
Abstract
The A118G single-nucleotide polymorphism (SNP rs1799971) in the μ-opioid receptor gene, OPRM1, has been much studied in relation to alcohol use disorders. The reported effects of allelic variation at this SNP on alcohol-related behaviors, and on opioid receptor antagonist treatments, have been inconsistent. We investigated the pharmacogenetic interaction between A118G variation and the effects of two μ-opioid receptor antagonists in a clinical lab setting. Fifty-six overweight and moderate–heavy drinkers were prospectively stratified by genotype (29 AA homozygotes, 27 carriers of at least 1 G allele) in a double-blind placebo-controlled, three-period crossover design with naltrexone (NTX; 25 mg OD for 2 days, then 50 mg OD for 3 days) and GSK1521498 (10 mg OD for 5 days). The primary end point was regional brain activation by the contrast between alcohol and neutral tastes measured using functional magnetic resonance imaging (fMRI). Secondary end points included other fMRI contrasts, subjective responses to intravenous alcohol challenge, and food intake. GSK1521498 (but not NTX) significantly attenuated fMRI activation by appetitive tastes in the midbrain and amygdala. GSK1521498 (and NTX to a lesser extent) significantly affected self-reported responses to alcohol infusion. Both drugs reduced food intake. Across all end points, there was less robust evidence for significant effects of OPRM1 allelic variation, or for pharmacogenetic interactions between genotype and drug treatment. These results do not support strong modulatory effects of OPRM1 genetic variation on opioid receptor antagonist attenuation of alcohol- and food-related behaviors. However, they do support further investigation of GSK1521498 as a potential therapeutic for alcohol use and eating disorders.
Similar content being viewed by others


INTRODUCTION
The A118G polymorphism in the μ-opioid receptor (MOR) gene (OPRM1) has attracted much attention as a putative modulator of opioid antagonist treatment response in alcohol use disorders. This single-nucleotide polymorphism (SNP) rs1799971 in exon 1 of OPRM1 causes an adenine to guanine substitution leading to the replacement of asparagine with aspartic acid at a putative N-glycosylation site (N40D) of the MOR (Huang et al, 2012). Although the minor G allele is often thought to result in a gain of function of the MOR, the evidence is equivocal.
Cellular assays (Beyer et al, 2004; Bond et al, 1998) and rodent models (Mague et al, 2009; Ramchandani et al, 2011) have demonstrated that G allele carriage causes both increased and decreased response to opioid agonists and antagonists. In macaques, the G allele of the C77G OPRM1 polymorphism was associated with increased β-endorphin binding affinity, greater alcohol preference and consumption, as well as greater decreases in preference and consumption following naltrexone (NTX) administration (Barr et al, 2007, 2010).
In humans, G allele carriage (G+) was associated with greater feelings of intoxication, stimulation, sedation, and happiness following pharmacokinetically controlled alcohol infusion (Ray and Hutchison, 2004). Heavy G+ drinkers had greater approach tendencies for alcohol (Wiers et al, 2009), greater self-reported alcohol craving in a cue reactivity task (van den Wildenberg et al, 2007), and greater intravenous self-administration of ethanol than AA homozygotes (Hendershot et al, 2014). In functional neuroimaging studies, G-carriers had greater brain activation to alcohol taste in reward regions, including the striatum and orbitofrontal cortex (Filbey et al, 2008), and reduced frontostriatal connectivity (Ray et al, 2013). PET imaging has shown greater striatal dopamine release to intravenous alcohol administration in G-carriers (Ramchandani et al, 2011).
However, G-carriage had no effects on drinking behavior measured in alcohol-dependent subjects (Anton et al, 2012) and effects on alcohol-related behaviors were moderated by dopamine transporter genes (Anton et al, 2012; Ray et al, 2014; Schacht et al, 2013). Even in studies that have reported positive evidence of some phenotypic associations with A118G allelic variation, the results have not been consistent across all the phenotypes measured (Ray et al, 2012). Initial findings of better treatment response to NTX in alcohol-dependent G+ patients from post hoc analyses of clinical trial data (Anton et al, 2008; Oslin et al, 2003) have not been consistently replicated (Arias et al, 2006; Gelernter et al, 2007). Most evidence suggests that the A118G SNP is not a risk factor for alcohol dependence, at least in Caucasian populations (Arias et al, 2006; Rouvinen-Lagerstrom et al, 2013; Schwantes-An et al, 2016); although there is stronger evidence for this in East Asian populations (Chen et al, 2012). Greater frequency of the G allele has been reported in obese binge eating disorder (BED) patients (Davis et al, 2009), and GG homozygous subjects demonstrated higher preference for sweet, fatty foods (Davis et al, 2011). The G allele has also been associated with a greater dose requirement for opioid analgesia (see the meta-analysis by Hwang et al, 2014 and references therein).
GSK1521498 is a novel μ-opioid receptor antagonist (Ignar et al, 2011; Kelly et al, 2015) with some inverse agonist properties that is being investigated for the treatment of disorders of compulsive consumption of food, alcohol, and drugs. GSK1521498 has been differentiated from NTX in a series of human, animal, and cellular models in terms of its opioid receptor pharmacology, its pharmacokinetics, and its greater pharmacodynamic efficacy on functional magnetic resonance imaging (fMRI) activation by fruit juice, on consumption of food and alcohol, and on behavioral models of drug seeking behavior for heroin, cocaine, and alcohol (Ignar et al, 2011; Rabiner et al, 2011; Kelly et al, 2015; Giuliano et al, 2012, 2013, 2015; Ripley et al, 2015). In humans, GSK1521498 reduced the pleasurable response to high-fat/high-sugar food samples (Nathan et al, 2012; Ziauddeen et al, 2013a), food intake (Ziauddeen et al, 2013a), attentional bias to foods (Chamberlain et al, 2012), and fMRI activation by food images (Cambridge et al, 2012). In a 28-day dosing study of patients with obesity and high levels of binge eating behaviors, GSK1521498 did not cause significant placebo-controlled weight loss; however, post hoc genetic analysis suggested greater weight loss in the small number of G+ patients compared with AA homozygotes (Ziauddeen et al, 2013a).
