
Abstract
Chronic kidney disease (CKD) is a complex condition with a prevalence of 10–15% worldwide. An inverse-graded relationship exists between cardiovascular events and mortality with kidney function which is independent of age, sex, and other risk factors. The proportion of deaths due to heart failure and sudden cardiac death increase with progression of chronic kidney disease with relatively fewer deaths from atheromatous, vasculo-occlusive processes. This phenomenon can largely be explained by the increased prevalence of CKD-associated cardiomyopathy with worsening kidney function. The key features of CKD-associated cardiomyopathy are increased left ventricular mass and left ventricular hypertrophy, diastolic and systolic left ventricular dysfunction, and profound cardiac fibrosis on histology. While these features have predominantly been described in patients with advanced kidney disease on dialysis treatment, patients with only mild to moderate renal impairment already exhibit structural and functional changes consistent with CKD-associated cardiomyopathy. In this review we discuss the key drivers of CKD-associated cardiomyopathy and the key role of hypertension in its pathogenesis. We also evaluate existing, as well as developing therapies in the treatment of CKD-associated cardiomyopathy.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD) and kidney failure with replacement therapy (KFRT) are complex chronic conditions with a combined prevalence of 10–15% worldwide [1,2,3,4,5]. Hypertension is an equally significant global problem and remains one of the most important preventable causes of mortality worldwide [6, 7], with the prevalence expected to rise to 1.56 billion by 2025 [6]. Affecting 67–92% of patients with CKD, hypertension is also the most common comorbidity with increasing prevalence and severity as kidney function progresses [8]. Its pathogenesis and that of CKD are tightly intertwined, with hypertension being both a complication of and driver of kidney disease progression [9,10,11].
An inverse graded relationship exists between cardiovascular events and mortality with estimated glomerular filtration rate (eGFR), which is independent of age, sex, and other risk factors [12,13,14,15,16,17,18]. The proportion of deaths due to heart failure and sudden cardiac death (SCD) increase with progression of CKD, with relatively fewer deaths from atheromatous processes [19,20,21,22,23]. This is also evidenced by trials which show benefit of lipid-lowering therapies in early CKD [24,25,26], but appear to be ineffective in patients with KFRT [26,27,28]. This is thought to be the result of the development of CKD-associated cardiomyopathy.
In this state-of-the-art review, we will discuss some of the key drivers of CKD-associated cardiomyopathy and the key role of hypertension in its pathogenesis, and evaluate existing, as well as developing therapies in the treatment of CKD-associated cardiomyopathy.
CKD-associated cardiomyopathy
The concept of CKD-associated cardiomyopathy (Fig. 1) first appeared in the 1980s following reports of common abnormalities in cardiac structure and function in patients with CKD and KFRT [29, 30]. The key features described were:
-
Increased left ventricular (LV) mass and left ventricular hypertrophy (LVH),
-
Diastolic and systolic LV dysfunction,
-
Profound myocardial fibrosis on histology [15, 31,32,33,34,35,36,37,38,39,40,41,42,43,44].

CKD-associated cardiomyopathy is characterised by structural remodelling of the heart. Diffuse interstitial fibrosis and cardiac hypertrophy give rise to cardiac electromechanical dysfunction and increased risk of sudden death.
Whilst these features have been predominantly identified in patients with KFRT, who are undoubtedly uraemic, those with mild to moderately reduced eGFR already exhibit structural and functional changes consistent with CKD-associated cardiomyopathy [35]. This is consistent with observational studies reporting a 20% higher risk of death and cardiovascular events in patients with eGFR between 45 and 59 ml/min/1.73 m2 compared to those with eGFR greater than 60 ml/min/1.73 m2 [14].
Increased left ventricular mass/left ventricular hypertrophy
The LV is a main target for end-organ damage in hypertension, resulting from a combination of cardiomyocyte hypertrophy and expansion of the extracellular space caused by interstitial myocardial fibrosis from activated fibroblasts (myofibroblasts; Fig. 2) [15]. In the general population, the prevalence of LVH is between 15 and 21% [45]. The prevalence is significantly greater in patients with hypertension. In a meta-analysis encompassing 37,700 hypertensive patients, LVH was detected by echocardiography in 36–41%, increasing to 58–77% in high-risk patients who had severe or refractory hypertension, type 2 diabetes mellitus, or a history of previous cardiovascular events [46]. However, it should be noted that the relationship between LV mass and blood pressure is continuous, with no true dichotomy, and that any definition of LVH is a useful, but arbitrary way of defining hypertensive target organ damage [47, 48].

Laser scanning confocal image of human induced pluripotent stem cell-derived cardiac fibroblasts activated by transforming growth factor-β stained with antibodies against α-smooth muscle actin (green) and vimentin (red). Every third to fourth cell in the left ventricle is a fibroblast. Most cardiac research has focused on the cardiomyocyte with research on cardiac fibroblasts proving more challenging. (Image provided by Ms Caitlin Hall, Institute of Cardiovascular Sciences, University of Birmingham).
LVH is an independent risk factor for cardiovascular morbidity and mortality [49]. Prospective data from large observational studies [45, 50] have demonstrated independent associations between echocardiogram/electrocardiogram-LVH, and an increased risk of cardiovascular events and all-cause mortality. In a large retrospective study of 35,602 patients referred for echocardiography, the presence of LVH increased the risk of all-cause mortality by 2-fold over a mean follow-up of 3.2 years [51]. In the Heart and Soul Study, a cohort of 1016 patients with stable coronary artery disease followed up for 3.5 years, evidence of echocardiographic LVH was associated with a higher overall mortality (25% v 11%) and SCD (6.7% v 2.2%) [52]. Analysed as a continuous variable, every 20-unit increase in LV mass increased the adjusted hazard of overall mortality by 22% and of SCD by 40% [52].
Ambulatory blood pressure measurements more closely correlate with LV geometry abnormalities compared to office blood pressure measurements [53, 54]. Patients with a non-dipping or reverse dipping pattern on 24-h ambulatory monitoring have a higher LV mass and increased prevalence of LVH [55, 56] as well as worse CV outcomes [57, 58]. These abnormal blood pressure circadian rhythms are also more prevalent in patients with CKD and KRFT and are also associated with worse CV outcomes [59].
Increased left ventricular mass/left ventricular hypertrophy in chronic kidney disease
The connection between CKD, hypertension and LVH was first described by several 19th century physicians. Pioneering British physician Richard Bright (1789–1858) depicted with meticulous detail first observations of LVH present on autopsies of patients with albuminuria [60]. Years later, his successor Sir Samuel Wilks (1824–1911) described pathological changes in the heart and arteries accompanying Bright’s disease [61]. Further publications in the 1850s by Ludwig Traube (1818–56) and William Kirkes (1822–64) introduced the concept of arteriosclerosis and high intra-arterial pressure as a driver of LVH in renal disease, laying the foundation for our understanding of CKD-associated cardiomyopathy [62, 63].
Recent imaging studies confirm that increased LV mass and LVH are common manifestations of CKD-associated cardiomyopathy (Fig. 3). LVH is found in 48–84% of patients with pre-dialysis CKD [64,65,66], and in up to 90% of patients with KFRT receiving haemodialysis [5, 38, 67]. Similarly, LV mass is a continuous variable with a graded association with adverse cardiovascular outcomes and mortality [45, 68,69,70,71,72]. In an analysis of 1,249 patients with predialysis CKD, LVH was associated with a mortality risk of 25 deaths per 1000 person-years [68]. In patients who have LVH at dialysis initiation, the risk of mortality was 1.75-fold greater than those without LVH at 6 years follow-up [5]. Conversely, LVH regression may improve outcomes in patients with KFRT [67].

Short axis stack image (A) and horizontal long axis image (B) of a patient on haemodialysis with concentric left ventricular hypertrophy on cardiac magnetic resonance imaging (maximum wall thickness, 15 mm; left ventricular mass 93 g/m2). Short axis stack image (C) and horizontal long axis image (D) of a patient on peritoneal dialysis with normal left ventricular dimensions on cardiac magnetic resonance imaging (maximum wall thickness, 9 mm; left ventricular mass 63 g/m2).
LVH is associated with an increased risk of arrhythmia, which may explain, in part, the increased incidence of cardiovascular death in CKD [73]. A meta-analysis of 10 studies (27,141 patients) found that patients had 3.4-fold and 2.8-fold greater odds of developing sustained supraventricular tachycardia and ventricular arrhythmias, respectively, in the presence of LVH [74]. Other studies reported a 20% increase in the risk of atrial fibrillation (AF) for every standard deviation increase in LV mass [70]. The presence of LVH in hypertensive patients was a predictor for progression from paroxysmal to permanent AF [71], whilst blood pressure reductions of 7.3/1.8 mmHg resulted in regression of LV mass index with a corresponding 7-fold decline in the prevalence of paroxysmal AF after a mean follow-up of 24 months [75].
Diastolic and systolic dysfunction
Diastolic dysfunction is highly prevalent in patients with CKD [76] with studies reporting an incidence of 71% in CKD stages 2–4 [64] and up to 85–90% in KFRT [77,78,79]. Excess myocardial deposition of collagen affects cardiac muscle viscoelasticity, leading to increased ventricular stiffness, impaired myocardial relaxation and diastolic recoil [80]. Diastolic dysfunction is strongly association with increased LV mass and LVH [19], as well as myocardial fibrosis [41, 81], and is associated with increased mortality [78, 81]. Indeed, the American Society of Echocardiography and the European Association of Cardiovascular Imaging considers LVH as an indicator of diastolic dysfunction [82]. Furthermore, the presence of diastolic dysfunction is considered to be a major cause for the frequent presentation of HD patients with pulmonary oedema or intradialytic hypotension with only minor changes in fluid status [19, 81]. Overt LV systolic dysfunction, as manifest by a reduced left ventricular ejection fraction, is relatively uncommon in pre-dialysis CKD, with a reported prevalence of 8% and no association with eGFR [36, 64]. However, several studies have shown changes in LV deformation in early stages of CKD indicating the presence of sub-normal LV systolic function [83,84,85]. In KFRT, LV systolic dysfunction is very common with a reported prevalence 10–30 times greater than in the general population [86,87,88,89].
The presence of LVH has also been associated with the risk of development of systolic dysfunction in hypertensive subjects [90,91,92]. The 2013 American College of Cardiology Foundation/American Heart Association Guideline for the Management of Heart Failure recognised hypertension and LVH as stage A and B heart failure. The guideline emphasised the progressive nature of heart failure, and the importance of long-term treatment of elevated blood pressure to prevent symptomatic heart failure [93].
Myocardial fibrosis
It has been suggested that myocardial fibrosis may be the unifying pathophysiological process underlying CKD-associated cardiomyopathy [94]. In the 1990s, a post-mortem study found that myocardial fibrosis was present in 91% of CKD/KFRT patients without significant flow-limiting coronary lesions. The severity of fibrosis was related to the length of time on dialysis, but independent of hypertension, blood pressure, diabetes or anaemia [31]. However, given a plethora of drivers of cardiac fibrosis, as demonstrated by numerous preclinical and clinical studies, we cannot discount the contributing roles of these diseases on the activation of cardiac fibroblasts [95]. Over a decade later, Aoki et al. performed endocardial biopsies in 40 KFRT patients with reduced LV ejection fraction without coronary artery disease [15]. The predominant pathologic findings were extensive fibrosis and cardiomyocyte hypertrophy similar to that seen in the dilated phase of hypertrophic cardiomyopathy, a condition associated with extremely high morbidity and mortality [96, 97].
A post-mortem study of LV tissue from patients with KFRT found reduced capillary density, increased cardiomyocyte cross-sectional area and expansion of interstitial matrix. The apparent myocyte-capillary mismatch brought about by the reduction in capillary supply within a hypertrophied myocardium increases the oxygen diffusion distance, thereby exposing the heart to the risk of ischaemic injury and subsequent cardiac dysfunction [98]. Recent advances in imaging technology have enabled the indirect assessment of coronary microvascular dysfunction (CMD). Coronary flow reserve (CFR) is the most widely used surrogate marker, and has been successfully utilised to indirectly measure CMD in conditions such as hypertrophic cardiomyopathy and heart failure with preserved ejection fraction, both of which are characterised by LVH and myocardial fibrosis, similar to CKD-associated cardiomyopathy [99, 100]. Although data are limited, imaging studies utilising positron emission tomography, angiography and echocardiography have reported a prevalence of 24–90% in CKD patients with a graded relationship with eGFR [101]. In a retrospective study of CKD patients, which included those on dialysis, reduced CFR below 1.5 was associated with 2.1-fold increased risk of cardiovascular mortality independent of risk factors and LV function [102]. The mortality risk remained elevated in a later analysis of the dialysis-dependent cohort [103].
Studying myocardial fibrosis in CKD/KFRT has been challenging given that myocardial biopsies are ethically difficult to justify [104, 105]. Contrast-enhanced cardiac magnetic resonance (CMR) imaging has been used for the assessment of myocardial fibrosis in conditions such as myocardial infarction [106], dilated [107] and hypertrophic cardiomyopathies [108]. Patients with KFRT demonstrate midwall patterns of late gadolinium enhancement consistent with replacement myocardial fibrosis not associated with large vessel coronary artery disease [36]. Non-contrast myocardial native T1 relaxation time, or T1 mapping, has emerged as a viable alternative for the assessment of myocardial fibrosis in CKD/KFRT [40] (Fig. 4). In cardiac tissues, T1 relaxation time outside regions of scarring has been shown to correlate with histologically measured interstitial fibrosis severity in a number of disease states [109]. In patients with KFRT, native T1 times are higher than age- and sex-matched controls, and correlate with increased LV mass [33, 34]. In parallel with the changes to LV mass and function, native T1 times have been shown to be increased in early CKD [32] and increase with worsening CKD stages [110] (Figs. 3 and 4). The contribution of myocardial water content to the increased T1 times remains contentious and to date, T1 times have not been validated with diffuse interstitial fibrosis in CKD-associated cardiomyopathy.

T1 mapping of the short axis stacked image of the mid left ventricular cavity (A) (Global T1 time 1385 ms) and (B) (Global T1 time 1170 ms) of 2 peritoneal dialysis patients consistent with high and low levels of cardiac fibrosis respectively.
Pathogenesis of CKD-associated cardiomyopathy
The pathogenesis of CKD-associated cardiomyopathy is likely to be multifactorial but can broadly be divided into three categories:
-
Increased afterload [111],
-
Increased preload [112],
-
Intrinsic factors not directly to afterload or preload [113].
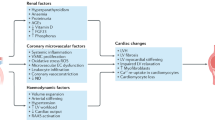
Although these categories are considered separately, it is important to note that there is considerable overlap between them [40, 41, 101, 114, 115]. Factors within these categories may indeed interact and exert their effects synergistically (Fig. 5).

An interplay of pathophysiological processes associated with the chronic kidney disease milieu contribute to the pathogenesis of CKD-associated cardiomyopathy. These can be broadly divided into factors which increase afterload, preload, and those which result from maladaptive perturbations of intrinsic factors. AGEs advanced glycation end-products, FGF23 fibroblast growth factor 23, PTH parathyroid hormone, RAAS renin-angiotensin-aldosterone system, SNS sympathetic nervous system.
