. The broader autism phenotype in infancy: when does it emerge? J. Am. Acad. Child Adolesc. Psychiatry 53, 398–407 (2014).
Landa, R. J., Gross, A. L., Stuart, E. A. & Bauman, M. Latent class analysis of early developmental trajectory in baby siblings of children with autism. J. Child Psychol. Psychiatry, Allied Discip. 53, 986–996 (2012).
Estes, A. et al. IBIS Network Behavioral, cognitive, and adaptive development in infants with autism spectrum disorder in the first 2 years of life. J. Neurodev. Disord. 7, 24 (2015).
Szatmari, P. et al. Prospective longitudinal studies of infant siblings of children with autism: lessons learned and future directions. J. Am. Acad. Child Adolesc. Psychiatry 55, 179–187 (2016).
BussuG., JonesE., J. H. CharmanT. JohnsonM. H.. & BuitelaarJ. K. BASIS Team Prediction of autism at 3 years from behavioural and developmental measures in high-risk infants: a longitudinal cross-domain classifier analysis. J Autism Dev Disord 48, 2418–2433 (2018).
Marrus, N. et al. Rapid video-referenced ratings of reciprocal social behavior in toddlers: a twin study. J. Child Psychol. Psychiatry 56, 1338–1346 (2015).
Constantino, J. N. et al. Infant head growth in male siblings of children with and without autism spectrum disorders. J. Neurodev. Disord. 2, 39–46 (2010).
West K. L., Leezenbaum N. B., Northrup J. B., Iverson J. M. The Relation Between Walking and Language in Infant Siblings of Children With Autism Spectrum Disorder. Child Dev 2017. Epub ahead of print.
Hilton, C. L., Zhang, Y., Whilte, M. R., Klohr, C. L. & Constantino, J. N. Motor impairment in sibling pairs concordant and discordant for autism spectrum disorders. Autism.: Int. J. Res. Pract. 16, 430–441 (2012).
Ament, K. et al. Evidence for specificity of motor impairments in catching and balance in children with autism. J. Autism Dev. Disord. 45, 742–751 (2015).
Mosconi, M. W. et al. Feedforward and feedback motor control abnormalities implicate cerebellar dysfunctions in autism spectrum disorder. J. Neurosci. 35, 2015–2025 (2015).
Mous, S. E., Jiang, A., Agrawal, A. & Constantino, J. N. Attention and motor deficits index non-specific background liabilities that predict autism recurrence in siblings. J. Neurodev. Disord. 9, 32 (2017).
Shephard E., et al BASIS Team. Early developmental pathways to childhood symptoms of attention-deficit hyperactivity disorder, anxiety and autism spectrum disorder. J Child Psychol Psychiatry 2018. Epub ahead of print.
Pierce, K. et al. Detecting, studying, and treating autism early: the one-year well-baby check-up approach. J. Pediatr. 159, 458–465 (2011).
Pruett J. R. Jr, et al. Impaired eye region search accuracy in children with autistic spectrum disorders. PLoS ONE 8, e581672013 (2013)
Sysoeva, O. V., Constantino, J. N. & Anokhin, A. P. Event-related potential (ERP) correlates of face processing in verbal children with autism spectrum disorders (ASD) and their first-degree relatives: a family study. Mol. Autism 9, 41 (2018).
Bosl, W. J., Tager-Flusberg, H. & Nelson, C. A. EEG analytics for early detection of autism spectrum disorder: a data-driven approach. Sci. Rep. 8, 6828 (2018).
Careaga, M. et al. Immune endophenotypes in children with autism spectrum disorder. Biol. Psychiatry 81, 434–441 (2017).
Eggebrecht, A. T. et al. Joint attention and brain functional connectivity in infants and toddlers. Cereb. Cortex 27, 1709–1720 (2017).
Hazlett, H. C. et al. IBIS Network Early brain development in infants at high risk for autism spectrum disorder. Nature 542, 348–351 (2017).