In this context, we aimed to assess the effects of allelic variation at the A118G SNP on a range of pharmacodynamic markers of rewarding consumption of alcohol and food, and to explore the potential pharmacogenetic interaction between OPRM1 genotype and μ-opioid receptor antagonism by both NTX and GSK1521498. We studied healthy overweight moderate–heavy drinkers in an experimental medicine study, prospectively balanced to measure genetic and drug effects in a sample comprising approximately equal numbers of AA and G+ participants. We predicted (1) that opioid receptor antagonists would attenuate markers of alcohol- and food-related reward processing; (2) that there would be a significant pharmacogenetic interaction between A118G genotype and opioid antagonist drug treatment; and (3) given that G allele enhances opioidergic effects (Ray and Hutchison, 2004; Ramchandani et al, 2011; Filbey et al, 2008), we predicted that G carriage would have functionally opposite effects to opioid antagonist drugs.
MATERIALS AND METHODS
Participants
A total of 56 volunteers (n=49 (88%) males) aged 18–65 years (mean±SD, 44.3±11.1 years) with body mass index ⩾22 kg/m2 (mean 28.9±3.5 kg/m2) were enrolled after providing written informed consent. The study was approved by the London Riverside Research Ethics Committee, UK, and registered with EudraCT (2012-002479-32) and ClinicalTrials.gov (NCT01738867).
A total of 27 participants (48% G+) carried at least one copy of the G allele at the OPRM1 A118G SNP and 29 participants (52% AA) were AA homozygotes (see Supplementary Table S1). The alcohol consumption criterion for inclusion was ⩾3 drinks (⩾2 for women) consumed on at least 2 days per week on average, or a score of ⩾6 (mean±SD, 8.0±3.5, AA 8.8±3.8, G+ 7.3±3.1) on the Alcohol Use Disorders Identification Test (AUDIT) (Supplementary Table S2).
Exclusion criteria were a history of axis-I disorders including substance dependence or abuse (assessed by the Mini-International Neuropsychiatric Interview (MINI); Sheehan et al, 1998); positive screen for illicit drugs; heavy tobacco smoking (>15 cigarettes/day); Beck Depression Inventory II (Beck et al, 1961) total score >13; and/or Columbia Suicide Severity Rating Scale (C-SSRS) (Posner et al, 2007) score >0.
Design
In a randomized, double-blind, placebo-controlled, 3-period crossover design, participants received 5 days of treatment with placebo, GSK1521498 10 mg, or NTX 50 mg (25 mg on first 2 days) in each treatment period. Participants were randomly assigned to one of the six possible sequences of treatment to counterbalance order effects. The double blind was maintained using a double-dummy design whereby participants received two tablets on each treatment day (GSK1521498 or GSK1521498-matched placebo and NTX or NTX-matched placebo). The doses were predicted to achieve comparable 24-h average receptor occupancy (~90%) for both drugs on day 5 based on previous studies (Rabiner et al, 2011; Ziauddeen et al, 2013a). The dosing regimen for NTX closely followed prior protocols (Schacht et al, 2013; Anton et al, 2012) and achieved the maximum recommended clinical dose (50 mg). The washout period between treatments was a minimum of 14 and maximum of 24 days; equivalent to >15 times the plasma half-life of GSK1521498 (20 h), NTX (6 h), or 6-β-naltrexol, its principal active metabolite (13 h) (Rabiner et al, 2011).
A sample size of 48 subjects (24 per genotype) in a three-way crossover design was calculated to provide 80% power to detect a pharmacogenetic interaction of standardized effect size (mean difference/between-subject SD) of 0.82 with two-tailed P=0.05, based on prior data (Hernandez-Avila et al, 2003; Ziauddeen et al, 2013a). The primary end points specified a priori were alcohol vs neutral taste contrasts estimated in anatomically defined regions of interest.
Procedures
The study design and testing schedule are summarized in Figure 1, top panel (see Supplementary Methods and Supplementary Table S3 for details).

Overview of study design (a) and study procedures (b). In each treatment period, participants were admitted to the GSK Clinical Unit Cambridge (CUC) on day 1 for an overnight stay. They underwent safety assessments, routine blood tests, and received their first dose (GSK1521498 10 mg, NTX 25 mg, or placebo). On day 2, participants received their second dose, repeated safety assessments, and were discharged with medication doses for days 3 and 4. They returned to the CUC on the afternoon of day 4 and underwent pain threshold assessments, an intravenous alcohol infusion challenge, and were served a buffet dinner. After an overnight fast, they received their final dose on day 5, then underwent fMRI scanning followed by hedonic taste response tests and an ad libitum snacking paradigm. Participants were discharged at the end of day 5 after final safety assessments. Participants attended a follow-up assessment 7–10 days after completion of the final washout period. (c) Overview of fMRI taste paradigm and trial structure.
fMRI taste paradigm
During scanning, small quantities of liquid (0.9 ml per trial) were delivered directly into participant’s mouths: two appetitive stimuli (the participant’s preferred alcoholic beverage and fruit juice) and a neutral taste control (25 mM KCl, 2 mM NaHCO3) (Claus et al, 2011; Filbey et al, 2008 Rabiner et al, 2011). In each trial (Figure 1 bottom panel), a letter cue was visually presented to indicate which taste was about to be delivered (eg, ‘A’ for alcohol). Each cue was presented 32 times and on 75% of trials it was followed by presentation of the word ‘TASTE’ and the delivery of the corresponding liquid. Participants were instructed to hold the liquid in their mouth until the word ‘SWALLOW’ was presented 3 s later. The same preferred beverage was administered in all three treatment periods for an individual participant, thus controlling the within-subject variability in alcohol consumption and exposure.
fMRI data acquisition and analysis
Gradient echo echoplanar imaging data were acquired on a 3-Tesla Siemens Trio scanner with a 225 mm field of view. For each session, 929 volumes were acquired using a T2*-weighted sequence with repetition time (TR)=2000 ms; echo times TE1=13 ms, TE2=31 ms; flip angle=80; and matrix size=64 × 64. A total of 36 slices, 3 mm thick with an interslice gap of 1 mm, provided whole brain coverage in an oblique axial orientation. The first six scans were discarded to allow for equilibration of the MR signal.