Increased afterload
Raised intra-arterial pressure induces cardiac hypertrophy through chronic haemodynamic stress on the myocardium. In vitro experimental models of cardiomyocyte and non-myocyte stretch resulted in cell hypertrophy and hyperplasia, induction of protein synthesis and increased expression of prohypertrophic proto-oncogenes such as fos, jun, myc, Ha-ras, and genes encoding atrial natriuretic factor and β-myosin heavy chain [116,117,118]. This was recapitulated in murine and porcine studies of hypertension and volume overload where transcription of prohypertrophic genes, and development of LVH were observed [119,120,121,122,123].
Arterial stiffness/decreased large vessel compliance/vascular calcification
Arterial stiffening occurs from the early stages of CKD and increases with the progression to KFRT [124,125,126]. The extent of arterial stiffness can be reliably estimated by non-invasive measurement of carotid-femoral pulse wave velocity (PWV), which can confer prognostic value in CKD patients independent of traditional risk factors [127, 128].
Arterial stiffness is both a cause and result of systolic hypertension, although extricating the bidirectional relationship can be challenging [129, 130]. Studies have demonstrated increased arterial stiffness as a consequence of increased distending vascular wall stress and remodelling from hypertension itself, so called pressure-dependent increases in PWV [131,132,133]. Other studies show that increased arterial stiffness, more specifically higher PWV, predicts future development of hypertension [134,135,136,137], possibly as a result of inherent pressure-independent vasculopathy. In patients with KFRT, PWV is independently associated with nonfatal stroke and myocardial infarction [126] and with cardiovascular and all-cause mortality [126, 138, 139]. Of those patients who died, it was found that the persistence of increased PWV despite a reduction in blood pressure by haemodialytic and pharmacological strategies (angiotensin converting enzyme inhibitor; ACEi) was a significant independent risk factor. This is in contrast to the surviving patients where a similar fall in blood pressure was mirrored by a reduction in PWV [139].
In addition to elevated blood pressure, there are several other postulated pathophysiological drivers of increased arterial stiffness in CKD such as activation of the renin-angiotensin-aldosterone system (RAAS), build-up of uraemic toxins, disordered bone and mineral metabolism (CKD-MBD), accumulation of advanced glycation end-products, chronic inflammation and oxidative stress which adversely affect endothelial function and calcific remodelling of the vessel wall [12, 129]. The reduced vessel compliance is manifested by hypertension and wide pulse pressure, and disruptions to end-organ perfusion, resulting in myocardial fibrosis, increased LV mass/LVH and diastolic dysfunction bearing the hallmarks of CKD-associated cardiomyopathy [140]. Taken together, blood pressure reduction alone without ‘de-stiffening’ of the arteries may be insufficient to reduce risk of death in patients with CKD/KRFT.
Increased preload
Intravascular volume expansion/hypervolaemia, secondary to salt and water loading, is a frequent occurrence in CKD/KRFT and a determinant of LV mass and mortality [141, 142]. Indeed, volume expansion is a major contributor to the hypertension observed in patients with CKD/KRFT [142, 143]. Reduced GFR, activation of the RAAS as well as superimposed cardiovascular disease all contribute to sodium and water retention in these patients [144,145,146].
As haemoglobin concentrations fall, complex haemodynamic compensatory mechanisms are activated to increase cardiac output and blood flow to compensate for tissue hypoxia [147]. These include a reduction in afterload secondary to reduced systemic vascular resistance, an increase in preload secondary to increased venous return, and increased LV function as a consequence of increased sympathetic tone [148, 149]. Treatment of anaemia in CKD with erythropoietin stimulating agents (ESAs) reduces stroke volume and cardiac output as a consequence of both reduced venous return and increased peripheral resistance [150,151,152,153]. These functional changes are paralleled by structural changes in the LV, with partial reduction of LVH [151, 152]. It should also be noted that treatment with ESA increases blood pressure by direct vasomotor effects [154,155,156].
The use of arteriovenous fistulas for haemodialysis is preferrable to vascular catheters due to improved dialysis quality and reduced infection rates [157,158,159]. However, the creation of a shunt between a high pressure arterial vasculature and a low resistance venous system increases cardiac output through the shunting itself, activation of the sympathetic nervous system, neurohormonal changes, and increased venous return [158]. Chronically increased preload and cardiac output lead to LVH and right ventricular remodelling, which in turn is associated with an 3.9-fold increased risk of death [160]. Several studies of fistulas being tied off after successful kidney transplantation show some evidence of LVH regression [161, 162]. However, although creation of an arteriovenous fistula decreases systemic blood pressure, ligation increases systolic blood pressure by an average of 5 mmHg [163]. The decision to proceed with fistula ligation should therefore balance, on an individualised basis, the benefits of cardiac remodelling regression against the potential adverse effect on blood pressure control.
Intrinsic factors
In addition to the haemodynamic factors already discussed, several other factors that become dysregulated in patients with CKD are involved in the pathogenesis of CKD-associated cardiomyopathy. These include activation of RAAS, sympathetic nervous system overactivity, increased transforming growth factor-beta (TGFß) signalling, insulin resistance, uraemic toxins (e.g., indoxyl sulfate, p-cresyl sulfate), increase in cardiotonic steroids, oxidative stress and factors associated with CKD-MBD namely hyperparathyroidism, vitamin D deficiency, increased circulating fibroblast growth factor-23 (FGF-23), decreased Klotho expression and hyperphosphataemia [41, 164,165,166]. Although often considered in isolation, there are significant overlaps and crosstalk between these pathways. For example, FGF-23 activates RAAS through suppression of ACE2 [167], which downregulates vasodilatory angiotensin- [1,2,3,4,5,6,7], and upregulates plasma/cardiac Angiotensin II and aldosterone [168,169,170]. In turn, aldosterone stimulates FGF-23 transcription in osteoblasts in vitro, an effect reversed by mineralocorticoid receptor blockade [171]. More recently, Böckmann et al. showed that FGF23 stimulated expression of RAAS genes and hypertrophic growth in cultured neonatal rat ventricular myocytes [172].
Treatment of CKD-associated cardiomyopathy
Given that CKD-associated cardiomyopathy, and its individual components, are powerful predictors of cardiovascular mortality in patients with CKD and KFRT, targeting the mechanisms involved seems a practical approach to improve outcomes. However, a meta-analysis assessing the validity of LV mass reduction as a surrogate marker of all-cause and cardiovascular-mortality in patients with CKD and KFRT concluded there was no clear association between intervention-induced change in LV mass and mortality [173]. It should be noted that, by the authors’ own admission, most of the included trials were of short duration with small sample sizes, with either an uncertain or high risk of bias, and sparse mortality data [173].
Dialysis treatments
Studies have shown that longer, frequent ‘intensive’ dialysis regimens (Fig. 6) are associated with reductions in LV mass, lower prevalence of LVH and reduced hospitalisations [174,175,176,177]. However, it is not clear if the reductions in LV mass are a direct consequence of the dialysis treatment - clearance of uraemic toxins, better phosphate and haemoglobin control; or improved blood pressure control [178,179,180]. An intriguing, but small, study of 54 incident haemodialysis patients found that lowering dialysate temperature by an average of 1 °C significantly reduced LV mass (treatment difference between groups 15.6, 95% CI 1.9–29.4 g) measured by CMR over a 12 month period with no differences in blood pressure between groups [181]. However, 10 of the 54 patients had their 12-month CMR results imputed. This study finding requires confirmation in larger trials.

Conventional haemodialysis regimens consist of a four-hour haemodialysis session delivered on alternate days, three times per week. Longer, more frequent dialysis regimens have been trialled, with benefits on blood pressure, left ventricular mass, mortality, hospitalisations, and quality of life. However, the implementation of such ‘intensive’ regimens significantly increases the demand on healthcare providers and may be associated with risks of infection, vascular access complications and loss of residual kidney function.
The previously discussed effects of intensive dialysis on reduction of hypertension and improvement in LV mass are encouraging, however it is still unclear as to whether this leads to better patient survival. A recent meta-analysis encompassing 70,506 patients found that intensive dialysis was associated with reduced mortality when compared to conventional dialysis, but the overall quality of the evidence was low [182]. Whilst intensive dialysis has clear health and quality of life benefits, it needs to be considered in conjunction with the potential risks of increased patient and caregiver burden, infection, loss of residual kidney function and vascular access complications [183].
Transplantation
The gold-standard for the treatment of KFRT is kidney transplantation [184]. The associated improvement in GFR reduces cardiovascular risk below that of those on waiting lists [185]. However, cardiovascular risk still remains higher than healthy individuals of the same age and sex with transplant recipients displaying a three-fold increased risk [186]. The restoration of kidney function associated with kidney transplantation improves many factors thought to cause CKD-associated cardiomyopathy. As a result it is generally assumed that kidney transplantation reduces LV mass, and improves diastolic and systolic function, and potentially reverses myocardial fibrosis [187, 188]. Indeed, review articles continue to state that CKD-associated cardiomyopathy is reversed by kidney transplantation. However, these articles frequently will either not cite any references [41], cite small, uncontrolled studies using either echocardiography [189] or radionucleotide ventriculography-gated blood pool scans [187, 190], or refer to other review articles [191].
A recent systematic review and meta-analysis has indeed confirmed that many echocardiographic studies reported significant reductions in LV mass after kidney transplantation [192]. However, this study highlighted key problems in the available literature. Few studies were blinded or had a control group thus were prone to bias; meta-analysis of the four studies containing a control group did not find any association between transplantation and LV mass. Echocardiography is unreliable for LV mass determination in dialysis patients [193]; few studies used CMR which is more reproducible in KFRT patients [40]. Indeed, none of the three CMR studies included in the review found a significant change in LV mass.
Intriguingly, a small study in 44 kidney transplant recipients found a significant reduction in native T1 time at 6 months after transplantation with no reduction in LV mass [194] A smaller study found no difference in global T1 times 6 weeks after transplantation suggesting changes may continue to progress over time [195]. Larger, controlled studies utilising CMR are required to further investigate this fundamental question.
Blood pressure treatment
Blood pressure target in chronic kidney disease
Prior to publication of the SPRINT trial results [196], most societies and guidelines recommend lowering blood pressure to below 140/90 mmHg [197,198,199,200,201] with some suggesting higher thresholds for the elderly [197, 198, 201] and lower thresholds for those at higher high risk including patients with diabetic mellitus and patients with CKD [197, 198, 201]. It is now recognised that more intensive blood pressure reduction strategies are effective and safe. The SPRINT investigators reported a 25% lower risk of major cardiovascular events such as myocardial infarction, heart failure and stroke and 27% lower risk of all-cause mortality when treating to a target systolic blood pressure of less than 120 mmHg compared to the standard target of 140 mmHg. This was associated with an increase in complications such as lower serum potassium, sodium, and syncopal episodes, but no increased risk of falls. Treatment of hypertension in those with CKD, which formed 28% of the SPRINT cohort, was associated with improved cardiovascular outcomes and all-cause mortality, with no adverse effects on kidney function or increased risk of dialysis. Elderly patients aged over 75 years old also sustained benefit from more intensive blood pressure control [196].
These data have resulted in lowering of both American and European blood pressure targets in their respective 2017 [202] and 2018 [203] hypertension guidelines. The recently published Kidney Disease Improving Global Outcomes (KDIGO) 2021 guideline on the management of blood pressure in CKD also recommends an intensive systolic blood pressure target of 120 mmHg or less in patients with CKD (excluding those with a functioning kidney transplant) with an emphasis on standardised office blood pressure measurement and lifestyle interventions such as salt restriction and moderate-intensity exercise [204] (Fig. 7).

The recently updated Kidney Diseases Improving Global Outcomes (KDIGO) 2021 Blood Pressure Guideline provides clear recommendations for the management and benefits of blood pressure control in patients with CKD (incl. transplant recipients).
Efficacy of blood pressure treatment on left ventricular hypertrophy
In the general population, regression of LVH is associated with a significant improvement in cardiovascular events [205]. Several landmark trials have confirmed that LV regression in response to treatment with antihypertensive medication is associated with significant reduction in cardiovascular events [206,207,208,209]. All classes of anti-hypertensive medications have been shown to regress LVH [210,211,212,213]. In general, meta-analyses have shown that ACE-inhibitors, angiotensin receptor blockers (ARBs) and calcium channel blockers are more effective than beta-blockers at regressing LVH [214, 215]. Multiple studies have shown that “CHIP” diuretics (chlorthalidone, indapamide and potassium-sparing diuretics/hydrochlorothiazide) are more effective than ACE-inhibitors/ARBs, as well as hydrochlorothiazide, at reducing LV mass [216,217,218] (Fig. 8).

Several landmark trials and meta-analyses have compared the efficacy of antihypertensive drug classes on cardiac functional, structural, and mortality endpoints.
Factors associated with persistent LVH and lack of regression of LVH with antihypertensive treatment include older age, higher body mass index, sub-optimal blood pressure control, duration of hypertension and crucially, the presence of CKD [210, 219, 220]. This, together with the fewer and much smaller studies available make the literature in patients with CKD/KRFT less clear. Most, but not all [221, 222] studies of ACEi/ARB compared with placebo or standard treatment in both patients with CKD [223] or on dialysis [224,225,226,227] have shown no significant reduction in LVM. The same is true for studies comparing ACEi/ARB with a CCB [228,229,230] or beta-blocker [231, 232] which have shown no difference between agents.
The confounding effect of blood pressure reduction on changes in LV mass are highlighted by a study examining the cardiovascular effects of spironolactone compared with placebo in early-stage CKD [233]. In 112 non-diabetic patients with CKD stages 2–3 and well-controlled blood pressure, the addition of spironolactone 25 mg once daily for 40 weeks reduced LV mass by 14 g compared with a non-significant change of 3 g with placebo. However, significantly greater falls in blood pressure were observed in the spironolactone group. A subsequent study in 154 patients, with similar characteristics to the original trial, found no significant difference in the reduction of LV mass observed with spironolactone compared with chlortalidone over a 40-week period [234]. Importantly, reductions in office and 24-hour blood pressure readings were not different between groups either.
Angiotensin receptor neprilysin inhibitor
Sacubitril/valsartan are the first in this new class of angiotensin receptor neprilysin inhibitors (ARNI), approved for the treatment of heart failure with reduced ejection fraction (HFrEF) in conjunction with other standard therapies [235]. The neprilysin inhibitor sacubitril enhances the activity of the natriuretic peptides which have a counter-regulatory role in conditions linked to RAAS activation such as heart failure and CKD through increased sodium & water excretion [236, 237]. Its use in combination with valsartan prevents reflex activation of RAAS [238]. Clinical studies have shown sacubitril/valsartan to reduce the risk of death and hospitalisation when compared to enalapril [239], and provide a further reduction to blood pressure versus valsartan alone [240]. In patients with CKD and HFrEF, a meta-analysis of randomised control trials comparing 3,460 patients on an ARNI versus a RAAS-blocker found that ARNIs significantly reduced blood pressure and N-terminal pro-brain natriuretic peptide (NT-proBNP, and was mildly renoprotective [241]. Patients with CKD can present with diastolic dysfunction only, or heart failure with preserved ejection fraction (HFpEF). The efficacy of ARNIs in HFpEF is less convincing. PARAGON-HF studied 4,796 patients with LV ejection fraction ≥45% and reported no significant change in heart failure hospitalisations and cardiovascular death when compared to valsartan alone [242]. In the PARALLAX trial, ARNI did not increase functional- or symptom-based scores despite a 16% reduction in NT-proBNP versus RAAS inhibition/placebo [243].