Shen, M. D. et al. Increased extra-axial cerebrospinal fluid in high-risk infants who later develop autism. Biol. Psychiatry 82, 186–193 (2017).
Swanson, M. R. et al. Subcortical brain and behavior phenotypes differentiate infants with autism versus language delay. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2, 664–672 (2017).
Jokiranta-Olkoniemi, E. et al. Risk of psychiatric and neurodevelopmental disorders among siblings of probands with autism spectrum disorders. JAMA Psychiatry 73, 622–629 (2016).
Lichtenstein, P., Carlström, E., Råstam, M., Gillberg, C. & Anckarsäter, H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am. J. Psychiatry 167, 1357–1363 (2010).
Floris, D. L. et al. Atypical lateralization of motor circuit functional connectivity in children with autism is associated with motor deficits. Mol. Autism 7, 35 (2016).
Reiersen, A. M., Constantino, J. N. & Todd, R. D. Co-occurrence of motor problems and autistic symptoms in attention-deficit/hyperactivity disorder. J. Am. Acad. Child Adolesc. Psychiatry 47, 662–672 (2008).
Constantino, J. N. & Frazier, T. W. Commentary: The observed association between autistic severity measured by the social responsiveness scale (SRS) and general psychopathology--a response to Hus et al.(2013). J. Child Psychol. Psychiatry 54, 695–697 (2013).
Hawks Z. W., Marrus N., Glowinski A. L., Constantino J. N. Early origins of autism comorbidity: neuropsychiatric traits correlated in childhood are independent in infancy. J Abnorm Child Psychol 2018. Epub ahead of print.
Lahey, B. B., Krueger, R. F., Rathouz, P. J., Waldman, I. D. & Zaid, D. H. A hierarchical causal taxonomy of psychopathology across the life span. Psychol. Bull. 143, 142–186 (2017).
Beauchaine T. P., Constantino J. N. Redefining the endophenotype concept to accommodate transdiagnostic vulnerabilities and etiological complexity. Biomark 11, 769–780 (2017).
Kamp, C. F., Sperlich, B. & Holmberg, H. C. Exercise reduces the symptoms of attention-deficit/hyperactivity disorder and improves social behaviour, motor skills, strength and neuropsychological parameters. Acta Paediatr. 103, 709–714 (2014).
Diamond, A. & Lee, K. Interventions shown to aid executive function development in children 4–12 years old. Science 333, 959–964 (2011).
Wagner R., et al Autism-Related Variation in Reciprocal Social Behavior: A Longitudinal Study. Child Dev In press.
Jones, W. & Klin, A. Attention to eyes is present but in decline in 2–6 month-olds later diagnosed with autism. Nature 504, 427–431 (2013).
Constantino, J. N. et al. Infant viewing of social scenes is under genetic control and is atypical in autism. Nature 547, 340–344 (2017).
Johnson, M. H., Jones, E. J. & Gliga, T. Brain adaptation and alternative developmental trajectories. Dev. Psychopathol. 27, 425–442 (2015).
Morris, S. M. et al. Disease burden and symptom structure of autism in neurofibromatosis type 1: A Study of the International NF1-ASD Consortium Team (INFACT).JAMA Psychiatry 73, 1276–1284 (2016).
Lewis A. K., et al. Attention to faces in social context in children with neurofibromatosis type 1. Dev Med Child Neurol 2018. Epub ahead of print.
Pantelis, P. C. & Kennedy, D. P. Deconstructing atypical eye gaze perception in autism spectrum disorder. Sci. Rep. 7, 14990 (2017).
Sifre, R. et al. A longitudinal investigation of preferential attention to biological motion in 2- to 24-month-old infants. Sci. Rep. 8, 2527 (2018).
Wang, Q., Campbell, D. J., Macari, S. L., Chawarska, K. & Shic, F. Operationalizing atypical gaze in toddlers with autism spectrum disorders: a cohesion-based approach. Mol. Autism 9, 25 (2018).