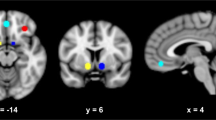
Data were analyzed by statistical parametric mapping using the SPM8 program (www.fil.ion.ucl.ac.uk). All images were realigned and regressed on the estimated 3D movement parameters, spatially normalized to a standard template, and smoothed with a 6 mm full-width-at-half-maximum Gaussian kernel. The time series were high-pass filtered (cutoff frequency of 1/120 Hz) and serial autocorrelations were estimated using an AR(1) model. At the first level analysis for each subject, the following events were modeled: cue (2 s, 32 per taste stimulus), taste delivery (3 s, 24 per stimulus), taste omission (3 s, 8 per stimulus), swallow (5 s), and urge scale (4 s). All events were modeled as boxcar functions convolved with the canonical hemodynamic response function and its temporal derivative, and covarying for motion realignment parameters. The key contrasts were alcohol versus neutral taste, and juice versus neutral taste, and were estimated only in the taste delivery trials (24 per stimulus) in the prior regions of interest (ROIs): the nucleus accumbens, the caudate nucleus, the putamen, the insula, the orbitofrontal cortex (OFC), and the amygdala bilaterally; and the midbrain (defined using the PickAtlas tool in SPM8; Maldjian et al, 2003). In addition, functional ROIs were defined by identifying the subset of voxels across all anatomical ROIs that were significantly activated by both taste contrasts on average over all treatments for all participants with familywise error of false positive activation controlled at PFWE<0.05.
Alcohol infusion challenge
This paradigm was implemented as specified previously (Ray and Hutchison, 2007; Ray et al, 2012). Ethanol (5%) was infused intravenously at rates of 0.166 and 0.126 ml/min × weight (kg) for males and females, respectively. At target breath alcohol concentrations (BrAC) of 0.02, 0.04, and 0.06 g/dl, measured instantaneously by breathalyzer, subjective responses, eg ‘high’, were examined using the Subjective High Assessment Scale (SHAS) (Schuckit, 1980), the Alcohol Rating Scale (ARS) (Ray and Hutchison, 2007), the Profile of Mood States-Brief (POMS-B) (McNair and Heuchert, 2005), and the Biphasic Alcohol Effects Scale (BAES) (Martin et al, 1993).
Hedonic and consummatory responses to food rewards
Eating behavior was measured as in previous studies of GSK1521498 (Ziauddeen et al, 2013a). Hedonic taste preference was self-reported on a nine-point scale in response to tasting small quantities of dairy products with varying fat and sugar content. Ad libitum calorie consumption was measured in a snacking paradigm, where participants could eat a selection of favorite snack foods, and separately in a buffet paradigm, where participants could choose to eat food items of variable (20, 40, or 60%) fat concentration.
Pressure pain threshold (PPT) and tolerance (PPTol)
Pain sensitivity was assessed at the left and right trapezius points using a validated algometer to apply pressure at a rate of ∼0.5 kg/cm2/s. Participants indicated when the sensation of pressure first became painful (PPT) and then intolerable (PPTol).
Cortisol
Peripheral venous blood was sampled on day 1 and day 5 before dose, at approximately the same time, and on day 5 after dose.
Pharmacokinetics (PK)
Pharmacokinetics (PK) samples were collected on day 4 (∼12 h after dose) and on day 5 (before dose and ∼1, 2, 3.5, 4.5, and 6 h after dose) and were analyzed by noncompartmental methods with Phoenix WinNonlin v. 6.3 (Pharsight, a Certara Company, Cary, NC). The maximum observed plasma concentration (Cmax), time to Cmax (Tmax), and area under the plasma concentration–time curve (AUC(0–t) and AUC(0–24)), were estimated.
Safety
Safety was assessed by cognitive measures of sedation and psychomotor function as described previously (Ziauddeen et al, 2013a, b; Bond and Lader, 1974), clinical questionnaires (POMS-B (McNair and Heuchert, 2005), Beck’s Anxiety Inventory [BAI] (Beck et al, 1988), Beck’s Depression Inventory [BDI-II] (Beck et al, 1961), C-SSRS (Posner et al, 2007)), cardiovascular parameters, adverse events, and clinical biochemistry.
Statistical Analysis
The primary and secondary end points (including fMRI contrasts in each anatomical ROI) were analyzed in accordance with a prior analysis plan. A repeated measures, mixed effects model was fit to the data for each end point with genotype, treatment, period, and genotype × treatment interaction as fixed effects for all end points; participants were modeled as random effects. For the fMRI analyses outliers were defined as measurements with a mean (absolute) standardized residual >2 after model fitting, or with a maximum standardized residual >4. All analyses were carried out using PROC MIXED in SAS and nlme in R. No formal corrections were made for multiple comparisons but fMRI analyses were restricted to a prior set of ROIs. All results are also presented descriptively as least square mean differences of drug treatment compared with placebo, or 118G+ compared with AA118.
RESULTS
fMRI Taste Paradigm: Anatomical ROIs
Following the removal of 8 outliers, data from 133 treatment sessions were included in the final analysis (AA118: 22 PBO, 23 NTX, 25 GSK1521498; 118G+: 19 PBO, 20 NTX, 24 GSK1521498). For each of the seven anatomical ROIs, fMRI activation was estimated for the contrast between alcohol and neutral tastes, and for the contrast between juice and neutral tastes. The alcohol vs neutral taste contrast was the primary end point per protocol; juice vs neutral and other taste-related contrasts were secondary end points. GSK1521498 significantly attenuated amygdala activation by the alcohol vs neutral taste contrast compared with placebo (t=2.42, df=75, P=0.05), but there were no significant effects of naltrexone or genotype or pharmacogenetic interaction on the primary end point at any of the anatomical ROIs (Supplementary Figure S3).