Mineralocorticoid receptor antagonists
It is well-established that aldosterone promotes cardiovascular damage and high concentrations are associated with LVH [244, 245] and myocardial fibrosis both in experimental and human studies [246, 247]. The mineralocorticoid receptor (MR) is expressed in vascular smooth muscle, as well as cardiomyocytes and myofibroblasts [248], and may directly stimulate proliferation of myofibroblasts [249]. Aldosterone directly induces cardiac hypertrophy, ventricular remodelling, arrhythmia and ischaemia, independently of its hemodynamic effects [250] and it appears that progression from LVH to cardiac failure is mediated by aldosterone through the MR [251]. Mineralocorticoid receptor antagonists (MRA) have consistently shown beneficial effects on left ventricular dilation, cardiac function, fibrosis or collagen content in preclinical studies [252,253,254] and also regress LVH in clinical trials [212]. MRA have consistently been shown to improve outcomes in patients with HFrEF and potentially in patients with HFpEF [255].
Whether the lowering of LV mass in CKD patients is dependent on the blood pressure lowering effect of MRAs is unclear [233, 234]. Recently two large placebo-controlled studies with the non-steroidal MRA finerenone have shown significant reductions in cardiovascular events and mortality in patients with diabetic nephropathy with little effect on blood pressure [256,257,258]. Consistent with the effect of blood pressure on stroke risk [203, 259] the incidence of stroke did not vary between groups [255]. The lower incidence of hyperkalaemia versus steroidal MRAs (spironolactone and eplerenone) makes finerenone an attractive option for the treatment of CKD-associated cardiomyopathy [255].
Sodium-glucose co-transporter-2 inhibitors
Sodium-glucose co-transporter-2 (SGLT2) inhibitors are a recent class of oral antidiabetic agents [260, 261]. They inhibit renal glucose reabsorption and several SGLT2-inhibitors are licensed in various countries for the treatment of diabetes mellitus. In addition SGLT2-inhibitors significantly reduce weight through glycosuria-associated calorie loss [260, 261]. They also decrease sodium-reabsorption exhibiting a mild natriuretic and diuretic effect [260, 261] (Fig. 9).

Sodium-glucose cotransporter type 2 inhibitors have traditionally been an effective antidiabetic therapy. Recent trials have demonstrated a multitude of beneficial cardiorenal, metabolic and vascular effects, which underpin the improvement in cardiovascular and renal outcomes independent of improved glycaemic control. Some of these mechanisms have been summarised.
A recent meta-analysis of large RCTs clearly demonstrated a 23% reduction in CV morbidity and mortality, especially heart failure hospitalisations in patients with diabetes mellitus [262]. Similar results are emerging in patients with CKD and with heart failure without diabetes [263, 264]. Short-term, mechanistic studies have shown that SGLT2-inhibitors reduce LV mass in diabetics with [265] and without LVH [266]. These early effects of SGLT2-inhibitors on LV remodelling are consistent with their rapid impact on cardiovascular death and heart failure hospitalisation observed in the trials [267]. In addition SGLT2-inhibitors have been shown to improve LV diastolic dysfunction [268, 269].
Several mechanisms have been proposed for the reduction in LV mass and improved diastolic dysfunction observed with SGLT2-inhibitors [267] (Fig. 9). Their effects on lowering intracellular sodium concentrations in the heart have however recently been disproved [270, 271]. It should be noted that SGLT2-inhibitors also significantly lower systolic and diastolic blood pressure by 3–5/1–2 mmHg [261], through their diuretic and natriuretic actions, as well as reported reductions in sympathetic activity, arterial stiffness and vascular resistance [260, 261] (Fig. 9). A significant correlation between blood pressure reduction and regression of LVH with SGLT2-inhibitor use has been reported [269]. SGLT2-inhibitors might therefore be useful agents to improve CKD-associated cardiomyopathy acting via both blood pressure reduction and blood pressure independent mechanisms.
Renal sympathetic denervation
Hypertension can be challenging to control in CKD, with the prevalence of apparent resistant hypertension reported to be as high as 40% [272]. Whilst the true prevalence is likely to be lower after accounting for medicines nonadherence and white coat syndrome, patients with true resistant hypertension are at increased risk of adverse cardiovascular outcomes compared with those who are treatment-responsive [273]. Following initial disappointment [274], several recent randomised sham-controlled trials have now shown that renal denervation therapy can effectively reduce blood pressure [275,276,277,278,279]. The latest analyses of 3-year data from the Global SYMPLICITY Registry provide encouraging news for patients with CKD, who experience a similar reduction in blood pressure following renal denervation to those without renal disease, and also demonstrate the safety of the procedure [280, 281]. The longer-term effects on renal function and cardiovascular events are yet to be determined, with trials currently ongoing (NCT01888315, NCT04264403).
Treating individual factors
Anaemia
The presence of anaemia is associated with the development of LVHH and heart failure in patients with CKD [282, 283], KFRT [284, 285], and kidney transplant recipients [286, 287]. Correction of severe anaemia with ESA is associated with a reduction in LV mass [288]. However, correction of moderate anaemia to target haemoglobins above 12 g/dL appears to have no effect on LV mass [288]. Interestingly, the correction/normalisation of anaemia to targets above 12 g/dL have consistently been shown to increase the risk of myocardial infarction, heart failure and all-cause mortality in patients with CKD and KFRT [289,290,291,292].
Factors associated with CKD-MBD
CKD-MBD encompasses the progressive deterioration in the homoeostasis of calcium, phosphate due to disruption in circulating hormones FGF23 and its co-receptor Klotho, PTH and vitamin D (Fig. 10). Higher circulating levels of phosphate are associated with higher LV mass and LVH in the general population, patients with CKD and in dialysis patients [164, 293, 294]. However, the relationship between serum phosphate and increased LV mass has not yet been proven to be causal although a small, recent study reported a significant association between reduction in serum phosphate and regression of LV mass [295].

Chronic kidney disease – mineral bone disorder (CKD-MBD) describes the alterations in circulating and tissue levels of calcium and phosphate because of changes in parathyroid hormone, vitamin D, fibroblast growth factor 23 (FGF23) and its coreceptor Klotho. These biochemical changes are associated with the cardiovascular remodelling and clinical manifestations observed in patients with CKD.
Low levels of vitamin D have been associated with higher blood pressure [296], a higher incidence of hypertension [297], and LVH, possibly mediated by parathyroid hormone [298, 299]. Patients with CKD often develop vitamin D (1,25-dihydroxyvitamin D3/calcitriol) deficiency because of a lack of its precursor, 25-hydroxyvitamin D3 and impaired activity of the kidney enzyme 1α-hydroxylase [300]. Observational studies have suggested a beneficial association between therapy with calcitriol or related analogues and reduced cardiovascular events [301,302,303,304,305] with experimental models suggesting that these actions are mediated by a reduction in LVH, and improved LV diastolic function [299, 306,307,308,309]. However, to date, randomised controlled trials have not shown a significant reduction in either blood pressure or LVH with vitamin D treatment [310, 311].
The phosphaturic hormone FGF23 is markedly increased in patients with CKD, and dialysis [165, 312,313,314] and has been causally linked to the development of LVH [315] and LV dysfunction [316] and arrythmias such as AF [317]. The calcimimetic agent cinacalcet reduces circulating FGF23 and has been shown to reduce cardiovascular death, sudden cardiac death and heart failure in patients on dialysis [318]. However, it should be noted that calcimimetics appear to have a consistent blood pressure lowering effect in experimental models of uraemia and patients with CKD and KFRT [295, 319,320,321,322].
The FGF23 co-receptor αKlotho is a protein severely downregulated in CKD [323] due to vitamin D deficiency [324, 325], RAAS overactivity [326, 327], inflammation [328] and also by FGF23 excess [329]. In haemodialysis patients, αKlotho deficiency is a strong predictor of cardiovascular events and mortality [330, 331]. In its secreted, circulating form, soluble αKlotho exerts a broad array of biological functions as evidenced in studies of αKlotho-deficient mice which manifested an ageing phenotype of phosphate imbalance, osteoporosis/osteopenia [332], vascular calcification, growth retardation, emphysema [333] and premature death [334], cardiac hypertrophy and fibrosis [335,336,337]. In addition, αKlotho has a role in blood pressure homoeostasis. In a study of 2774 subjects, higher αKlotho was associated with a lower risk of incident hypertension [338]. Preclinical studies have shed light on the potential underlying mechanisms, including αKlotho inhibition of inflammation-associated renal sodium retention [339, 340] and endothelial dysfunction through reduced oxidative stress and increased nitric oxide availability [341,342,343] in murine and in vitro studies. Klotho supplementation in uraemic and diabetic mice protected against uraemic toxin [344], angiotensin II [345], inflammation [345, 346], oxidative stress [346] and myocardial FGF21-induced LVH [347]. The use of recombinant αKlotho in the treatment of hypertension and CKD-associated cardiomyopathy remains a promising therapeutic option [348] although yet to trialled in larger mammals.
Weight loss
Obesity, defined as a body mass index of ≥30 kg/m2, is a strong predictor of metabolic syndrome and plays a critical role in the development of cardiovascular and kidney disease [349,350,351,352,353]. In a meta-analysis of 1022 obese patients with preserved systolic function, surgical bariatric interventions resulted in a decrease in LV mass and improved LV diastolic function during a mean follow-up period of 16 months [354]. Whilst lifestyle interventions can be effective in the short term, bariatric procedures have been proven to result in more persistent weight loss compared to non-surgical approaches [355]. A randomised controlled trial of 41 hypertensive, obese patients found that a mean weight loss of 8.3 kg achieved through lifestyle changes led to a 16% reduction (adjusted for body surface area) in LV mass, independent of blood pressure changes [356]. This is particularly intriguing as weight loss has also been associated with reduction in proteinuria and improved renal function [357,358,359]. However, data on the longer-term outcomes of lifestyle interventions in obese, diabetic individuals demonstrate no significant difference in cardiovascular events at 10 [360] and 20 [361] years of follow-up. Although few in number, recent observational studies reported significantly lower rates of cardiovascular events and death in obese patients 5–15 years following bariatric surgery [362,363,364]. How these data can be extrapolated to patients with kidney disease remains to be investigated.
Conclusions and future perspectives
The pathophysiology of CKD-associated cardiomyopathy is extremely complex and involves multiple inter-related mechanisms. Increasing evidence linking individual factors to its aetiology may well lead to novel treatments in the future. However, the current evidence continues to suggest that the diagnosis, characterisation, and optimisation of treatment of hypertension is the cornerstone of treatment for now. To date, specific, blood pressure independent, beneficial treatment effects on either LV mass or hard clinical end points have not been convincingly demonstrated. While further research into potential treatments targeting molecular aetiological pathways needs to continue, research into the optimal blood pressure targets, monitoring strategies and antihypertensive regimens, for individuals at different stages of CKD and with KFRT needs to progress in parallel.
References
Bruck K, Stel VS, Gambaro G, Hallan S, Volzke H, Arnlov J, et al. CKD prevalence varies across the european general population. J Am Soc Nephrol. 2016;27:2135–47.
Levin A, Tonelli M, Bonventre J, Coresh J, Donner JA, Fogo AB, et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet. 2017;390:1888–917.
Thomas B, Matsushita K, Abate KH, Al-Aly Z, Arnlov J, Asayama K, et al. Global cardiovascular and renal outcomes of reduced GFR. J Am Soc Nephrol. 2017;28:2167–79.
Hill NR, Fatoba ST, Oke JL, Hirst JA, O’Callaghan CA, Lasserson DS, et al. Global prevalence of chronic kidney disease - a systematic review and meta-analysis. PLoS One. 2016;11:e0158765.
Parfrey PS, Foley RN, Harnett JD, Kent GM, Murray DC, Barre PE. Outcome and risk factors for left ventricular disorders in chronic uraemia. Nephrol Dial Transpl. 1996;11:1277–85.
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005;365:217–23.
Bromfield S, Muntner P. High blood pressure: the leading global burden of disease risk factor and the need for worldwide prevention programs. Curr Hypertens Rep. 2013;15:134–6.
Muntner P, Anderson A, Charleston J, Chen Z, Ford V, Makos G, et al. Hypertension awareness, treatment, and control in adults with CKD: results from the Chronic Renal Insufficiency Cohort (CRIC) Study. Am J Kidney Dis. 2010;55:441–51.
Kestenbaum B, Rudser KD, de Boer IH, Peralta CA, Fried LF, Shlipak MG, et al. Differences in kidney function and incident hypertension: the multi-ethnic study of atherosclerosis. Ann Intern Med. 2008;148:501–8.
Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44:595–601.
Brantsma AH, Bakker SJ, de Zeeuw D, de Jong PE, Gansevoort RT. Urinary albumin excretion as a predictor of the development of hypertension in the general population. J Am Soc Nephrol. 2006;17:331–5.
Chue CD, Townend JN, Steeds RP, Ferro CJ. Arterial stiffness in chronic kidney disease: causes and consequences. Heart. 2010;96:817–23.
Moody WE, Edwards NC, Chue CD, Ferro CJ, Townend JN. Arterial disease in chronic kidney disease. Heart. 2013;99:365–72.
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl J Med. 2004;351:1296–305.
Aoki J, Ikari Y, Nakajima H, Mori M, Sugimoto T, Hatori M, et al. Clinical and pathologic characteristics of dilated cardiomyopathy in hemodialysis patients. Kidney Int. 2005;67:333–40.
Chronic Kidney Disease Prognosis C, Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, et al. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375:2073–81.
Steenkamp R, Rao A, Roderick P. UK renal registry 17th annual report: chapter 5 survival and cause of death in UK adult patients on renal replacement therapy in 2013: National and Centre-specific Analyses. Nephron 2015;129:99–129.
U.S. Renal Data System. USRDS 2015 annual data report: atlas of end-stage renal disease in the United States. Bethseda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2015.
Wanner C, Amann K, Shoji T. The heart and vascular system in dialysis. Lancet. 2016;388:276–84.
Kalra PA, Green D, Poulikakos D. Arrhythmia in hemodialysis patients and its relation to sudden death. Kidney Int. 2018;93:781–3.
Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, et al. US Renal Data System 2016 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis. 2017;69:A7–A8.
Methven S, Steenkamp R, Fraser S. UK Renal Registry 19th Annual Report: Chapter 5 Survival and Causes of Death in UK Adult Patients on Renal Replacement Therapy in 2015: National and Centre-specific Analyses. Nephron. 2017;137:117–50.
