
Abstract
Critical illness is generally hallmarked by activation of the hypothalamic–pituitary–adrenal axis. The development of very high levels of cortisol has been associated with severe illness and a raised risk of death. Likewise, a response that is inadequate relative to the degree of stress, termed relative adrenal insufficiency (also known as critical-illness-related corticosteroid insufficiency) has been associated with increased mortality. Much controversy exists with regard to the definition and biochemical testing of an adequate adrenal response to critical illness, which hampers diagnosis. High doses of glucocorticoids have been shown to have no effect in this setting and might be harmful. Moderate doses have been advocated, however, for critically ill patients with inflammatory conditions, such as acute respiratory distress syndrome and septic shock syndrome. Initial results from proof-of-concept studies were promising but thus far have not been reproduced in large, multicenter trials, although the latter were underpowered to yield definite conclusions. The role of glucocorticoid therapy in intensive care, therefore, remains uncertain. Until the debate has been settled, we recommend that use of glucocorticoid therapy in critically ill patients should continue to be based on the clinician's judgment and that routine adjuvant use should be avoided.
Key Points
-
An appropriate response of the hypothalamic–pituitary–adrenal axis to the severe stress of critical illness is essential for survival, as both very high cortisol responses and low responses (so-called relative adrenal insufficiency) have been associated with increased mortality
-
The adrenocorticotropic hormone stimulation test is widely used to assess activation of the hypothalamic–pituitary–adrenal axis, albeit without a confirmed optimum dose
-
The concept of relative adrenal insufficiency remains enigmatic as there is no consensus about the accurate biochemical diagnostic criteria by which to identify this condition, particularly since total cortisol response does not necessarily reflect response at the free hormone level
-
Initial studies on moderate-dose glucocorticoid therapy in acute respiratory distress syndrome or septic shock syndrome showed major clinical benefits; larger, multicenter trials did not confirm these results or even suggested harm, but statistical power or design issues hamper solid conclusions
-
Demonstrating a survival benefit for glucocorticoid therapy in critically ill patients is challenging due to the enormous number of patients that would be needed in order to obtain sufficient statistical power, but trial consortia might enable such studies
-
Until the issues surrounding the role of glucocorticoid therapy in critical illness are resolved, physicians should not use these drugs as routine adjuvant therapy, although use as a rescue strategy at the clinician's discretion is acceptable
Similar content being viewed by others

Introduction
The response of the hypothalamic–pituitary–adrenal (HPA) axis to the sustained stress of severe illness is biphasic, as is the case for the other neuroendocrine axes.1,2 The hypothalamic–pituitary axes have central roles in the endocrine regulation of metabolic and immunological homeostasis. The severe alterations in response to critical illness or after trauma have long been known to contribute to a high risk of morbidity and mortality. Therapeutic interventions designed to correct these alterations—with the aim of improving survival—have been studied with varying success.
Much controversy surrounds the topic of alterations in the HPA axis. Here, we explain the normal physiology of this system and its response to stress in critically ill patients before discussing issues relating to the definition of an appropriate response to underlying illness and the possible benefits of treatment with glucocorticoids.
The adrenal axis and critical illness
Physiology of the adrenal cortex
The adrenal cortex is responsible for the production of three major steroid hormones: cortisol, the main and most physiologically active glucocorticoid in humans; aldosterone, a mineralocorticoid; and dehydroepiandrosterone, an androgen.
In a stress-free human, cortisol is secreted in a diurnal pattern, with levels reaching a peak early in the morning and being lowest in the late evening. The synthesis and secretion of cortisol are stimulated by adrenocorticotropic hormone (ACTH). ACTH release by the anterior pituitary is mainly regulated by hypothalamic corticotropin-releasing hormone (CRH), but vasopressin is also a weak ACTH secretagogue and acts in synergy with CRH. Cortisol exerts negative feedback control at the level of ACTH, CRH and vasopressin release. More than 90% of circulating cortisol is bound to proteins, predominantly to corticosteroid-binding globulin and, to a lesser extent, to albumin; only free cortisol, however, is biologically active. Conversion between inactive cortisone and active cortisol is regulated by type 1 and type 2 11β-hydroxysteroid dehydrogenases. Cortisol acts through the glucocorticoid receptor. It also has mineralocorticoid activity via its ability to bind to the mineralocorticoid receptor. In many target tissues, however, this action of cortisol is blocked by the coexpression of 11β-hydroxysteroid dehydrogenase type 2, which converts cortisol to inactive cortisone.3,4
Both dehydroepiandrosterone, considered the only biologically active form of this androgen, and its derivative dehydroepiandrosterone sulfate are synthesized and secreted by the adrenals, a process stimulated by ACTH. Aldosterone production is also regulated by ACTH, but a more important regulator of this hormone is the renin–angiotensin system.