For a more extensive analysis of the fMRI data, we used a linear model to estimate and test the main factorial effects of genotype (AA, G+), treatment (GSK1521498, NTX, PBO), taste (alcohol vs neutral or juice vs neutral), and period (sessions 1, 2, or 3). We also estimated the two-way interactions between genotype × treatment, treatment × taste, and genotype × taste, and the three-way interaction between treatment × genotype × taste. Inclusion of the eight outliers did not change the findings. The results are reported in full for all fixed effects and regions in Supplementary Table S4 and least square mean differences averaged over contrasts are summarized graphically in Figure 2.

Treatment and genotype effects on fMRI measures of brain activation by alcohol and fruit juice tastes compared with neutral taste. (Top left) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) for anatomical ROIs, on average over genotype. (Top right) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) for functional ROI in amygdala, on average over genotype. (Bottom left) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) for anatomical ROIs, plotted separately by genotype (AA or G+). (Bottom right) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) for functional ROI in amygdala, plotted separately by genotype (AA or G+). Statistically significant effects are highlighted: *P<0.05, **P<0.01 for treatment versus placebo; #P<0.05 for NTX versus GSK1521498. There were no significant treatment by genotype interactions. However, in the midbrain ROI and the amygdala fROI, the overall treatment effect was driven by the G-carriers.
The main effect of treatment was significant in the amygdala (F2, 202=4.77, P=0.01) and in the midbrain (F2, 202=6.15, P=0.002). After conservatively using the Bonferroni correction for multiple comparisons, the treatment effect in the midbrain remained significant. There were no significant effects of treatment in any other anatomical ROIs. The main effects of taste and genotype were not significant in any ROI. The treatment × genotype interaction was not significant in any ROI, although it was on the cusp of nominal significance (F2, 202=3.04, P=0.05) for the caudate nucleus (Figure 2, left-hand panels). There were no significant treatment × contrast or treatment × genotype × taste interactions.
The post hoc analysis of pairwise differences between treatment effects in the amygdala and the midbrain demonstrated that GSK1521498 treatment significantly attenuated appetitive brain activation, ie, the average over both alcohol vs neutral and juice vs neutral taste contrasts, compared both with placebo (amygdala, t=2.51, df= 202, P=0.03; midbrain, t=3.37, df=202, P=0.003) and naltrexone (amygdala, t=2.7, df=208, P=0.02; midbrain, t=2.6, df=202, P=0.03; Figure 2).
fMRI Taste Paradigm: Functional ROIs
Two clusters of appetitively activated voxels defined a functional ROI in the left and right amygdala (PFWE<0.05). There was a significant effect of treatment on amygdala activation (F2, 202=3.88, P=0.02). The genotype × treatment interaction was close to nominal significance (F2, 202, P=0.057). The main effect of genotype was not significant (Figure 2).
Alcohol Infusion Challenge
Across nearly all measures of the subjective response to alcohol (ARS, SHAS, BAES, and POMS-B), there were significant effects of increasing breath alcohol concentration (BrAC) and period. On average over all BrAC levels, there were significant treatment effects on the ARS (F2, 550=3.74, P=0.024; PBO>GSK1521498, t=2.71, P=0.019), the SHAS (F2, 549=5.295, P=0.005; PBO>GSK1521498, t=2.44, P=0.039, NTX>GSK1521498, t=2.79, P=0.015), the BAES stimulant subscale (F2, 550=3.16, P=0.043; PBO>NTX, t=2.82, P=0.014), and the POMS-B vigor subscale (F2, 550=8.82, P<0.001; PBO>GSK1521498, t=2.47, P=0.036, PBO>NTX, t=4.34, P<0.001). There was a significant effect of genotype (F2, 53=8.1,P=0.006) and treatment × genotype interaction (F2, 550=3.83, P=0.022) only on the POMS-B confusion subscale (see Figure 3 and Supplementary Tables S5–S9.

Results of subjective rating scales from the alcohol infusion challenge: the plots show the least square means for each treatment compared with placebo across both genotypes at each breath alcohol concentration; error bars are SE of the LS means. GSK1521498 significantly attenuated the hedonic response to alcohol as measured by the Alcohol Rating Scale (ARS) and the Subjective High Assessment Scale (SHAS), ie, alcohol was overall less pleasurable (see top panel). NTX decreased the experienced stimulating effects of alcohol (BAES stimulant items) such as elation and excitement as well as feelings of vigor and energy (POMS-B vigor). All significant effects are main effects of treatment across all breath alcohol concentrations. *P<0.05 versus placebo, #p<0.05 NTX vs GSK1521498. ***p<0.001.
Eating Behaviors
There was a significant effect of treatment on total calorie consumption in both the buffet dinner (F2, 93=9.34, P=0.0002) and the snacking session (F2, 98=7.84, P=0.0007). The post hoc analysis of pairwise differences between treatments demonstrated that, compared with placebo, both GSK1521498 and NTX reduced consumption of the buffet by ∼270 calories (GSK1521498, t=4.33, df=90, P=0.0001; NTX, t=4.47, df=90, P=0.0001), and both drugs also significantly reduced snacking (GSK1521498, t=2.71, df=98, P=0.022; NTX, t=3.95, df=98, P=0.0004; Figure 4). There were no significant effects of genotype or treatment × genotype interaction on eating behavior.

Treatment and genotype effects on ad libitum calorie consumption. (Top left) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) in the ad libitum buffet paradigm on average over genotype. (Top right) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) in the ad libitum snacking paradigm on average over genotype. (Bottom left) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) for the buffet paradigm, plotted separately by genotype (AA or G+). (Bottom right) Treatment effects of GSK1521498 and NTX (least square mean difference compared with placebo; y axis) for the snacking paradigm, plotted separately by genotype (AA or G+). Statistically significant effects are highlighted: *P<0.05, **P<0.01, ***P<0.001 for treatment versus placebo.
There were no significant effects of treatment, genotype, or treatment × genotype interaction on hedonic evaluation of fatty and sugary food samples; the period effect was highly significant on many of these scales (see Supplementary Tables S10–S12).