Thompson S, James M, Wiebe N, Hemmelgarn B, Manns B, Klarenbach S, et al. Cause of death in patients with reduced kidney function. J Am Soc Nephrol. 2015;26:2504–11.
Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet. 2011;377:2181–92.
Tonelli M, Isles C, Curhan GC, Tonkin A, Pfeffer MA, Shepherd J, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation. 2004;110:1557–63.
Ferro CJ, Mark PB, Kanbay M, Sarafidis P, Heine GH, Rossignol P, et al. Lipid management in patients with chronic kidney disease. Nat Rev Nephrol. 2018;14:727–49.
Wanner C, Krane V, Marz W, Olschewski M, Asmus HG, Kramer W, et al. Randomized controlled trial on the efficacy and safety of atorvastatin in patients with type 2 diabetes on hemodialysis (4D study): demographic and baseline characteristics. Kidney Blood Press Res. 2004;27:259–66.
Fellstrom BC, Jardine AG, Schmieder RE, Holdaas H, Bannister K, Beutler J, et al. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360:1395–407.
Drueke T, Le Pailleur C, Zingraff J, Jungers P. Uremic cardiomyopathy and pericarditis. Adv Nephrol Necker Hosp. 1980;9:33–70.
Hung J, Harris PJ, Uren RF, Tiller DJ, Kelly DT. Uremic cardiomyopathy-effect of hemodialysis on left ventricular function in end-stage renal failure. N Engl J Med. 1980;302:547–51.
Mall G, Huther W, Schneider J, Lundin P, Ritz E. Diffuse intermyocardiocytic fibrosis in uraemic patients. Nephrol Dial Transpl. 1990;5:39–44.
Hayer MK, Price AM, Liu B, Baig S, Ferro CJ, Townend JN, et al. Diffuse myocardial interstitial fibrosis and dysfunction in early chronic kidney disease. Am J Cardiol. 2018;121:656–60.
Rutherford E, Talle MA, Mangion K, Bell E, Rauhalammi SM, Roditi G, et al. Defining myocardial tissue abnormalities in end-stage renal failure with cardiac magnetic resonance imaging using native T1 mapping. Kidney Int. 2016;90:845–52.
Graham-Brown MP, March DS, Churchward DR, Stensel DJ, Singh A, Arnold R, et al. Novel cardiac nuclear magnetic resonance method for noninvasive assessment of myocardial fibrosis in hemodialysis patients. Kidney Int. 2016;90:835–44.
Edwards NC, Moody WE, Yuan M, Hayer MK, Ferro CJ, Townend JN, et al. Diffuse interstitial fibrosis and myocardial dysfunction in early chronic kidney disease. Am J Cardiol. 2015;115:1311–7.
Mark PB, Johnston N, Groenning BA, Foster JE, Blyth KG, Martin TN, et al. Redefinition of uremic cardiomyopathy by contrast-enhanced cardiac magnetic resonance imaging. Kidney Int. 2006;69:1839–45.
Chen L, Kuang P, Liu H, Wei Q, Cui H, Fang J, et al. Sodium fluoride (NaF) induces inflammatory responses via activating MAPKs/NF-kappab signaling pathway and reducing anti-inflammatory cytokine expression in the mouse liver. Biol Trace Elem Res. 2019;189:157–71.
Foley RN, Parfrey PS, Harnett JD, Kent GM, Martin CJ, Murray DC, et al. Clinical and echocardiographic disease in patients starting end-stage renal disease therapy. Kidney Int. 1995;47:186–92.
Parfrey PS, Harnett JD, Griffiths SM, Taylor R, Hand J, King A, et al. The clinical course of left ventricular hypertrophy in dialysis patients. Nephron. 1990;55:114–20.
Edwards NC, Moody WE, Chue CD, Ferro CJ, Townend JN, Steeds RP. Defining the natural history of uremic cardiomyopathy in chronic kidney disease: the role of cardiovascular magnetic resonance. JACC Cardiovasc Imaging. 2014;7:703–14.
Wang X, Shapiro JI. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nat Rev Nephrol. 2019;15:159–75.
Drueke T, Le Pailleur C, Sigal-Saglier M, Zingraff J. Angiocardiographic and haemodynamic studies in chronic haemodialysis patients with cardiomegaly. Proc Eur Dial Transpl Assoc. 1979;16:244–51.
Drueke T, Le Pailleur C, Meilhac B, Koutoudis C, Zingraff J, Di Matteo J, et al. Congestive cardiomyopathy in uraemic patients on long term haemodialysis. Br Med J. 1977;1:350–3.
London GM, Marchais SJ, Guerin AP, Metivier F. Contributive factors to cardiovascular hypertrophy in renal failure. Am J Hypertens. 1989;2:261S–3S.
Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–6.
Cuspidi C, Sala C, Negri F, Mancia G, Morganti A. Italian Society of H. Prevalence of left-ventricular hypertrophy in hypertension: an updated review of echocardiographic studies. J Hum Hypertens. 2012;26:343–9.
Gardin JM, Wagenknecht LE, Anton-Culver H, Flack J, Gidding S, Kurosaki T, et al. Relationship of cardiovascular risk factors to echocardiographic left ventricular mass in healthy young black and white adult men and women. The CARDIA study. Coronary Artery Risk Development in Young Adults. Circulation. 1995;92:380–7.
Akasheh A, Wu Y, Li Y, Dustin LD, Wong ND, Gardin JM, et al. Association of blood pressure with left ventricular mass in untreated hypertensives in rural Yunnan Province. Am J Hypertens. 2009;22:730–4.
Lavie CJ, Patel DA, Milani RV, Ventura HO, Shah S, Gilliland Y. Impact of echocardiographic left ventricular geometry on clinical prognosis. Prog Cardiovasc Dis. 2014;57:3–9.
Bang CN, Soliman EZ, Simpson LM, Davis BR, Devereux RB, Okin PM, et al. Electrocardiographic left ventricular hypertrophy predicts cardiovascular morbidity and mortality in hypertensive patients: the ALLHAT study. Am J Hypertens. 2017;30:914–22.
Milani RV, Lavie CJ, Mehra MR, Ventura HO, Kurtz JD, Messerli FH. Left ventricular geometry and survival in patients with normal left ventricular ejection fraction. Am J Cardiol. 2006;97:959–63.
Turakhia MP, Schiller NB, Whooley MA. Prognostic significance of increased left ventricular mass index to mortality and sudden death in patients with stable coronary heart disease (from the Heart and Soul Study). Am J Cardiol. 2008;102:1131–5.
Mancia G, Zanchetti A, Agabiti-Rosei E, Benemio G, De Cesaris R, Fogari R, et al. Ambulatory blood pressure is superior to clinic blood pressure in predicting treatment-induced regression of left ventricular hypertrophy. SAMPLE Study Group. Study on Ambulatory Monitoring of Blood Pressure and Lisinopril Evaluation. Circulation. 1997;95:1464–70.
Bliziotis IA, Destounis A, Stergiou GS. Home versus ambulatory and office blood pressure in predicting target organ damage in hypertension: a systematic review and meta-analysis. J Hypertens. 2012;30:1289–99.
Verdecchia P, Schillaci G, Guerrieri M, Gatteschi C, Benemio G, Boldrini F, et al. Circadian blood pressure changes and left ventricular hypertrophy in essential hypertension. Circulation. 1990;81:528–36.
Abdalla M, Caughey MC, Tanner RM, Booth JN, 3rd, Diaz KM, Anstey DE, et al. Associations of blood pressure dipping patterns with left ventricular mass and left ventricular hypertrophy in blacks: the Jackson Heart study. J Am Heart Assoc. 2017;6:e004847.
Ben-Dov IZ, Kark JD, Ben-Ishay D, Mekler J, Ben-Arie L, Bursztyn M. Predictors of all-cause mortality in clinical ambulatory monitoring: unique aspects of blood pressure during sleep. Hypertension. 2007;49:1235–41.
Fagard RH, Thijs L, Staessen JA, Clement DL, De Buyzere ML, De Bacquer DA. Night-day blood pressure ratio and dipping pattern as predictors of death and cardiovascular events in hypertension. J Hum Hypertens. 2009;23:645–53.
Velasquez MT, Beddhu S, Nobakht E, Rahman M, Raj DS. Ambulatory blood pressure in chronic kidney disease: ready for prime time? Kidney Int Rep. 2016;1:94–104.
Ferro CJ, Steeds RP, Townend JN. Hypertension, arterial haemodynamics and left ventricular disease: historical observations. QJM. 2012;105:709–16.
Pearce JM. Sir Samuel Wilks (1824-1911): ʻthe most philosophical of english physiciansʼ. Eur Neurol. 2009;61:119–23.
Conti AA. Nineteenth century “Traube’s pulse” and current “Cardiac alternans”: significant features in the history of cardiology. Clin Ter. 2012;163:e71–2.
Cameron JS, Hicks J. High blood pressure and the kidney: the forgotten contribution of William Senhouse Kirkes. Kidney Int. 2000;57:724–34.
Park M, Hsu CY, Li Y, Mishra RK, Keane M, Rosas SE, et al. Associations between kidney function and subclinical cardiac abnormalities in CKD. J Am Soc Nephrol. 2012;23:1725–34.
Levin A, Singer J, Thompson CR, Ross H, Lewis M. Prevalent left ventricular hypertrophy in the predialysis population: identifying opportunities for intervention. Am J Kidney Dis. 1996;27:347–54.
Paoletti E, Bellino D, Cassottana P, Rolla D, Cannella G. Left ventricular hypertrophy in nondiabetic predialysis CKD. Am J Kidney Dis. 2005;46:320–7.
London GM, Pannier B, Guerin AP, Blacher J, Marchais SJ, Darne B, et al. Alterations of left ventricular hypertrophy in and survival of patients receiving hemodialysis: follow-up of an interventional study. J Am Soc Nephrol. 2001;12:2759–67.
Shlipak MG, Fried LF, Cushman M, Manolio TA, Peterson D, Stehman-Breen C, et al. Cardiovascular mortality risk in chronic kidney disease: comparison of traditional and novel risk factors. JAMA J Am Med Assoc. 2005;293:1737–45.
Haider AW, Larson MG, Benjamin EJ, Levy D. Increased left ventricular mass and hypertrophy are associated with increased risk for sudden death. J Am Coll Cardiol. 1998;32:1454–9.
Verdecchia P, Reboldi G, Gattobigio R, Bentivoglio M, Borgioni C, Angeli F, et al. Atrial fibrillation in hypertension: predictors and outcome. Hypertension. 2003;41:218–23.
Erkuner O, Dudink E, Nieuwlaat R, Rienstra M, Van Gelder IC, Camm AJ, et al. Effect of systemic hypertension with versus without left ventricular hypertrophy on the progression of atrial fibrillation (from the Euro Heart Survey). Am J Cardiol. 2018;122:578–83.
Zoccali C, Benedetto FA, Mallamaci F, Tripepi G, Giacone G, Stancanelli B, et al. Left ventricular mass monitoring in the follow-up of dialysis patients: prognostic value of left ventricular hypertrophy progression. Kidney Int. 2004;65:1492–8.
Shenasa M, Shenasa H, El-Sherif N. Left ventricular hypertrophy and arrhythmogenesis. Card Electrophysiol Clin. 2015;7:207–20.
Chatterjee S, Bavishi C, Sardar P, Agarwal V, Krishnamoorthy P, Grodzicki T, et al. Meta-analysis of left ventricular hypertrophy and sustained arrhythmias. Am J Cardiol. 2014;114:1049–52.
Hennersdorf MG, Schueller PO, Steiner S, Strauer BE. Prevalence of paroxysmal atrial fibrillation depending on the regression of left ventricular hypertrophy in arterial hypertension. Hypertens Res. 2007;30:535–40.
Otsuka T, Suzuki M, Yoshikawa H, Sugi K. Left ventricular diastolic dysfunction in the early stage of chronic kidney disease. J Cardiol. 2009;54:199–204.
de Bie MK, Ajmone Marsan N, Gaasbeek A, Bax JJ, Groeneveld M, Gabreels BA, et al. Left ventricular diastolic dysfunction in dialysis patients assessed by novel speckle tracking strain rate analysis: prevalence and determinants. Int J Nephrol. 2012;2012:963504.
Farshid A, Pathak R, Shadbolt B, Arnolda L, Talaulikar G. Diastolic function is a strong predictor of mortality in patients with chronic kidney disease. BMC Nephrol. 2013;14:280.
Pabst S, Hammerstingl C, Hundt F, Gerhardt T, Grohe C, Nickenig G, et al. Pulmonary hypertension in patients with chronic kidney disease on dialysis and without dialysis: results of the PEPPER-study. PLoS One. 2012;7:e35310.
Moreo A, Ambrosio G, De Chiara B, Pu M, Tran T, Mauri F, et al. Influence of myocardial fibrosis on left ventricular diastolic function: noninvasive assessment by cardiac magnetic resonance and echo. Circ Cardiovasc Imaging. 2009;2:437–43.
Pecoits-Filho R, Bucharles S, Barberato SH. Diastolic heart failure in dialysis patients: mechanisms, diagnostic approach, and treatment. Semin Dial. 2012;25:35–41.
Nagueh SF, Smiseth OA, Appleton CP, Byrd BF 3rd, Dokainish H, Edvardsen T, et al. Recommendations for the evaluation of left ventricular diastolic function by echocardiography: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2016;17:1321–60.
Rakhit DJ, Zhang XH, Leano R, Armstrong KA, Isbel NM, Marwick TH. Prognostic role of subclinical left ventricular abnormalities and impact of transplantation in chronic kidney disease. Am Heart J. 2007;153:656–64.
Edwards NC, Ferro CJ, Townend JN, Steeds RP. Aortic distensibility and arterial-ventricular coupling in early chronic kidney disease: a pattern resembling heart failure with preserved ejection fraction. Heart. 2008;94:1038–43.
Hensen LCR, Goossens K, Delgado V, Abou R, Rotmans JI, Jukema JW, et al. Prevalence of left ventricular systolic dysfunction in pre-dialysis and dialysis patients with preserved left ventricular ejection fraction. Eur J Heart Fail. 2018;20:560–8.
Stack AG, Bloembergen WE. A cross-sectional study of the prevalence and clinical correlates of congestive heart failure among incident US dialysis patients. Am J Kidney Dis. 2001;38:992–1000.
Collins AJ, Li S, Gilbertson DT, Liu J, Chen SC, Herzog CA. Chronic kidney disease and cardiovascular disease in the Medicare population. Kidney Int Suppl. 2003:S24-31. https://doi.org/10.1046/j.1523-1755.64.s87.5.x.
Jassal SV, Trpeski L, Zhu N, Fenton S, Hemmelgarn B. Changes in survival among elderly patients initiating dialysis from 1990 to 1999. CMAJ. 2007;177:1033–8.
Sood MM, Pauly RP, Rigatto C, Komenda P. Left ventricular dysfunction in the haemodialysis population. NDT. 2008;1:199–205.
Drazner MH, Rame JE, Marino EK, Gottdiener JS, Kitzman DW, Gardin JM, et al. Increased left ventricular mass is a risk factor for the development of a depressed left ventricular ejection fraction within five years: the Cardiovascular Health Study. J Am Coll Cardiol. 2004;43:2207–15.