Physiologic actions of glucocorticoids
Glucocorticoids have a major influence on metabolism via their counter-regulatory actions on glucose metabolism, which result in insulin resistance and hyperglycemia. They also activate lipolysis in adipose tissue, have a catabolic effect on muscle by inhibiting protein synthesis and activating proteolysis, and are involved in bone and mineral metabolism.5
Glucocorticoids have cardiovascular effects and play a part in maintenance of myocardial contractility, vascular tone, and blood pressure by modulating synthesis of and/or cardiovascular reactivity to angiotensin II and the catecholamines epinephrine and norepinephrine, regulating vascular permeability, and decreasing production of nitric oxide and other vasodilators.5
The immune system is also affected by glucocorticoids. Actions include stimulation of anti-inflammatory cytokine production and inhibition of proinflammatory cytokine production, inflammatory cell migration, and expression of inflammatory mediators (e.g. phospholipase 2, prostaglandin synthase 2, cyclo-oxygenase 2 and inducible nitric oxide synthase).5,6 Three distinct mechanisms account for these effects: genomic actions by direct binding of the liganded glucocorticoid receptor to DNA; interaction of the ligand-bound receptor with other transcription factors, in which interference with NF-κB signaling plays a major part; and nongenomic actions mediated by second messenger systems, activation of kinase pathways, and ion fluxes.6,7
Response of the adrenal axis to critical illness
Any type of acute illness or trauma results in loss of the diurnal variation in cortisol secretion.8 In the early phase of critical illness cortisol levels frequently rise, either directly, in response to increased release of CRH and ACTH, or via resistance to or inhibition of negative-feedback control, exerted by cortisol.8,9 Specific cytokines, concentrations of which are elevated in critical illness, have been shown to activate the HPA axis and to modulate the activity of the 11β-hydroxysteroid dehydrogenases and the number, affinity or both of glucocorticoid receptors.10,11,12 Levels of corticosteroid-binding globulin are substantially depleted,13,14 due primarily to decreased hepatic production15 but also due in part to elastase-induced cleavage,16 which could represent a mechanism of cortisol delivery to sites of inflammation.17 Also, severely reduced albumin levels have been observed in critically ill patients.18 The loss of cortisol-binding proteins results in proportionally much higher increases in the levels of the free hormone relative to the total cortisol level.13,14,18
In the chronic phase of critical illness, high cortisol levels are generally sustained. In contrast to the acute phase, ACTH levels are low, indicating that pathways not mediated by this hormone are involved.19,20 In sustained critical illness, levels of corticosteroid-binding globulin gradually increase.13,14 Cortisol levels decrease only slowly, reaching normal levels in the recovery phase of illness.1
Both very high and very low cortisol levels have been associated with increased mortality from critical illness.21 This finding suggests that appropriate activation of the HPA axis is a determining factor for survival. High cortisol levels reflect severe stress, whereas low levels, at baseline and/or upon ACTH stimulation, could point to an inability to sufficiently respond to stress.22 This effect is termed relative adrenal insufficiency (also known as critical-illness-related corticosteroid insufficiency). The hypercortisolism induced by acute stress is crucial, as it fosters the acute provision of energy by altering carbohydrate, fat and protein metabolism, protects against excessive inflammation by suppressing the inflammatory response, and improves hemodynamic status by inducing fluid retention and sensitization of the vasopressor response to catecholamines.1,11 Whether the persistent elevation of cortisol levels is beneficial in sustained critical illness is unclear as, in theory, it could contribute to increased susceptibility to infectious complications.