Plasma Cortisol
There was a significant effect of treatment on plasma cortisol concentration before dose on day 5 (F2, 96=9.48, P=0.0002). The post hoc analysis of pairwise treatment differences demonstrated that compared with placebo both GSK1521498 and NTX significantly increased plasma cortisol (GSK1521498, t=3.26, df=96, P=0.004; NTX, t=4.16, df=4.16, P=0.0002). There were no significant effects of genotype or treatment × genotype (see Supplementary Table S13).
Pressure Pain Threshold
There were no significant effects of treatment, genotype, or treatment × genotype interaction on pressure pain tolerance or threshold (see Supplementary Table S14).
Descriptive Analysis of Treatment and Genotype Effects on all End Points
To compare treatment and genotype effects across all end points, the experimental data were Z-transformed and the estimated fixed effects of genotype and treatment plotted across all measures (see Figure 5). These data demonstrate that G-carriage was associated with numerically greater hedonic response to fatty and sugary foods, and greater self-reported effects of alcohol infusion. However, the effects of genotype were generally estimated with less precision than the effects of treatment.

Treatment and genotype effects across all end points. The effects of treatment (NTX and GSK1521498 bars represent LS mean differences between NTX and placebo, and GSK1521498 and placebo, averaged over genotype) and the effects of genotype (A118G bar represent LS mean difference between G+ and AA, averaged over treatments) are shown for the normalized fMRI, alcohol challenge, eating behavior, and other end points. The SHAS (Subjective High Assessment Scale), ARS (Alcohol Rating Scale), and the BAES (Biphasic Alcohol Effects Scale) sedative items and stimulant items subscales are self-report measures from the alcohol infusion challenge. For most end points apart from cortisol, where the effects are in opposite directions, genotype effects represented by a bar extending to the right of the midline indicate a numerically greater effect in the G+ group compared with the AA homozygotes; treatment effects represented by a bar extending to the left of the midline indicate a numerically greater effect in the GSK1521498 or NTX groups compared with placebo. Statistically significant effects are highlighted: *P<0.05, **P<0.01, ***P<0.001 for treatment versus placebo; #P<0.05 for NTX versus GSK1521498.
Pharmacokinetics
The PK parameters of both drugs are summarized in more detail in Supplementary Table S15. Mean exposures and between-subject variability for both GSK1521498 and NTX were consistent with prior reports (Mason et al, 2002; Nathan et al, 2012; Rabiner et al, 2011) and did not differ between AA and G+ subjects.
For GSK1521498 treatment, PK parameters of area under the curve of time versus plasma concentration, AUC(0−t), and peak plasma concentration, Cmax, were both positively correlated with drug effects on appetitive activation in the amygdala (AUC r=0.3, P=0.035, Cmax r=0.36, P=0.012) and in the midbrain (AUC r=0.36, P=0.012, Cmax r=0.31, P=0.028). There were no significant correlations between any pharmacodynamic effects of NTX and the pharmacokinetic parameters of NTX or 6-β naltrexol (its principal metabolite).
Safety
The most commonly reported adverse events (AEs) across all treatments were headache (percentage of participants experiencing at least one episode: 23% PBO, 50% NTX, 40% GSK1521498), fatigue (12% PBO, 22% NTX, 18% GSK1521498), and nausea (6% PBO, 32% NTX, 16% GSK1521498). The incidence of AEs was higher for both NTX and GSK1521498 compared with placebo, but was somewhat lower for GSK1521498 than for NTX (see Supplementary Table S16 for details). One serious adverse event (SAE) of asthenia was reported on day 2 of NTX treatment period and was considered to be treatment related. The participant was withdrawn from the study and the SAE resolved 9 h later.
There were no clinically significant effects of treatment or treatment × genotype interaction on cardiovascular parameters, cognitive tests, or routine bloods (see Supplementary Tables S17–S20). There were some significant effects of genotype on cognitive function: AA homozygotes had longer response latencies for motor response on attentional tests (see Supplementary Table S19).
DISCUSSION
This study is the first randomized, double-blind, genetically stratified, balanced crossover experimental medicine study to test the effect of allelic variation in the μ-opioid receptor gene, the effects of two μ-opioid receptor antagonist drugs, and the pharmacogenetic interactions between them on a range of alcohol- and food-related end points measured in a clinical lab setting.
In a sample of predominantly male, overweight moderate–heavy drinkers, we demonstrated that GSK1521498 treatment significantly attenuated the primary end point of fMRI activation by the alcohol vs neutral taste contrast in the amygdala. More generally, we used mixed models to estimate the main effects of genotype, treatment, taste, and period; and the two-way interactions between these factors. The main effect of taste was not significant for any ROI, ie, fMRI activation by the alcohol vs neutral contrast was not different to activation by the juice vs neutral contrast. The main effect of treatment (on both appetitive taste contrasts) was significant in two of the a priori anatomically defined ROIs. In both the amygdala and the midbrain, this treatment effect reflected significant attenuation of appetitive activation by GSK1521498 compared with both placebo and NTX; and these pharmacodynamic effects of GSK1521498 were positively correlated with PK measures of peripheral exposure. These results are convergent with prior data (Rabiner et al, 2011), demonstrating significant attenuation by μ-opioid receptor antagonist drugs of amygdala activation by an experimentally very similar juice vs neutral contrast; however, the taste stimuli and contrasts tested here are not so directly comparable to the methods of previous studies that have used either fruit juice (Filbey et al, 2007, 2008) or distilled water (Ray et al, 2014), as the neutral taste contrasted with alcohol taste. It is important to acknowledge that such pharmacological fMRI signals may be confounded by drug effects on cerebral blood flow; and that these results in a male-dominated sample may not generalize to the pharmacogenetics of OPRM1 in women. In addition, there is a potential limitation of assessing responses to alcohol in the morning (a time of day not normally associated with nondependent alcohol consumption) and all the fMRI sessions took place in the morning. However, GSK1521498 also demonstrated significant effects on subjective changes on the ARS and SHAS scales following controlled alcohol infusion. On both these measures, GSK1521498 significantly reduced feelings of intoxication and high from, and liking for, alcohol compared with placebo, and on the SHAS scale compared with NTX as well. Both GSK1521498 and NTX significantly reduced feelings of vigor (POMS-B vigor subscale). NTX significantly reduced the experienced stimulating effects of alcohol (BAES stimulant items subscale) as shown previously (King et al, 1997).