Milani RV, Drazner MH, Lavie CJ, Morin DP, Ventura HO. Progression from concentric left ventricular hypertrophy and normal ejection fraction to left ventricular dysfunction. Am J Cardiol. 2011;108:992–6.
Krishnamoorthy A, Brown T, Ayers CR, Gupta S, Rame JE, Patel PC, et al. Progression from normal to reduced left ventricular ejection fraction in patients with concentric left ventricular hypertrophy after long-term follow-up. Am J Cardiol. 2011;108:997–1001.
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., Drazner MH, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–239.
Zannad F, Rossignol P. Cardiorenal syndrome revisited. Circulation. 2018;138:929–44.
Hall C, Gehmlich K, Denning C, Pavlovic D. Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J Am Heart Assoc. 2021;10:e019338.
Pickup LC, Law JP, Townend JN, Ferro CJ. Sudden cardiac death in chronic renal disease: aetiology and risk reduction strategies. Nephrol Dial Transpl. 2021;36:1386–8.
Brown PF, Miller C, Di Marco A, Schmitt M. Towards cardiac MRI based risk stratification in idiopathic dilated cardiomyopathy. Heart. 2019;105:270–5.
Amann K, Breitbach M, Ritz E, Mall G. Myocyte/capillary mismatch in the heart of uremic patients. J Am Soc Nephrol. 1998;9:1018–22.
Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG. Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003;349:1027–35.
Nelson MD, Wei J, Bairey, Merz CN. Coronary microvascular dysfunction and heart failure with preserved ejection fraction as female-pattern cardiovascular disease: the chicken or the egg? Eur Heart J. 2018;39:850–2.
Radhakrishnan A, Pickup LC, Price AM, Law JP, Edwards NC, Steeds RP, et al. Coronary microvascular dysfunction: a key step in the development of uraemic cardiomyopathy? Heart. 2019;105:1302–9.
Murthy VL, Naya M, Foster CR, Hainer J, Gaber M, Dorbala S, et al. Coronary vascular dysfunction and prognosis in patients with chronic kidney disease. JACC Cardiovasc Imaging. 2012;5:1025–34.
Shah NR, Charytan DM, Murthy VL, Skali Lami H, Veeranna V, Cheezum MK, et al. Prognostic Value of Coronary Flow Reserve in Patients with Dialysis-Dependent ESRD. J Am Soc Nephrol. 2016;27:1823–9.
Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, et al. The role of endomyocardial biopsy in the management of cardiovascular disease: a scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Circulation. 2007;116:2216–33.
Becker AE, Heijmans CD, Essed CE. Chronic non-ischaemic congestive heart disease and endomyocardial biopsies. Worth the extra?. Eur Heart J. 1991;12:218–23.
Simonetti OP, Kim RJ, Fieno DS, Hillenbrand HB, Wu E, Bundy JM, et al. An improved MR imaging technique for the visualization of myocardial infarction. Radiology. 2001;218:215–23.
McCrohon JA, Moon JC, Prasad SK, McKenna WJ, Lorenz CH, Coats AJ, et al. Differentiation of heart failure related to dilated cardiomyopathy and coronary artery disease using gadolinium-enhanced cardiovascular magnetic resonance. Circulation. 2003;108:54–9.
Moon JC, Reed E, Sheppard MN, Elkington AG, Ho SY, Burke M, et al. The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;43:2260–4.
Bull S, White SK, Piechnik SK, Flett AS, Ferreira VM, Loudon M, et al. Human non-contrast T1 values and correlation with histology in diffuse fibrosis. Heart. 2013;99:932–7.
Hayer M, Price A, Liu B, Baig S, Ferro CJ, Townend JN, et al. Progression of myocaradial fibrosis in chronic kidney disease. Nephrol Dial Transpl. 2019;34:i35–i6.
Mominadam S, Ozkahya M, Kayikcioglu M, Toz H, Asci G, Duman S, et al. Interdialytic blood pressure obtained by ambulatory blood pressure measurement and left ventricular structure in hypertensive hemodialysis patients. Hemodial Int. 2008;12:322–7.
Braunwald E. Heart disease 3rd ed. United States: WB Saunders Co; 1988.
Ritz E. Left ventricular hypertrophy in renal disease: beyond preload and afterload. Kidney Int. 2009;75:771–3.
Gross ML, Ritz E. Hypertrophy and fibrosis in the cardiomyopathy of uremia-beyond coronary heart disease. Semin Dialysis. 2008;21:308–18.
Ritz E, Wanner C. The challenge of sudden death in dialysis patients. Clin J Am Soc Nephrol. 2008;3:920–9.
Komuro I, Kaida T, Shibazaki Y, Kurabayashi M, Katoh Y, Hoh E, et al. Stretching cardiac myocytes stimulates protooncogene expression. J Biol Chem. 1990;265:3595–8.
Sadoshima J, Jahn L, Takahashi T, Kulik TJ, Izumo S. Molecular characterization of the stretch-induced adaptation of cultured cardiac cells. An in vitro model of load-induced cardiac hypertrophy. J Biol Chem. 1992;267:10551–60.
Komuro I, Kurabayashi M, Takaku F, Yazaki Y. Expression of cellular oncogenes in the myocardium during the developmental stage and pressure-overloaded hypertrophy of the rat heart. Circ Res. 1988;62:1075–9.
Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, et al. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA. 1991;88:8277–81.
Fiorillo C, Nediani C, Ponziani V, Giannini L, Celli A, Nassi N, et al. Cardiac volume overload rapidly induces oxidative stress-mediated myocyte apoptosis and hypertrophy. Biochim Biophys Acta. 2005;1741:173–82.
Boguslavskyi A, Pavlovic D, Aughton K, Clark JE, Howie J, Fuller W, et al. Cardiac hypertrophy in mice expressing unphosphorylatable phospholemman. Cardiovascular Res. 2014;104:72–82.
Bastug-Ozel Z, Wright PT, Kraft AE, Pavlovic D, Howie J, Froese A, et al. Heart failure leads to altered beta2-adrenoceptor/cyclic adenosine monophosphate dynamics in the sarcolemmal phospholemman/Na,K ATPase microdomain. Cardiovasc Res. 2019;115:546–55.
He S, Kou K, O’Shea C, Chen T, Mu UMR, Dong R, et al. A dataset of dual calcium and voltage optical mapping in healthy and hypertrophied murine hearts. Sci Data. 2021;8:314.
Briet M, Bozec E, Laurent S, Fassot C, London GM, Jacquot C, et al. Arterial stiffness and enlargement in mild-to-moderate chronic kidney disease. Kidney Int. 2006;69:350–7.
Ferreira JP, Girerd N, Pannier B, Rossignol P, London GM. High pulse-wave velocity defines a very high cardiovascular risk cohort of dialysis patients under age 60. Am J Nephrol. 2017;45:72–81.
Sarafidis PA, Loutradis C, Karpetas A, Tzanis G, Piperidou A, Koutroumpas G, et al. Ambulatory pulse wave velocity is a stronger predictor of cardiovascular events and all-cause mortality than office and ambulatory blood pressure in hemodialysis patients. Hypertension. 2017;70:148–57.
Briet M, Boutouyrie P, Laurent S, London GM. Arterial stiffness and pulse pressure in CKD and ESRD. Kidney Int. 2012;82:388–400.
Pannier B, Guerin AP, Marchais SJ, Safar ME, London GM. Stiffness of capacitive and conduit arteries: prognostic significance for end-stage renal disease patients. Hypertension. 2005;45:592–6.
Pickup L, Radhakrishnan A, Townend JN, Ferro CJ. Arterial stiffness in chronic kidney disease: a modifiable cardiovascular risk factor? Curr Opin Nephrol Hypertens. 2019;28:527–36.
Mourad JJ, Girerd X, Boutouyrie P, Laurent S, Safar M, London G. Increased stiffness of radial artery wall material in end-stage renal disease. Hypertension. 1997;30:1425–30.
Cecelja M, Chowienczyk P. Dissociation of aortic pulse wave velocity with risk factors for cardiovascular disease other than hypertension: a systematic review. Hypertension. 2009;54:1328–36.
McEniery CM, Yasmin, Maki-Petaja KM, McDonnell BJ, Munnery M, Hickson SS, et al. The impact of cardiovascular risk factors on aortic stiffness and wave reflections depends on age: the Anglo-Cardiff Collaborative Trial (ACCT III). Hypertension. 2010;56:591–7.
Stewart AD, Jiang B, Millasseau SC, Ritter JM, Chowienczyk PJ. Acute reduction of blood pressure by nitroglycerin does not normalize large artery stiffness in essential hypertension. Hypertension. 2006;48:404–10.
Zheng X, Jin C, Liu Y, Zhang J, Zhu Y, Kan S, et al. Arterial stiffness as a predictor of clinical hypertension. J Clin Hypertens. 2015;17:582–91.
Liao D, Arnett DK, Tyroler HA, Riley WA, Chambless LE, Szklo M, et al. Arterial stiffness and the development of hypertension. The ARIC study. Hypertension. 1999;34:201–6.
Dernellis J, Panaretou M. Aortic stiffness is an independent predictor of progression to hypertension in nonhypertensive subjects. Hypertension. 2005;45:426–31.
Salaymeh KJ, Banerjee A. Evaluation of arterial stiffness in children with Williams syndrome: does it play a role in evolving hypertension? Am Heart J. 2001;142:549–55.
Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation. 1999;99:2434–9.
Guerin AP, Blacher J, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness attenuation on survival of patients in end-stage renal failure. Circulation. 2001;103:987–92.
Belz GG. Elastic properties and Windkessel function of the human aorta. Cardiovasc Drugs Ther. 1995;9:73–83.
Zoccali C, Moissl U, Chazot C, Mallamaci F, Tripepi G, Arkossy O, et al. Chronic fluid overload and mortality in ESRD. J Am Soc Nephrol. 2017;28:2491–7.
Hur E, Usta M, Toz H, Asci G, Wabel P, Kahvecioglu S, et al. Effect of fluid management guided by bioimpedance spectroscopy on cardiovascular parameters in hemodialysis patients: a randomized controlled trial. Am J Kidney Dis. 2013;61:957–65.
Hung SC, Kuo KL, Peng CH, Wu CH, Lien YC, Wang YC, et al. Volume overload correlates with cardiovascular risk factors in patients with chronic kidney disease. Kidney Int. 2014;85:703–9.
Martin LC, Franco RJ, Gavras I, Matsubara BB, Garcia S, Caramori JT, et al. Association between hypervolemia and ventricular hypertrophy in hemodialysis patients. Am J Hypertens. 2004;17:1163–9.
Di Lullo L, Floccari F, Polito P. Right ventricular diastolic function in dialysis patients could be affected by vascular access. Nephron Clin Pr. 2011;118:c257–61.
Di Lullo L, Gorini A, Russo D, Santoboni A, Ronco C. Left ventricular hypertrophy in chronic kidney disease patients: from pathophysiology to treatment. Cardiorenal Med. 2015;5:254–66.
Metivier F, Marchais SJ, Guerin AP, Pannier B, London GM. Pathophysiology of anaemia: focus on the heart and blood vessels. Nephrol Dial Transpl. 2000;15:14–8.
Beard JL, Tobin BW, Smith SM. Effects of iron repletion and correction of anemia on norepinephrine turnover and thyroid metabolism in iron deficiency. Proc Soc Exp Biol Med. 1990;193:306–12.
Muller R, Steffen HM, Brunner R, Saric J, Pollok M, Baldamus CA, et al. Changes in the alpha adrenergic system and increase in blood pressure with recombinant human erythropoietin (rHuEpo) therapy for renal anemia. Clin Invest Med. 1991;14:614–22.
London GM, Zins B, Pannier B, Naret C, Berthelot JM, Jacquot C, et al. Vascular changes in hemodialysis patients in response to recombinant human erythropoietin. Kidney Int. 1989;36:878–82.
Cannella G, La Canna G, Sandrini M, Gaggiotti M, Nordio G, Movilli E, et al. Reversal of left ventricular hypertrophy following recombinant human erythropoietin treatment of anaemic dialysed uraemic patients. Nephrol Dial Transpl. 1991;6:31–7.
Pascual J, Teruel JL, Moya JL, Liano F, Jimenez-Mena M, Ortuno J. Regression of left ventricular hypertrophy after partial correction of anemia with erythropoietin in patients on hemodialysis: a prospective study. Clin Nephrol. 1991;35:280–7.
Fellner SK, Lang RM, Neumann A, Korcarz C, Borow KM. Cardiovascular consequences of correction of the anemia of renal failure with erythropoietin. Kidney Int. 1993;44:1309–15.
Heidenreich S, Rahn KH, Zidek W. Direct vasopressor effect of recombinant human erythropoietin on renal resistance vessels. Kidney Int. 1991;39:259–65.
Vaziri ND, Zhou XJ, Naqvi F, Smith J, Oveisi F, Wang ZQ, et al. Role of nitric oxide resistance in erythropoietin-induced hypertension in rats with chronic renal failure. Am J Physiol. 1996;271:E113–22.
Wada Y, Matsuoka H, Tamai O, Kohno K, Okuda S, Imaizumi T. Erythropoietin impairs endothelium-dependent vasorelaxation through cyclooxygenase-dependent mechanisms in humans. Am J Hypertens. 1999;12:980–7.
Dhingra RK, Young EW, Hulbert-Shearon TE, Leavey SF, Port FK. Type of vascular access and mortality in U.S. hemodialysis patients. Kidney Int. 2001;60:1443–51.
Pastan S, Soucie JM, McClellan WM. Vascular access and increased risk of death among hemodialysis patients. Kidney Int. 2002;62:620–6.
Xue JL, Dahl D, Ebben JP, Collins AJ. The association of initial hemodialysis access type with mortality outcomes in elderly Medicare ESRD patients. Am J Kidney Dis. 2003;42:1013–9.
Reddy YNV, Obokata M, Dean PG, Melenovsky V, Nath KA, Borlaug BA. Long-term cardiovascular changes following creation of arteriovenous fistula in patients with end stage renal disease. Eur Heart J. 2017;38:1913–23.
van Duijnhoven EC, Cheriex EC, Tordoir JH, Kooman JP, van Hooff JP. Effect of closure of the arteriovenous fistula on left ventricular dimensions in renal transplant patients. Nephrol Dial Transpl. 2001;16:368–72.
Unger P, Velez-Roa S, Wissing KM, Hoang AD, van de Borne P. Regression of left ventricular hypertrophy after arteriovenous fistula closure in renal transplant recipients: a long-term follow-up. Am J Transpl. 2004;4:2038–44.
Scholz SS, Vukadinovic D, Lauder L, Ewen S, Ukena C, Townsend RR, et al. Effects of arteriovenous fistula on blood pressure in patients with end-stage renal disease: a systematic meta-analysis. J Am Heart Assoc. 2019;8:e011183.
Chue CD, Edwards NC, Moody WE, Steeds RP, Townend JN, Ferro CJ. Serum phosphate is associated with left ventricular mass in patients with chronic kidney disease: a cardiac magnetic resonance study. Heart. 2012;98:219–24.