Doherty, B. R. et al. Visual search and autism symptoms: What young children search for and co-occurring ADHD matter. Dev Sci 21, e12661 (2018).
Lawson, R. P., Rees, G. & Friston, K. J. An aberrant precision account of autism. Front Hum. Neurosci. 8, 302 (2014).
von der Lühe T., et al. Interpersonal predictive coding, not action perception, is impaired in autism. Philos Trans R Soc Lond B Biol Sci 371 2016.
Clements, C.C. et al. Evaluation of the social motivation hypothesis of autism: a systematic review and meta-analysis. JAMA Psychiatry 75, 797–808 (2018).
Tryfon, A., Foster, N. E. V., Sharda, M. & Hyde, K. L. Speech perception in autism spectrum disorder: an activation likelihood estimation meta-analysis. Behav. Brain Res 338, 118–127 (2018).
Edwards, L. A., Wagner, J. B., Tager-Flusberg, H. & Nelson, C. A. Differences in neural correlates of speech perception in 3 month olds at high and low risk for autism spectrum disorder. J. Autism Dev. Disord. 47, 3125–3138 (2017).
Valnegri, P. et al. RNF8/UBC13 ubiquitin signaling suppresses synapse formation in the mammalian brain. Nat. Commun. 8, 1271 (2017).
Nyström, P. et al. Enhanced pupillary light reflex in infancy is associated with autism diagnosis in toddlerhood. Nat. Commun. 2018, 9 (1678).
Constantino, J. N. Recurrence rates in autism spectrum disorders. JAMA 312, 1154–1155 (2014).
Gockley, J. et al. The female protective effect in autism spectrum disorder is not mediated by a single genetic locus. Mol. Autism 6, 25 (2015).
Constantino, J. N. Data from the baby siblings research consortium confirm and specify the nature of the female protective effect in autism: a commentary on Messinger et al. Mol. Autism 7, 32 (2016).
Jacquemont, S. et al. A higher mutational burden in females supports a “female protective model” in neurodevelopmental disorders. Am. J. Hum. Genet 94, 415–425 (2014).
Palmer, N. et al. Association of sex with recurrence of autism spectrum disorder among siblings. JAMA Pediatr. 171, 1107–1112 (2017).
Bedford, R. et al. Sex differences in the association between infant markers and later autistic traits. Mol. Autism 7, 21 (2016).
Frazier, T. W. et al. Confirmatory factor analytic structure and measurement invariance of quantitative autistic traits measured by the social responsiveness scale-2. Autism 18, 31–44 (2014).
Piven, J., Elison, J. T. & Zylka, M. J. Toward a conceptual framework for early brain and behavior development in autism. Mol. Psychiatry 22, 1385–1394 (2017).
Funding
This work was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number U54 HD087011 to the Intellectual and Developmental Disabilities Research Center at Washington University in St. Louis. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
J.N.C. receives royalties from Western Psychological Services for the commercial distribution of the Social Responsiveness Scale, a quantitative measure of autistic traits for individuals age 30 months through adulthood.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Constantino, J.N. Early behavioral indices of inherited liability to autism. Pediatr Res 85, 127–133 (2019). https://doi.org/10.1038/s41390-018-0217-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-018-0217-3
This article is cited by
-
Using the Infant Sibling-Design to Explore Associations Between Autism and ADHD Traits in Probands and Temperament in the Younger Siblings
Journal of Autism and Developmental Disorders (2023)
-
New guidance to seekers of autism biomarkers: an update from studies of identical twins
Molecular Autism (2021)
-
Brain function distinguishes female carriers and non-carriers of familial risk for autism
Molecular Autism (2020)
-
On the Nature of Monozygotic Twin Concordance and Discordance for Autistic Trait Severity: A Quantitative Analysis
Behavior Genetics (2020)