There were significant treatment effects on total calories consumed ad libitum in a buffet and a snacking paradigm. The effects of both opioid antagonists on reduced calorie consumption are consistent with prior results for GSK1521498 (Ziauddeen et al, 2013a) and NTX (Yeomans and Gray, 2002). The finding that both antagonists significantly increased plasma cortisol concentrations is also consistent with prior data (Hernandez-Avila et al, 2003) and theory implicating MOR signaling as an inhibitor of ACTH secretion by the pituitary.
In short, the primary and other end points of the study demonstrate reasonably robust and significant effects of drug treatment. However, the pharmacogenetic interaction was only on the cusp of significance for three of the many end points tested: the anatomical ROI for caudate (P=0.05), the functional ROI for amygdala (P=0.057), and one of the self-reported questionnaire subscales (POMS-B confusion) in the alcohol infusion experiment (P=0.02).
Two key methodological issues might have detracted from a clearer demonstration of hypothetically predicted effects of the A118G SNP on MOR antagonism. First, we sampled moderate–heavy drinkers rather than patients with alcohol dependence. There is some prior evidence (van den Wildenberg et al, 2007; Wiers et al, 2009; but see Anton et al, 2012; Schacht et al, 2013) to suggest that the pharmacogenetic interaction of OPRM1 and opioid receptor antagonist drugs may be greater in alcohol-dependent patients (rather than nondependent, frequent alcohol drinkers). Second, the sample size may have been too small to detect a pharmacogenetic effect. However, the sample size was defined by a prior calculation that predicted 80% power to refute the null hypothesis if it was false. We note that our final sample included a larger number of G+ participants (27) than several studies that have demonstrated a significant pharmacogenetic interaction with unbalanced genotype groups (Ray and Hutchison, 2004 (23AA, 15G+); Ray et al, 2013 (23AA, 20G+); Filbey et al, 2008 (33AA, 11G+)). Two other studies that have more evenly balanced the genotype groups (Anton et al, 2012 (40AA, 43G+); Schacht et al, 2013 (38AA, 36G+)) have not replicated a significant pharmacogenetic interaction. A recent randomized control trial of NTX in 221 alcohol-dependent subjects, which included one of the largest samples of G-carriers in a prospectively designed experiment (82G+), also did not demonstrate a significant pharmacogenetic interaction (Oslin et al, 2015).
We conclude that these data provide further evidence for GSK1521498 as a potential therapeutic for disorders of alcohol or food consumption. However, they do not provide such strong support for hypothetically predicted effects of a significant interaction between μ-opioid receptor antagonists and allelic variation at the A118G SNP on the OPRM1 gene.
FUNDING AND DISCLOSURE
HZ, PCF, and NS are employed by the University of Cambridge and have received consultancy fees from GSK. HZ was previously jointly funded by GSK and the Wellcome Trust on the Translational Medicine and Therapeutics PhD program. ETB is employed half-time by the University of Cambridge and half-time by GSK; he holds stock in GSK. PJN, BKS, CD, LW, and LJN were former employees of GSK. SRM, LKH, BW, DW, LVJ, DF, KM, AK, and DBR are employees of GSK and hold stock in GSK.
References
Anton RF, Oroszi G, O'Malley S, Couper D, Swift R, Pettinati H et al (2008). An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: results from the Combined Pharmacotherapies and Behavioral Interventions for Alcohol Dependence (COMBINE) study. Arch Gen Psychiatry 65: 135–144.
Anton RF, Voronin KK, Randall PK, Myrick H, Tiffany A (2012). Naltrexone modification of drinking effects in a subacute treatment and bar-lab paradigm: influence of OPRM1 and dopamine transporter (SLC6A3) genes. Alcohol Clin Exp Res 36: 2000–2007.
Arias A, Feinn R, Kranzler HR (2006). Association of an Asn40Asp (A118G) polymorphism in the μ-opioid receptor gene with substance dependence: a meta- analysis. Drug Alcohol Depend 83: 262–268.
Barr CS, Chen SA, Schwandt ML, Lindell SG, Sun H, Suomi SJ et al (2010). Suppression of alcohol preference by naltrexone in the rhesus macaque: a critical role of genetic variation at the μ-opioid receptor gene locus. Biol Psychiatry 67: 78–80.
Barr CS, Schwandt M, Lindell SG, Chen SA, Goldman D, Suomi SJ et al (2007). Association of a functional polymorphism in the μ-opioid receptor gene with alcohol response and consumption in male rhesus macaques. Arch Gen Psychiatry 64: 369–376.
Beck AT, Epstein N, Brown G, Steer RA (1988). An inventory for measuring clinical anxiety: psychometric properties. J Consult Clin Psychol 56: 893–897.
Beck AT, Ward CH, Mendelson J, Mock J, Erbaugh J (1961). An inventory for measuring depression. Arch Gen Psychiatry 4: 561–571.
Beyer A, Koch T, Schröder H, Schulz S, Höllt V (2004). Effect of the A118G polymorphism on binding affinity, potency and agonist-mediated endocytosis, desensitization, and resensitization of the human mu-opioid receptor. J Neurochem 89: 553–560.
Bond A, Lader M (1974). The use of analogue scales in rating subjective feelings. Br J Med Psychol 47: 211–218.
Bond C, LaForge KS, Tian MT, Melia D, Zhang SW, Borg L et al (1998). Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: Possible implications for opiate addiction. Proc Natl Acad Sci USA 95: 9608–9613.
Cambridge VC, Ziauddeen H, Nathan PJ, Subramaniam N, Dodds C, Chamberlain SR et al (2012). Neural and behavioral effects of a novel mu opioid receptor antagonist in binge-eating obese people. Biol Psychiatry 73: 887–894.