Law JP, Price AM, Pickup L, Radhakrishnan A, Weston C, Jones AM, et al. Clinical potential of targeting fibroblast growth factor-23 and alphaKlotho in the treatment of uremic cardiomyopathy. J Am Heart Assoc. 2020;9:e016041.
Pavlovic D. Endogenous cardiotonic steroids and cardiovascular disease, where to next? Cell Calcium. 2020;86:102156.
Dai B, David V, Martin A, Huang J, Li H, Jiao Y, et al. A comparative transcriptome analysis identifying FGF23 regulated genes in the kidney of a mouse CKD model. PLoS One. 2012;7:e44161.
Trask AJ, Groban L, Westwood BM, Varagic J, Ganten D, Gallagher PE, et al. Inhibition of angiotensin-converting enzyme 2 exacerbates cardiac hypertrophy and fibrosis in Ren-2 hypertensive rats. Am J Hypertens. 2010;23:687–93.
Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8.
Santos RA, Ferreira AJ. Angiotensin-(1-7) and the renin-angiotensin system. Curr Opin Nephrol Hypertens. 2007;16:122–8.
Zhang B, Umbach AT, Chen H, Yan J, Fakhri H, Fajol A, et al. Up-regulation of FGF23 release by aldosterone. Biochem Biophys Res Commun. 2016;470:384–90.
Bockmann I, Lischka J, Richter B, Deppe J, Rahn A, Fischer DC, et al. FGF23-mediated activation of local RAAS promotes cardiac hypertrophy and fibrosis. Int J Mol Sci. 2019;20:4634.
Badve SV, Palmer SC, Strippoli GFM, Roberts MA, Teixeira-Pinto A, Boudville N, et al. The validity of left ventricular mass as a surrogate end point for all-cause and cardiovascular mortality outcomes in people with CKD: a systematic review and meta-analysis. Am J Kidney Dis. 2016;68:554–63.
Ly J, Chan CT. Impact of augmenting dialysis frequency and duration on cardiovascular function. ASAIO J. 2006;52:e11–4.
Fagugli RM, Pasini P, Pasticci F, Ciao G, Cicconi B, Buoncristiani U. Effects of short daily hemodialysis and extended standard hemodialysis on blood pressure and cardiac hypertrophy: a comparative study. J Nephrol. 2006;19:77–83.
Weinreich T, De los Rios T, Gauly A, Passlick-Deetjen J. Effects of an increase in time vs. frequency on cardiovascular parameters in chronic hemodialysis patients. Clin Nephrol. 2006;66:433–9.
Susantitaphong P, Koulouridis I, Balk EM, Madias NE, Jaber BL. Effect of frequent or extended hemodialysis on cardiovascular parameters: a meta-analysis. Am J Kidney Dis. 2012;59:689–99.
Powell JR, Oluwaseun O, Woo YM, Padmanabhan N, Narasinghan E, Latta C, et al. Ten years experience of in-center thrice weekly long overnight hemodialysis. Clin J Am Soc Nephrol. 2009;4:1097–101.
Chan CT, Floras JS, Miller JA, Richardson RM, Pierratos A. Regression of left ventricular hypertrophy after conversion to nocturnal hemodialysis. Kidney Int. 2002;61:2235–9.
Wong B, Collister D, Muneer M, Storie D, Courtney M, Lloyd A, et al. In-center nocturnal hemodialysis versus conventional hemodialysis: a systematic review of the evidence. Am J Kidney Dis. 2017;70:218–34.
Odudu A, Eldehni MT, McCann GP, McIntyre CW. Randomized controlled trial of individualized dialysate cooling for cardiac protection in hemodialysis patients. Clin J Am Soc Nephrol. 2015;10:1408–17.
Mathew A, McLeggon JA, Mehta N, Leung S, Barta V, McGinn T, et al. Mortality and hospitalizations in intensive dialysis: a systematic review and meta-analysis. Can J Kidney Health Dis. 2018;5:2054358117749531.
Kraus MA, Kansal S, Copland M, Komenda P, Weinhandl ED, Bakris GL, et al. Intensive hemodialysis and potential risks with increasing treatment. Am J Kidney Dis. 2016;68:S51–S8.
Al Rahbi F, Al Salmi I. Commercial kidney transplantation: attitude, knowledge, perception, and experience of recipients. Kidney Int Rep. 2017;2:626–33.
Neale J, Smith AC. Cardiovascular risk factors following renal transplant. World J Transpl. 2015;5:183–95.
Devine PA, Courtney AE, Maxwell AP. Cardiovascular risk in renal transplant recipients. J Nephrol. 2019;32:389–99.
Alhaj E, Alhaj N, Rahman I, Niazi TO, Berkowitz R, Klapholz M. Uremic cardiomyopathy: an underdiagnosed disease. Congest Heart Fail. 2013;19:E40–5.
Patel H, Madanieh R, Kosmas CE, Vatti SK, Vittorio TJ. Reversible cardiomyopathies. Clin Med Insights Cardiol. 2015;9:7–14.
Kaesler N, Babler A, Floege J, Kramann R. Cardiac remodeling in chronic kidney disease. Toxins. 2020;12:161.
Hawwa N, Schreiber MJ Jr., Tang WH. Pharmacologic management of chronic reno-cardiac syndrome. Curr Heart Fail Rep. 2013;10:54–62.
Lekawanvijit S, Adrahtas A, Kelly DJ, Kompa AR, Wang BH, Krum H. Does indoxyl sulfate, a uraemic toxin, have direct effects on cardiac fibroblasts and myocytes? Eur Heart J. 2010;31:1771–9.
Pickup LC, Law JP, Radhakrishnan A, Price AM, Loutradis C, Smith TO, et al. Changes in left ventricular structure and function associated with renal transplantation: a systematic review and meta-analysis. ESC Heart Fail. 2021;8:2045–57.
Stewart GA, Foster J, Cowan M, Rooney E, McDonagh T, Dargie HJ, et al. Echocardiography overestimates left ventricular mass in hemodialysis patients relative to magnetic resonance imaging. Kidney Int. 1999;56:2248–53.
Contti MM, Barbosa MF, Del Carmen Villanueva Mauricio A, Nga HS, Valiatti MF, Takase HM, et al. Kidney transplantation is associated with reduced myocardial fibrosis. A cardiovascular magnetic resonance study with native T1 mapping. J Cardiovasc Magn Reson. 2019;21:21.
Hayer MK, Radhakrishnan A, Price AM, Baig S, Liu B, Ferro CJ, et al. Early effects of kidney transplantation on the heart - a cardiac magnetic resonance multi-parametric study. Int J Cardiol. 2019;293:272–7.
Group SR, Wright JT Jr., Williamson JD, Whelton PK, Snyder JK, Sink KM, et al. A randomized trial of intensive versus standard blood-pressure control. N. Engl J Med. 2015;373:2103–16.
Boffa RJ, Constanti M, Floyd CN, Wierzbicki AS, Guideline Committee. Hypertension in adults: summary of updated NICE guidance. BMJ. 2019;367:l5310.
Weber MA, Schiffrin EL, White WB, Mann S, Lindholm LH, Kenerson JG, et al. Clinical practice guidelines for the management of hypertension in the community: a statement by the American Society of Hypertension and the International Society of Hypertension. J Clin Hypertens. 2014;16:14–26.
James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA J Am Med Assoc. 2014;311:507–20.
Board JBS. Joint British Societies’ consensus recommendations for the prevention of cardiovascular disease (JBS3). Heart. 2014;100:ii1–ii67.
Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Bohm M, et al. 2013 ESH/ESC Guidelines for the management of arterial hypertension: the Task Force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). J Hypertens. 2013;31:1281–357.
Whelton PK, Carey RM, Aronow WS, Casey DE Jr., Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2018;71:e127–e248.
Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur Heart J. 2018;39:3021–104.
Cheung AK, Chang TI, Cushman WC, Furth SL, Hou FF, Ix JH, et al. Executive summary of the KDIGO 2021 Clinical Practice Guideline for the Management of Blood Pressure in Chronic Kidney Disease. Kidney Int. 2021;99:559–69.
Levy D, Salomon M, D’Agostino RB, Belanger AJ, Kannel WB. Prognostic implications of baseline electrocardiographic features and their serial changes in subjects with left ventricular hypertrophy. Circulation. 1994;90:1786–93.
Mathew J, Sleight P, Lonn E, Johnstone D, Pogue J, Yi Q, et al. Reduction of cardiovascular risk by regression of electrocardiographic markers of left ventricular hypertrophy by the angiotensin-converting enzyme inhibitor ramipril. Circulation. 2001;104:1615–21.
Prineas RJ, Rautaharju PM, Grandits G, Crow R, Group MR. Independent risk for cardiovascular disease predicted by modified continuous score electrocardiographic criteria for 6-year incidence and regression of left ventricular hypertrophy among clinically disease free men: 16-year follow-up for the multiple risk factor intervention trial. J Electrocardiol. 2001;34:91–101.
Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003.
Soliman EZ, Ambrosius WT, Cushman WC, Zhang ZM, Bates JT, Neyra JA, et al. Effect of intensive blood pressure lowering on left ventricular hypertrophy in patients with hypertension: SPRINT (Systolic Blood Pressure Intervention Trial). Circulation. 2017;136:440–50.
Yildiz M, Oktay AA, Stewart MH, Milani RV, Ventura HO, Lavie CJ. Left ventricular hypertrophy and hypertension. Prog Cardiovasc Dis. 2020;63:10–21.
Devereux RB, Palmieri V, Sharpe N, De Quattro V, Bella JN, de Simone G, et al. Effects of once-daily angiotensin-converting enzyme inhibition and calcium channel blockade-based antihypertensive treatment regimens on left ventricular hypertrophy and diastolic filling in hypertension: the prospective randomized enalapril study evaluating regression of ventricular enlargement (preserve) trial. Circulation. 2001;104:1248–54.
Pitt B, Reichek N, Willenbrock R, Zannad F, Phillips RA, Roniker B, et al. Effects of eplerenone, enalapril, and eplerenone/enalapril in patients with essential hypertension and left ventricular hypertrophy: the 4E-left ventricular hypertrophy study. Circulation. 2003;108:1831–8.
Investigators AES, Group AC. Effects of perindopril-indapamide on left ventricular diastolic function and mass in patients with type 2 diabetes: the ADVANCE Echocardiography Substudy. J Hypertens. 2011;29:1439–47.
Klingbeil AU, Schneider M, Martus P, Messerli FH, Schmieder RE. A meta-analysis of the effects of treatment on left ventricular mass in essential hypertension. Am J Med. 2003;115:41–6.
Fagard RH, Celis H, Thijs L, Wouters S. Regression of left ventricular mass by antihypertensive treatment: a meta-analysis of randomized comparative studies. Hypertension. 2009;54:1084–91.
Roush GC, Abdelfattah R, Song S, Ernst ME, Sica DA, Kostis JB. Hydrochlorothiazide vs chlorthalidone, indapamide, and potassium-sparing/hydrochlorothiazide diuretics for reducing left ventricular hypertrophy: a systematic review and meta-analysis. J Clin Hypertens. 2018;20:1507–15.
Roush GC, Abdelfattah R, Song S, Kostis JB, Ernst ME, Sica DA. Hydrochlorothiazide and alternative diuretics versus renin-angiotensin system inhibitors for the regression of left ventricular hypertrophy: a head-to-head meta-analysis. J Hypertens. 2018;36:1247–55.
Mehta RT, Pareek A, Dharmadhikari S. Compelling therapy of LVH: straight (and not-so-straight) inferences from evidence. Clin Hypertens. 2019;25:25.
de Simone G, Devereux RB, Izzo R, Girfoglio D, Lee ET, Howard BV, et al. Lack of reduction of left ventricular mass in treated hypertension: the strong heart study. J Am Heart Assoc. 2013;2:e000144.
Lonnebakken MT, Izzo R, Mancusi C, Gerdts E, Losi MA, Canciello G, et al. Left ventricular hypertrophy regression during antihypertensive treatment in an outpatient clinic (the Campania Salute Network). J Am Heart Assoc. 2017;6:e004152.
Kanno Y, Kaneko K, Kaneko M, Kotaki S, Mimura T, Takane H, et al. Angiotensin receptor antagonist regresses left ventricular hypertrophy associated with diabetic nephropathy in dialysis patients. J Cardiovasc Pharm. 2004;43:380–6.
Mitsuhashi H, Tamura K, Yamauchi J, Ozawa M, Yanagi M, Dejima T, et al. Effect of losartan on ambulatory short-term blood pressure variability and cardiovascular remodeling in hypertensive patients on hemodialysis. Atherosclerosis. 2009;207:186–90.
Suzuki H, Kanno Y, Ikeda N, Nakamoto H, Okada H, Sugahara S. Selection of the dose of angiotensin converting enzyme inhibitor for patients with diabetic nephropathy depends on the presence or absence of left ventricular hypertrophy. Hypertens Res. 2002;25:865–73.
Robson R, Collins J, Johnson R, Kitching R, Searle M, Walker R, et al. Effects of simvastatin and enalapril on serum lipoprotein concentrations and left ventricular mass in patients on dialysis. The Perfect Study Collaborative Group. J Nephrol. 1997;10:33–40.
Suzuki H, Nakamoto H, Okada H, Sugahara S, Kanno Y. A selective angiotensin receptor antagonist, Valsartan, produced regression of left ventricular hypertrophy associated with a reduction of arterial stiffness. Adv Perit Dial. 2003;19:59–66.
Matsumoto N, Ishimitsu T, Okamura A, Seta H, Takahashi M, Matsuoka H. Effects of imidapril on left ventricular mass in chronic hemodialysis patients. Hypertens Res. 2006;29:253–60.
Yu WC, Lin YP, Lin IF, Chuang SY, Chen CH. Effect of ramipril on left ventricular mass in normotensive hemodialysis patients. Am J Kidney Dis. 2006;47:478–84.
London GM, Pannier B, Guerin AP, Marchais SJ, Safar ME, Cuche JL. Cardiac hypertrophy, aortic compliance, peripheral resistance, and wave reflection in end-stage renal disease. Comparative effects of ACE inhibition and calcium channel blockade. Circulation. 1994;90:2786–96.
Shibasaki Y, Masaki H, Nishiue T, Nishikawa M, Matsubara H, Iwasaka T. Angiotensin II type 1 receptor antagonist, losartan, causes regression of left ventricular hypertrophy in end-stage renal disease. Nephron. 2002;90:256–61.
Yilmaz R, Altun B, Kahraman S, Ozer N, Akinci D, Turgan C. Impact of amlodipine or ramipril treatment on left ventricular mass and carotid intima-media thickness in nondiabetic hemodialysis patients. Ren Fail. 2010;32:903–12.
Zeltner R, Poliak R, Stiasny B, Schmieder RE, Schulze BD. Renal and cardiac effects of antihypertensive treatment with ramipril vs metoprolol in autosomal dominant polycystic kidney disease. Nephrol Dial Transpl. 2008;23:573–9.