Chamberlain SR, Mogg K, Bradley BP, Koch A, Dodds CM, Tao WX et al (2012). Effects of mu opioid receptor antagonism on cognition in obese binge-eating individuals. Psychopharmacology 224: 501–509.
Chen D, Liu L, Xiao Y, Peng Y, Yang C, Wang Z (2012). Ethnic-specific meta-analyses of association between the OPRM1 A118G polymorphism and alcohol dependence among Asians and Caucasians. Drug Alcohol Depend 123: 1–6.
Claus ED, Ewing SWF, Filbey FM, Sabbineni A, Hutchison KE (2011). Identifying neurobiological phenotypes associated with alcohol use disorder severity. Neuropsychopharmacology 36: 2086–2096.
Davis C, Zai C, Levitan RD, Kaplan AS, Carter JC, Reid-Westoby C et al (2011). Opiates, overeating and obesity: a psychogenetic analysis. Int J Obes (Lond) 35: 1347–1354.
Davis CA, Levitan RD, Reid C, Carter JC, Kaplan AS, Patte KA et al (2009). Dopamine for ‘wanting’ and opioids for ‘liking’: a comparison of obese adults with and without binge eating. Obesity (Silver Spring) 17: 1220–1225.
Filbey FM, Claus E, Audette AR, Niculescu M, Banich MT, Tanabe J et al (2007). Exposure to the taste of alcohol elicits activation of the mesocorticolimbic neurocircuitry. Neuropsychopharmacology 33: 1391–1401.
Filbey FM, Ray L, Smolen A, Claus ED, Audette A, Hutchison KE (2008). Differential neural response to alcohol priming and alcohol taste cues is associated with DRD4 VNTR and OPRM1 genotypes. Alcohol Clin Exp Res 32: 1113–1123.
Gelernter J, Gueorguieva R, Kranzler HR, Zhang H, Cramer J, Rosenheck R et al (2007). Opioid receptor gene (OPRM1, OPRK1, and OPRD1) variants and response to naltrexone treatment for alcohol dependence: results from the VA Cooperative Study. Alcohol Clin Exp Res 31: 555–563.
Giuliano C, Robbins TW, Nathan PJ, Bullmore ET, Everitt BJ (2012). Inhibition of opioid transmission at the μ-opioid receptor prevents both food seeking and binge-like eating. Neuropsychopharmacology 37: 2643–2652.
Giuliano C, Robbins TW, Wille DR, Bullmore ET, Everitt BJ (2013). Attenuation of cocaine and heroin seeking by μ-opioid receptor antagonism. Psychopharmacology 227: 137–147.
Giuliano C, Goodlett CR, Economidou D, García-Pardo MP, Belin D, Robbins TW et al (2015). The novel μ-opioid receptor antagonist GSK1521498 decreases both alcohol seeking and drinking: evidence from a new preclinical model of alcohol seeking. Neuropsychopharmacology 40: 2981–2992.
Hendershot CS, Claus ED, Ramchandani VA (2016). Associations of OPRM1 A118G and alcohol sensitivity with intravenous alcohol self-administration in young adults. Addiction Biology 21: 125–135.
Hernandez-Avila CA, Wand G, Luo X, Gelernter J, Kranzler HR (2003). Association between the cortisol response to opioid blockade and the Asn40Asp polymorphism at the mu-opioid receptor locus (OPRM1). Am J Med Genet B Neuropsychiatr Genet 118B(1): 60–65.
Huang P, Chen C, Mague SD, Blendy JA, Liu-Chen L-Y (2012). A common single nucleotide polymorphism A118G of the μ opioid receptor alters its N-glycosylation and protein stability. Biochem J 441: 379–386.
Hwang IC, Park J-Y, Myung S-K, Ahn HY, Fukuda K-I, Liao Q (2014). OPRM1 A118G gene variant and postoperative opioid requirement: a systematic review and meta-analysis. Anesthesiology 121: 825–834.
Ignar DM, Goetz AS, Noble KN, Carballo LH, Stroup AE, Fisher JC et al (2011). Regulation of ingestive behaviors in the rat by GSK1521498, a novel -opioid receptor-selective inverse agonist. J Pharmacol Exp Ther 339: 24–34.
Kelly E, Mundell SJ, Sava A, Roth AL, Felici A, Maltby K et al (2015). The opioid receptor pharmacology of GSK1521498 compared to other ligands with differential effects on compulsive reward-related behaviours. Psychopharmacology 232: 305–314.
King AC, Volpicelli JR, Frazer A, O'Brien CP (1997). Effect of naltrexone on subjective alcohol response in subjects at high and low risk for future alcohol dependence. Psychopharmacology 129: 15–22.
Mague SD, Isiegas C, Huang P, Liu-Chen L-Y, Lerman C, Blendy JA (2009). Mouse model of OPRM1 (A118G) polymorphism has sex-specific effects on drug-mediated behavior. Proc Natl Acad Sci USA 106: 10847–10852.
Maldjian JA, Laurienti PJ, Kraft RA, Burdette JH (2003). An automated method for neuroanatomic and cytoarchitectonic atlas-based interrogation of fMRI data sets. Neuroimage 19: 1233–1239.
Martin CS, Earleywine M, Musty RE, Perrine MW, Swift RM (1993). Development and validation of the Biphasic Alcohol Effects Scale. Alcohol Clin Exp Res 17: 140–146.
Mason BJ, Goodman AM, Dixon RM, Hameed MH, Hulot T, Wesnes K et al (2002). A pharmacokinetic and pharmacodynamic drug interaction study of acamprosate and naltrexone. Neuropsychopharmacology 27: 596–606.
McNair DM, Heuchert JWP (2005) Profile of Mood States technical update. North Tonawanda, NY: Multi-Health Systems.
Nathan PJ, Bush MA, Tao WX, Koch A, Davies KM, Maltby K et al (2012). Multiple-dose safety, pharmacokinetics, and pharmacodynamics of the μ-opioid receptor inverse agonist GSK1521498. J Clin Pharmacol 52: 1456–1467.
Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR et al (2003). A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology 28: 1546–1552.