Agarwal R, Sinha AD, Pappas MK, Abraham TN, Tegegne GG. Hypertension in hemodialysis patients treated with atenolol or lisinopril: a randomized controlled trial. Nephrol Dial Transpl. 2014;29:672–81.
Edwards NC, Steeds RP, Stewart PM, Ferro CJ, Townend JN. Effect of spironolactone on left ventricular mass and aortic stiffness in early-stage chronic kidney disease: a randomized controlled trial. J Am Coll Cardiol. 2009;54:505–12.
Edwards NC, Price AM, Mehta S, Hiemstra TF, Kaur A, Greasley PJ, et al. Effects of spironolactone and chlorthalidone on cardiovascular structure and function in chronic kidney disease: a randomized, open-label trial. Clin J Am Soc Nephrol. 2021;16:1491–501.
Writing Committee M, Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr., et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines and the Heart Failure Society of America. Circulation. 2016;134:e282–93.
Lang CC, Motwani J, Coutie WJ, Struthers AD. Influence of candoxatril on plasma brain natriuretic peptide in heart failure. Lancet. 1991;338:255.
Good JM, Peters M, Wilkins M, Jackson N, Oakley CM, Cleland JG. Renal response to candoxatrilat in patients with heart failure. J Am Coll Cardiol. 1995;25:1273–81.
Ferro CJ, Spratt JC, Haynes WG, Webb DJ. Inhibition of neutral endopeptidase causes vasoconstriction of human resistance vessels in vivo. Circulation. 1998;97:2323–30.
McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993–1004.
Ruilope LM, Dukat A, Bohm M, Lacourciere Y, Gong J, Lefkowitz MP. Blood-pressure reduction with LCZ696, a novel dual-acting inhibitor of the angiotensin II receptor and neprilysin: a randomised, double-blind, placebo-controlled, active comparator study. Lancet. 2010;375:1255–66.
Kang H, Zhang J, Zhang X, Qin G, Wang K, Deng Z, et al. Effects of sacubitril/valsartan in patients with heart failure and chronic kidney disease: A meta-analysis. Eur J Pharm. 2020;884:173444.
Solomon SD, McMurray JJV, Anand IS, Ge J, Lam CSP, Maggioni AP, et al. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. 2019;381:1609–20.
Pieske B, Wachter R, Shah SJ, Baldridge A, Szeczoedy P, Ibram G, et al. Effect of sacubitril/valsartan vs standard medical therapies on plasma NT-proBNP concentration and submaximal exercise capacity in patients with heart failure and preserved ejection fraction: the PARALLAX randomized clinical trial. JAMA J Am Med Assoc. 2021;326:1919–29.
Vasan RS, Evans JC, Benjamin EJ, Levy D, Larson MG, Sundstrom J, et al. Relations of serum aldosterone to cardiac structure: gender-related differences in the Framingham Heart Study. Hypertension. 2004;43:957–62.
Redfield MM. Heart failure with preserved ejection fraction. N Engl J Med. 2016;375:1868–77.
Chun TY, Pratt JH. Aldosterone increases plasminogen activator inhibitor-1 synthesis in rat cardiomyocytes. Mol Cell Endocrinol. 2005;239:55–61.
Freel EM, Mark PB, Weir RA, McQuarrie EP, Allan K, Dargie HJ, et al. Demonstration of blood pressure-independent noninfarct myocardial fibrosis in primary aldosteronism: a cardiac magnetic resonance imaging study. Circ Cardiovasc Imaging. 2012;5:740–7.
Lombes M, Oblin ME, Gasc JM, Baulieu EE, Farman N, Bonvalet JP. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ Res. 1992;71:503–10.
Neumann S, Huse K, Semrau R, Diegeler A, Gebhardt R, Buniatian GH, et al. Aldosterone and D-glucose stimulate the proliferation of human cardiac myofibroblasts in vitro. Hypertension. 2002;39:756–60.
Bauersachs J, Jaisser F, Toto R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension. 2015;65:257–63.
Kuster GM, Kotlyar E, Rude MK, Siwik DA, Liao R, Colucci WS, et al. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation. 2005;111:420–7.
Suzuki G, Morita H, Mishima T, Sharov VG, Todor A, Tanhehco EJ, et al. Effects of long-term monotherapy with eplerenone, a novel aldosterone blocker, on progression of left ventricular dysfunction and remodeling in dogs with heart failure. Circulation. 2002;106:2967–72.
Fraccarollo D, Galuppo P, Hildemann S, Christ M, Ertl G, Bauersachs J. Additive improvement of left ventricular remodeling and neurohormonal activation by aldosterone receptor blockade with eplerenone and ACE inhibition in rats with myocardial infarction. J Am Coll Cardiol. 2003;42:1666–73.
Wang D, Liu YH, Yang XP, Rhaleb NE, Xu J, Peterson E, et al. Role of a selective aldosterone blocker in mice with chronic heart failure. J Card Fail. 2004;10:67–73.
Ortiz A, Ferro CJ, Balafa O, Burnier M, Ekart R, Halimi JM, et al. Mineralocorticoid receptor antagonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. Nephrol Dial Transplant. 2021:gfab167. https://doi.org/10.1093/ndt/gfab167. Epub ahead of print.
Pitt B, Filippatos G, Agarwal R, Anker SD, Bakris GL, Rossing P, et al. Cardiovascular events with finerenone in kidney disease and type 2 diabetes. N Engl J Med. 2021;385:2252–63.
Bakris GL, Agarwal R, Filippatos G, Group F-DS. Finerenone and chronic kidney disease outcomes in type 2 diabetes. Reply. N Engl J Med. 2021;384:e42.
Agarwal R, Filippatos G, Pitt B, Anker SD, Rossing P, Joseph A, et al. Cardiovascular and kidney outcomes with finerenone in patients with type 2 diabetes and chronic kidney disease: the FIDELITY pooled analysis. Eur Heart J. 2022;43:474–84.
Ettehad D, Emdin CA, Kiran A, Anderson SG, Callender T, Emberson J, et al. Blood pressure lowering for prevention of cardiovascular disease and death: a systematic review and meta-analysis. Lancet. 2016;387:957–67.
Sarafidis P, Ferro CJ, Morales E, Ortiz A, Malyszko J, Hojs R, et al. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. A consensus statement by the EURECA-m and the DIABESITY working groups of the ERA-EDTA. Nephrol Dial Transplant. 2019;34:208–30.
Sarafidis P, Ortiz A, Ferro CJ, Halimi JM, Kreutz R, Mallamaci F, et al. Sodium-glucose co-transporter-2 inhibitors for patients with diabetic and nondiabetic chronic kidney disease: a new era has already begun. J Hypertens. 2021;39:1090–7.
Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. 2019;393:31–9.
Herrington W, Preiss D, Haynes R, von Eynatten M, Staplin N, Hauske SJ, et al. The potential for improving cardio-renal outcomes by sodium-glucose co-transporter-2 inhibition in people with chronic kidney disease: a rationale for the EMPA-KIDNEY study. Clin Kidney J. 2018;11:749–61.
Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383:1436–46.
Brown AJM, Gandy S, McCrimmon R, Houston JG, Struthers AD, Lang CC. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: the DAPA-LVH trial. Eur Heart J. 2020;41:3421–32.
Verma S, Mazer CD, Yan AT, Mason T, Garg V, Teoh H, et al. Effect of empagliflozin on left ventricular mass in patients with type 2 diabetes mellitus and coronary artery disease: the EMPA-HEART CardioLink-6 randomized clinical trial. Circulation. 2019;140:1693–702.
Paneni F, Costantino S, Hamdani N. Regression of left ventricular hypertrophy with SGLT2 inhibitors. Eur Heart J. 2020;41:3433–6.
Pabel S, Wagner S, Bollenberg H, Bengel P, Kovacs A, Schach C, et al. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail. 2018;20:1690–700.
Brown A, Gandy S, Mordi IR, McCrimmon R, Ramkumar PG, Houston JG, et al. Dapagliflozin improves left ventricular myocardial longitudinal function in patients with type 2 diabetes. JACC Cardiovasc Imaging. 2021;14:503–4.
Chung YJ, Park KC, Tokar S, Eykyn TR, Fuller W, Pavlovic D, et al. SGLT2 inhibitors and the cardiac Na+/H+ exchanger-1: the plot thickens. Cardiovasc Res. 2021;117:2702–4.
Chung YJ, Park KC, Tokar S, Eykyn TR, Fuller W, Pavlovic D, et al. Off-target effects of sodium-glucose co-transporter 2 blockers: empagliflozin does not inhibit Na+/H+ exchanger-1 or lower [Na+]i in the heart. Cardiovasc Res. 2021;117:2794–806.
Thomas G, Xie D, Chen HY, Anderson AH, Appel LJ, Bodana S, et al. Prevalence and prognostic significance of apparent treatment resistant hypertension in chronic kidney disease: report from the chronic renal insufficiency cohort study. Hypertension. 2016;67:387–96.
Daugherty SL, Powers JD, Magid DJ, Tavel HM, Masoudi FA, Margolis KL, et al. Incidence and prognosis of resistant hypertension in hypertensive patients. Circulation. 2012;125:1635–42.
Bhatt DL, Kandzari DE, O’Neill WW, D’Agostino R, Flack JM, Katzen BT, et al. A controlled trial of renal denervation for resistant hypertension. N Engl J Med. 2014;370:1393–401.
Kandzari DE, Bohm M, Mahfoud F, Townsend RR, Weber MA, Pocock S, et al. Effect of renal denervation on blood pressure in the presence of antihypertensive drugs: 6-month efficacy and safety results from the SPYRAL HTN-ON MED proof-of-concept randomised trial. Lancet. 2018;391:2346–55.
Azizi M, Schmieder RE, Mahfoud F, Weber MA, Daemen J, Davies J, et al. Endovascular ultrasound renal denervation to treat hypertension (RADIANCE-HTN SOLO): a multicentre, international, single-blind, randomised, sham-controlled trial. Lancet. 2018;391:2335–45.
Bohm M, Kario K, Kandzari DE, Mahfoud F, Weber MA, Schmieder RE, et al. Efficacy of catheter-based renal denervation in the absence of antihypertensive medications (SPYRAL HTN-OFF MED Pivotal): a multicentre, randomised, sham-controlled trial. Lancet. 2020;395:1444–51.
Azizi M, Sanghvi K, Saxena M, Gosse P, Reilly JP, Levy T, et al. Ultrasound renal denervation for hypertension resistant to a triple medication pill (RADIANCE-HTN TRIO): a randomised, multicentre, single-blind, sham-controlled trial. Lancet. 2021;397:2476–86.
Weber MA, Kirtane AJ, Weir MR, Radhakrishnan J, Das T, Berk M, et al. The REDUCE HTN: REINFORCE: randomized, sham-controlled trial of bipolar radiofrequency renal denervation for the treatment of hypertension. JACC Cardiovasc Inter. 2020;13:461–70.
Ott C, Mahfoud F, Mancia G, Narkiewicz K, Ruilope LM, Fahy M, et al. Renal denervation in patients with versus without chronic kidney disease: results from the Global SYMPLICITY Registry with follow-up data of 3 years. Nephrol Dial Transpl. 2022;37:304–10.
Mahfoud F, Bohm M, Schmieder R, Narkiewicz K, Ewen S, Ruilope L, et al. Effects of renal denervation on kidney function and long-term outcomes: 3-year follow-up from the Global SYMPLICITY Registry. Eur Heart J. 2019;40:3474–82.
Walker AM, Schneider G, Yeaw J, Nordstrom B, Robbins S, Pettitt D. Anemia as a predictor of cardiovascular events in patients with elevated serum creatinine. J Am Soc Nephrol. 2006;17:2293–8.
Levin A, Thompson CR, Ethier J, Carlisle EJ, Tobe S, Mendelssohn D, et al. Left ventricular mass index increase in early renal disease: impact of decline in hemoglobin. Am J Kidney Dis. 1999;34:125–34.
Harnett JD, Foley RN, Kent GM, Barre PE, Murray D, Parfrey PS. Congestive heart failure in dialysis patients: prevalence, incidence, prognosis and risk factors. Kidney Int. 1995;47:884–90.
Foley RN, Parfrey PS, Harnett JD, Kent GM, Murray DC, Barre PE. The impact of anemia on cardiomyopathy, morbidity, and and mortality in end-stage renal disease. Am J Kidney Dis. 1996;28:53–61.
Rigatto C, Parfrey P, Foley R, Negrijn C, Tribula C, Jeffery J. Congestive heart failure in renal transplant recipients: risk factors, outcomes, and relationship with ischemic heart disease. J Am Soc Nephrol. 2002;13:1084–90.
Rigatto C, Foley R, Jeffery J, Negrijn C, Tribula C, Parfrey P. Electrocardiographic left ventricular hypertrophy in renal transplant recipients: prognostic value and impact of blood pressure and anemia. J Am Soc Nephrol. 2003;14:462–8.
Parfrey PS, Lauve M, Latremouille-Viau D, Lefebvre P. Erythropoietin therapy and left ventricular mass index in CKD and ESRD patients: a meta-analysis. Clin J Am Soc Nephrol. 2009;4:755–62.
Besarab A, Bolton WK, Browne JK, Egrie JC, Nissenson AR, Okamoto DM, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339:584–90.
Drueke TB, Locatelli F, Clyne N, Eckardt KU, Macdougall IC, Tsakiris D, et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. 2006;355:2071–84.
Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085–98.
Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019–32.
Foley RN, Collins AJ, Herzog CA, Ishani A, Kalra PA. Serum phosphate and left ventricular hypertrophy in young adults: the coronary artery risk development in young adults study. Kidney Blood Press Res. 2009;32:37–44.
Patel RK, Oliver S, Mark PB, Powell JR, McQuarrie EP, Traynor JP, et al. Determinants of left ventricular mass and hypertrophy in hemodialysis patients assessed by cardiac magnetic resonance imaging. Clin J Am Soc Nephrol. 2009;4:1477–83.
Eddington H, Chinnadurai R, Alderson H, Ibrahim ST, Chrysochou C, Green D, et al. A randomised controlled trial to examine the effects of cinacalcet on bone and cardiovascular parameters in haemodialysis patients with advanced secondary hyperparathyroidism. BMC Nephrol. 2021;22:106.
Scragg R, Sowers M, Bell C. Serum 25-hydroxyvitamin D, ethnicity, and blood pressure in the Third National Health and Nutrition Examination Survey. Am J Hypertens. 2007;20:713–9.
Forman JP, Giovannucci E, Holmes MD, Bischoff-Ferrari HA, Tworoger SS, Willett WC, et al. Plasma 25-hydroxyvitamin D levels and risk of incident hypertension. Hypertension. 2007;49:1063–9.
Saleh FN, Schirmer H, Sundsfjord J, Jorde R. Parathyroid hormone and left ventricular hypertrophy. Eur Heart J. 2003;24:2054–60.
Simpson RU, Hershey SH, Nibbelink KA. Characterization of heart size and blood pressure in the vitamin D receptor knockout mouse. J Steroid Biochem Mol Biol. 2007;103:521–4.
Quarles LD. Endocrine functions of bone in mineral metabolism regulation. J Clin Invest. 2008;118:3820–8.
Teng M, Wolf M, Lowrie E, Ofsthun N, Lazarus JM, Thadhani R. Survival of patients undergoing hemodialysis with paricalcitol or calcitriol therapy. N Engl J Med. 2003;349:446–56.
Teng M, Wolf M, Ofsthun MN, Lazarus JM, Hernan MA, Camargo CA Jr., et al. Activated injectable vitamin D and hemodialysis survival: a historical cohort study. J Am Soc Nephrol. 2005;16:1115–25.
Kalantar-Zadeh K, Kuwae N, Regidor DL, Kovesdy CP, Kilpatrick RD, Shinaberger CS, et al. Survival predictability of time-varying indicators of bone disease in maintenance hemodialysis patients. Kidney Int. 2006;70:771–80.
Drechsler C, Pilz S, Obermayer-Pietsch B, Verduijn M, Tomaschitz A, Krane V, et al. Vitamin D deficiency is associated with sudden cardiac death, combined cardiovascular events, and mortality in haemodialysis patients. Eur Heart J. 2010;31:2253–61.
Drechsler C, Verduijn M, Pilz S, Dekker FW, Krediet RT, Ritz E, et al. Vitamin D status and clinical outcomes in incident dialysis patients: results from the NECOSAD study. Nephrol Dial Transpl. 2011;26:1024–32.
Wu J, Garami M, Cheng T, Gardner DG. 1,25(OH)2 vitamin D3, and retinoic acid antagonize endothelin-stimulated hypertrophy of neonatal rat cardiac myocytes. J Clin Invest. 1996;97:1577–88.
Bodyak N, Ayus JC, Achinger S, Shivalingappa V, Ke Q, Chen YS, et al. Activated vitamin D attenuates left ventricular abnormalities induced by dietary sodium in Dahl salt-sensitive animals. Proc Natl Acad Sci USA. 2007;104:16810–5.
Przybylski R, McCune S, Hollis B, Simpson RU. Vitamin D deficiency in the spontaneously hypertensive heart failure [SHHF] prone rat. Nutr Metab Cardiovasc Dis. 2010;20:641–6.
Bae S, Yalamarti B, Ke Q, Choudhury S, Yu H, Karumanchi SA, et al. Preventing progression of cardiac hypertrophy and development of heart failure by paricalcitol therapy in rats. Cardiovasc Res. 2011;91:632–9.
Thadhani R, Appelbaum E, Pritchett Y, Chang Y, Wenger J, Tamez H, et al. Vitamin D therapy and cardiac structure and function in patients with chronic kidney disease: the PRIMO randomized controlled trial. JAMA J Am Med Assoc. 2012;307:674–84.
Witham MD, Ireland S, Houston JG, Gandy SJ, Waugh S, Macdonald TM, et al. Vitamin D therapy to reduce blood pressure and left ventricular hypertrophy in resistant hypertension: randomized, controlled trial. Hypertension. 2014;63:706–12.
Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011;79:1370–8.
Levin A, Rigatto C, Barrett B, Madore F, Muirhead N, Holmes D, et al. Biomarkers of inflammation, fibrosis, cardiac stretch and injury predict death but not renal replacement therapy at 1 year in a Canadian chronic kidney disease cohort. Nephrol Dial Transpl. 2014;29:1037–47.
Block GA, Bushinsky DA, Cheng S, Cunningham J, Dehmel B, Drueke TB, et al. Effect of etelcalcetide vs cinacalcet on serum parathyroid hormone in patients receiving hemodialysis with secondary hyperparathyroidism: a randomized clinical trial. JAMA J Am Med Assoc. 2017;317:156–64.
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. 2011;121:4393–408.
Poelzl G, Trenkler C, Kliebhan J, Wuertinger P, Seger C, Kaser S, et al. FGF23 is associated with disease severity and prognosis in chronic heart failure. Eur J Clin Invest. 2014;44:1150–8.
Chua W, Law JP, Cardoso VR, Purmah Y, Neculau G, Jawad-Ul-Qamar M, et al. Quantification of fibroblast growth factor 23 and N-terminal pro-B-type natriuretic peptide to identify patients with atrial fibrillation using a high-throughput platform: a validation study. PLoS Med. 2021;18:e1003405.
Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R, et al. Cinacalcet, fibroblast growth factor-23, and cardiovascular disease in hemodialysis: the evaluation of cinacalcet HCl therapy to lower cardiovascular events (EVOLVE) trial. Circulation. 2015;132:27–39.
Ogata H, Ritz E, Odoni G, Amann K, Orth SR. Beneficial effects of calcimimetics on progression of renal failure and cardiovascular risk factors. J Am Soc Nephrol. 2003;14:959–67.
Odenwald T, Nakagawa K, Hadtstein C, Roesch F, Gohlke P, Ritz E, et al. Acute blood pressure effects and chronic hypotensive action of calcimimetics in uremic rats. J Am Soc Nephrol. 2006;17:655–62.
Wetmore JB, Liu S, Krebill R, Menard R, Quarles LD. Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin J Am Soc Nephrol. 2010;5:110–6.
Sprague SM, Wetmore JB, Gurevich K, Da Roza G, Buerkert J, Reiner M, et al. Effect of cinacalcet and vitamin D analogs on fibroblast growth factor-23 during the treatment of secondary hyperparathyroidism. Clin J Am Soc Nephrol. 2015;10:1021–30.
Pavik I, Jaeger P, Ebner L, Wagner CA, Petzold K, Spichtig D, et al. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: a sequence suggested from a cross-sectional study. Nephrol Dial Transpl. 2013;28:352–9.
Forster RE, Jurutka PW, Hsieh JC, Haussler CA, Lowmiller CL, Kaneko I, et al. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem Biophys Res Commun. 2011;414:557–62.
Tsujikawa H, Kurotaki Y, Fujimori T, Fukuda K, Nabeshima Y. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17:2393–403.
de Borst MH, Vervloet MG, ter Wee PM, Navis G. Cross talk between the renin-angiotensin-aldosterone system and vitamin D-FGF-23-klotho in chronic kidney disease. J Am Soc Nephrol. 2011;22:1603–9.
Tang R, Zhou QL, Ao X, Peng WS, Veeraragoo P, Tang TF. Fosinopril and losartan regulate klotho gene and nicotinamide adenine dinucleotide phosphate oxidase expression in kidneys of spontaneously hypertensive rats. Kidney Blood Press Res. 2011;34:350–7.
Moreno JA, Izquierdo MC, Sanchez-Nino MD, Suarez-Alvarez B, Lopez-Larrea C, Jakubowski A, et al. The inflammatory cytokines TWEAK and TNFalpha reduce renal klotho expression through NFkappaB. J Am Soc Nephrol. 2011;22:1315–25.
Marsell R, Krajisnik T, Goransson H, Ohlsson C, Ljunggren O, Larsson TE, et al. Gene expression analysis of kidneys from transgenic mice expressing fibroblast growth factor-23. Nephrol Dial Transpl. 2008;23:827–33.
Memmos E, Sarafidis P, Pateinakis P, Tsiantoulas A, Faitatzidou D, Giamalis P, et al. Soluble Klotho is associated with mortality and cardiovascular events in hemodialysis. BMC Nephrol. 2019;20:217.
Marcais C, Maucort-Boulch D, Drai J, Dantony E, Carlier MC, Blond E, et al. Circulating Klotho associates with cardiovascular morbidity and mortality during hemodialysis. J Clin Endocrinol Metab. 2017;102:3154–61.
Kawaguchi H, Manabe N, Miyaura C, Chikuda H, Nakamura K, Kuro-o M. Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J Clin Invest. 1999;104:229–37.
Suga T, Kurabayashi M, Sando Y, Ohyama Y, Maeno T, Maeno Y, et al. Disruption of the klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am J Respir Cell Mol Biol. 2000;22:26–33.
Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45–51.
Hu MC, Shi M, Cho HJ, Adams-Huet B, Paek J, Hill K, et al. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol. 2015;26:1290–302.
Chen K, Wang S, Sun QW, Zhang B, Ullah M, Sun Z. Klotho deficiency causes heart aging via impairing the Nrf2-GR pathway. Circ Res. 2021;128:492–507.
Xie J, Yoon J, An SW, Kuro-o M, Huang CL. Soluble Klotho protects against uremic cardiomyopathy independently of fibroblast growth factor 23 and phosphate. J Am Soc Nephrol. 2015;26:1150–60.
Drew DA, Katz R, Kritchevsky S, Ix JH, Shlipak MG, Newman AB, et al. Soluble Klotho and incident hypertension. Clin J Am Soc Nephrol. 2021;16:1502–11.
Zhou X, Chen K, Lei H, Sun Z. Klotho gene deficiency causes salt-sensitive hypertension via monocyte chemotactic protein-1/CC chemokine receptor 2-mediated inflammation. J Am Soc Nephrol. 2015;26:121–32.
Citterio L, Delli Carpini S, Lupoli S, Brioni E, Simonini M, Fontana S, et al. Klotho gene in human salt-sensitive hypertension. Clin J Am Soc Nephrol. 2020;15:375–83.
Wang Y, Kuro-o M, Sun Z. Klotho gene delivery suppresses Nox2 expression and attenuates oxidative stress in rat aortic smooth muscle cells via the cAMP-PKA pathway. Aging Cell. 2012;11:410–7.
Saito Y, Nakamura T, Ohyama Y, Suzuki T, Iida A, Shiraki-Iida T, et al. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem Biophys Res Commun. 2000;276:767–72.
Wang Y, Sun Z. Klotho gene delivery prevents the progression of spontaneous hypertension and renal damage. Hypertension. 2009;54:810–7.
Yang K, Wang C, Nie L, Zhao X, Gu J, Guan X, et al. Klotho protects against indoxyl sulphate-induced myocardial hypertrophy. J Am Soc Nephrol. 2015;26:2434–46.
Ding J, Tang Q, Luo B, Zhang L, Lin L, Han L, et al. Klotho inhibits angiotensin II-induced cardiac hypertrophy, fibrosis, and dysfunction in mice through suppression of transforming growth factor-beta1 signaling pathway. Eur J Pharm. 2019;859:172549.
Guo Y, Zhuang X, Huang Z, Zou J, Yang D, Hu X, et al. Klotho protects the heart from hyperglycemia-induced injury by inactivating ROS and NF-kappaB-mediated inflammation both in vitro and in vivo. Biochim Biophys Acta Mol Basis Dis. 2018;1864:238–51.
Suassuna PGA, Cherem PM, de Castro BB, Maquigussa E, Cenedeze MA, Lovisi JCM, et al. alphaKlotho attenuates cardiac hypertrophy and increases myocardial fibroblast growth factor 21 expression in uremic rats. Exp Biol Med. 2020;245:66–78.
Hu MC, Shi M, Gillings N, Flores B, Takahashi M, Kuro OM, et al. Recombinant alpha-Klotho may be prophylactic and therapeutic for acute to chronic kidney disease progression and uremic cardiomyopathy. Kidney Int. 2017;91:1104–14.
Berthoux F, Mariat C, Maillard N. Overweight/obesity revisited as a predictive risk factor in primary IgA nephropathy. Nephrol Dial Transpl. 2013;28:iv160–6.
United States Renal Data System. 2013 USRDS Annual Data Report: Atlas of chronic kidney disease and endstage renal disease in the United States. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD, 2013.
Guh DP, Zhang W, Bansback N, Amarsi Z, Birmingham CL, Anis AH. The incidence of co-morbidities related to obesity and overweight: a systematic review and meta-analysis. BMC Public Health. 2009;9:88.
Shihab HM, Meoni LA, Chu AY, Wang NY, Ford DE, Liang KY, et al. Body mass index and risk of incident hypertension over the life course: the Johns Hopkins Precursors Study. Circulation. 2012;126:2983–9.
Burton JO, Gray LJ, Webb DR, Davies MJ, Khunti K, Crasto W, et al. Association of anthropometric obesity measures with chronic kidney disease risk in a non-diabetic patient population. Nephrol Dial Transpl. 2012;27:1860–6.
Cuspidi C, Rescaldani M, Tadic M, Sala C, Grassi G. Effects of bariatric surgery on cardiac structure and function: a systematic review and meta-analysis. Am J Hypertens. 2014;27:146–56.
Colquitt JL, Pickett K, Loveman E, Frampton GK. Surgery for weight loss in adults. Cochrane Database Syst Rev. 2014;2014:CD003641.
MacMahon SW, Wilcken DE, Macdonald GJ. The effect of weight reduction on left ventricular mass. A randomized controlled trial in young, overweight hypertensive patients. N Engl J Med. 1986;314:334–9.
Van Huffel L, Tomson CR, Ruige J, Nistor I, Van Biesen W, Bolignano D. Dietary restriction and exercise for diabetic patients with chronic kidney disease: a systematic review. PLoS One. 2014;9:e113667.
Chagnac A, Weinstein T, Herman M, Hirsh J, Gafter U, Ori Y. The effects of weight loss on renal function in patients with severe obesity. J Am Soc Nephrol. 2003;14:1480–6.
Straznicky NE, Grima MT, Lambert EA, Eikelis N, Dawood T, Lambert GW, et al. Exercise augments weight loss induced improvement in renal function in obese metabolic syndrome individuals. J Hypertens. 2011;29:553–64.
Uusitupa M, Peltonen M, Lindstrom J, Aunola S, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, et al. Ten-year mortality and cardiovascular morbidity in the Finnish Diabetes Prevention Study-secondary analysis of the randomized trial. PLoS One. 2009;4:e5656.
Li G, Zhang P, Wang J, Gregg EW, Yang W, Gong Q, et al. The long-term effect of lifestyle interventions to prevent diabetes in the China Da Qing Diabetes Prevention Study: a 20-year follow-up study. Lancet. 2008;371:1783–9.
Scott JD, Johnson BL, Blackhurst DW, Bour ES. Does bariatric surgery reduce the risk of major cardiovascular events? A retrospective cohort study of morbidly obese surgical patients. Surg Obes Relat Dis. 2013;9:32–9.
Johnson BL, Blackhurst DW, Latham BB, Cull DL, Bour ES, Oliver TL, et al. Bariatric surgery is associated with a reduction in major macrovascular and microvascular complications in moderately to severely obese patients with type 2 diabetes mellitus. J Am Coll Surg. 2013;216:545–56. discussion 56-8
Sjostrom L, Peltonen M, Jacobson P, Sjostrom CD, Karason K, Wedel H, et al. Bariatric surgery and long-term cardiovascular events. JAMA J Am Med Assoc. 2012;307:56–65.
Author information
Authors and Affiliations
Contributions
JPL and CJF devised and wrote the first version of the manuscript. LP, JNT, and DP contributed to the production of the figures and sections to subsequent versions of the manuscript. All authors have critically reviewed and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Law, J.P., Pickup, L., Pavlovic, D. et al. Hypertension and cardiomyopathy associated with chronic kidney disease: epidemiology, pathogenesis and treatment considerations. J Hum Hypertens 37, 1–19 (2023). https://doi.org/10.1038/s41371-022-00751-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41371-022-00751-4