Oslin DW, Leong SH, Lynch KG, Berrettini W, O'Brien CP, Gordon AJ et al (2015). Naltrexone vs placebo for the treatment of alcohol dependence: a randomized clinical trial. JAMA Psychiatry 72: 430–437.
Posner K, Oquendo M, Gould M, Stanley B, Davies M (2007). Columbia Classification Algorithm of Suicide Assessment (C-CASA): classification of suicidal events in the FDA’s pediatric suicidal risk analysis of antidepressants. Am J Psychiatry 164: 1035–1043.
Rabiner EA, Beaver J, Makwana A, Searle G, Long C, Nathan PJ et al (2011). Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward-related brain activation in humans. Mol Psychiatry 16: 826–785.
Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H et al (2011). A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry 16: 809–817.
Ray LA, Bujarski S, Mackillop J, Courtney KE, Monti PM, Miotto K (2012). Subjective response to alcohol among alcohol-dependent individuals: effects of the mu-opioid receptor (OPRM1) gene and alcoholism severity. Alcohol Clin Exp Res 37(Suppl 1): E116–E124.
Ray LA, Bujarski S, Squeglia LM, Ashenhurst JR, Anton RF (2014). Interactive effects of OPRM1 and DAT1 genetic variation on subjective responses to alcohol. Alcohol Alcohol 49: 261–270.
Ray LA, Courtney KE, Hutchison KE, Mackillop J, Galvan A, Ghahremani DG (2013). Initial evidence that OPRM1 genotype moderates ventral and dorsal striatum functional connectivity during alcohol cues. Alcohol Clin Exp Res 38: 78–89.
Ray LA, Hutchison KE (2004). A polymorphism of the mu-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcohol Clin Exp Res 28: 1789–1795.
Ray LA, Hutchison KE (2007). Effects of naltrexone on alcohol sensitivity and genetic moderators of medication response: a double-blind placebo-controlled study. Arch Gen Psychiatry 64: 1069–1077.
Ripley TL, Sanchez-Roige S, Bullmore ET, Mugnaini M, Maltby K, Miller SR et al (2015). The novel mu-opioid antagonist, GSK1521498, reduces ethanol consumption in C57BL/6J mice. Psychopharmacology 232: 3431–3441.
Rouvinen-Lagerstrom N, Lahti J, Alho H, Kovanen L, Aalto M, Partonen T et al (2013). Opioid receptor gene (OPRM1) polymorphism A118G: lack of association in Finnish populations with alcohol dependence or alcohol consumption. Alcohol Alcohol 48: 519–525.
Schacht JP, Anton RF, Voronin KE, Randall PK, Li X, Henderson S et al (2013). Interacting effects of naltrexone and OPRM1 and DAT1 variation on the neural response to alcohol cues. Neuropsychopharmacology 38: 414–422.
Schuckit MA (1980). Self-rating of alcohol intoxication by young men with and without family histories of alcoholism. J Stud Alcohol 41: 242–249.
Schwantes-An T-H, Zhang J, Chen L-S, Hartz SM, Culverhouse RC, Chen X et al (2016). Association of the OPRM1 variant rs1799971 (A118G) with non-specific liability to substance dependence in a collaborative de novo meta-analysis of European-ancestry cohorts. Behav Genet 46: 151–169.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E et al (1998). The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry 59(Suppl 20): 22-33; quiz 34–57.
van den Wildenberg E, Wiers RW, Dessers J, Janssen RGJH, Lambrichs EH, Smeets HJM et al (2007). A functional polymorphism of the μ-opioid receptor gene (OPRM1) influences cue-induced craving for alcohol in male heavy drinkers. Alcohol Clin Exp Res 31: 1–10.
Wiers RW, Rinck M, Dictus M, van den Wildenberg E (2009). Relatively strong automatic appetitive action-tendencies in male carriers of the OPRM1 G-allele. Genes Brain Behav 8: 101–106.
Yeomans MR, Gray RW (2002). Opioid peptides and the control of human ingestive behaviour. Neurosci Biobehav Rev 26: 713–728.
Ziauddeen H, Chamberlain SR, Nathan PJ, Koch A, Maltby K, Bush M et al (2013a). Effects of the mu-opioid receptor antagonist GSK1521498 on hedonic and consummatory eating behaviour: a proof of mechanism study in binge-eating obese subjects. Mol Psychiatry 18: 1287–1293.
Ziauddeen H, Nathan PJ, Dodds C, Maltby K, Miller SR, Waterworth D et al (2013b). The effects of alcohol on the pharmacokinetics and pharmacodynamics of the selective mu-opioid receptor antagonist GSK1521498 in healthy subjects. J Clin Pharmacol 53: 1078–1090.
Acknowledgements
This study was sponsored and funded by GlaxoSmithKline (GSK) that has a commercial interest in GSK1521498.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Neuropsychopharmacology website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Ziauddeen, H., Nestor, L., Subramaniam, N. et al. Opioid Antagonists and the A118G Polymorphism in the μ-Opioid Receptor Gene: Effects of GSK1521498 and Naltrexone in Healthy Drinkers Stratified by OPRM1 Genotype. Neuropsychopharmacol 41, 2647–2657 (2016). https://doi.org/10.1038/npp.2016.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2016.60
This article is cited by
-
An analysis of the effect of mu-opioid receptor gene (OPRM1) promoter region DNA methylation on the response of naltrexone treatment of alcohol dependence
The Pharmacogenomics Journal (2020)
-
Improving translation of animal models of addiction and relapse by reverse translation
Nature Reviews Neuroscience (2020)
-
The frequency of DRD2 rs1076560 and OPRM1 rs1799971 in substance use disorder patients from the United Arab Emirates
Annals of General Psychiatry (2018)
-
Evidence for a Long-Lasting Compulsive Alcohol Seeking Phenotype in Rats
Neuropsychopharmacology (2018)
-
Lack of associations of the opioid receptor mu 1 (OPRM1) A118G polymorphism (rs1799971) with alcohol dependence: review and meta-analysis of retrospective controlled studies
BMC Medical Genetics (2017)