Dehydroepiandrosterone sulfate levels decrease in response to critical illness, as documented for severe sepsis and septic shock.19,23,24 Nevertheless, levels of dehydroepiandrosterone have been found to increase in acute illness. The most severely ill patients and nonsurvivors have a high ratio of cortisol to dehydroepiandrosterone. This ratio has, therefore, been proposed as a novel potential prognostic marker in septic shock.23
The concept of relative adrenal insufficiency
In a substantial fraction of critically ill patients, cortisol response is believed to be inadequate relative to the severity of illness and degree of stress. Although the concept of relative adrenal insufficiency has been related to increased morbidity and mortality in several studies, its actual existence remains controversial. The reported incidence of this condition varies widely, depending on the patient population studied and diagnostic criteria used.25 The variation in expected cortisol levels according to type and severity of disease hampers the creation of a standardized definition of normal biochemical response to illness. Several criteria have been proposed—based on random cortisol levels, stimulation tests to assess functionality of the whole or parts of the adrenal axis, or both—but no consensus has been reached. Proposed thresholds are 276–938 nmol/l for minimum baseline total cortisol levels, 497 nmol/l for stimulated total cortisol levels, and 193–248 nmol/l for total cortisol increment after stimulation, or certain combinations of thresholds for baseline cortisol and increment after stimulation.6,22,25,26,27 Thresholds for free cortisol levels of 49.7 nmol/l before and 85.6 nmol/l after stimulation have also been proposed.18
Such cut-off levels all emerged from empirical studies of factors associated with poor outcome. For instance, in the study by Rothwell et al.27 all patients with a cortisol response below 250 nmol/l died, versus 32% of those with a higher response. A three-level prognostic classification for patients with septic shock was determined by Annane et al.22 based on the association of cortisol levels with mortality at 28 days after diagnosis of septic shock. A reasonable prognosis (mortality 26%) was observed in patients with baseline cortisol levels of 938 nmol/l or lower and a maximum increment after ACTH stimulation of more than 250 nmol/l; a poor prognosis (mortality 82%) was indicated by baseline cortisol levels higher than 938 nmol/l and a maximum increment of 250 nmol/l or lower.
Confounding factors in the biochemical diagnosis of relative adrenal insufficiency
Several diagnostic tests are available for evaluating cortisol response in a patient who is not in an intensive care unit, including ACTH stimulation, the metyrapone test and the insulin-tolerance (hypoglycemia) test, all of which have inherent limitations. The insulin-tolerance test assesses the integrity of the whole HPA axis and has traditionally been regarded the gold standard. It is, however, unreliable, and unsafe to perform in critically ill patients given the risk of developing severe hypoglycemia. Decreasing cortisol concentrations by administration of metyrapone in order to evaluate the compensatory response might also expose the patients to severe risk.
Stimulation with ACTH is preferentially used to assess cortisol response in critically ill patients, but the optimum ACTH dosage continues to be debated. Pharmacological dosing with 250 µg ACTH leads to supraphysiologic stimulation and might elicit an adequate response despite inadequate adrenal reserve, as such a high dose could overcome adrenal resistance to ACTH. Physiologic testing with 1 µg ACTH has been performed to identify patients with relative adrenal insufficiency who would have been overlooked when using the high-dose test,28,29 but the value of the findings has been brought into question.30,31
The adrenals of critically ill patients might already be maximally stimulated at baseline. Diagnostic criteria based on a minimum rise in cortisol after ACTH are probably invalid in such individuals and the use of stimulated cortisol level is supported. The latter approach could, however, be affected by disturbances in cortisol metabolism and by the marked hourly variation in cortisol that continues to occur despite loss of the circadian rhythm.32 Furthermore, the response to a short stimulation test might not reliably reflect the response to chronic stress.
Routine cortisol assays measure total levels of the hormone, despite only the free hormone being biologically active. This issue is important given the depletion of cortisol-binding globulins during critical illness.13,14 Accordingly, appropriate levels and responses of free cortisol have been observed in patients despite abnormalities in total cortisol levels,18 partly invalidating the use of total hormone levels in the diagnosis of relative adrenal insufficiency. Direct determination of free cortisol levels is, however, labor-intensive and impractical for routine clinical use. Alternative surrogate markers are the free cortisol index,19 free cortisol levels calculated from total cortisol and cortisol-binding globulins,14,33,34 and salivary cortisol levels.25
Another confounding factor relates to the wide variety of total cortisol assays that are commercially available. Given the differences in specificity, sensitivity, accuracy, precision and reproducibility, many of these assays overestimate or underestimate actual cortisol levels35 and, as such, hamper the correct diagnosis of relative adrenal insufficiency.
Potential development of (tissue-specific) glucocorticoid resistance might hinder accurate diagnosis of relative adrenal insufficiency, warranting adjustment of the criteria for appropriate cortisol levels in this condition. Another important consideration is that patients whose initial responses appear adequate could develop adrenal insufficiency at a later stage.
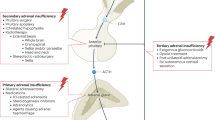
Clinical suspicion might increase the sensitivity and specificity of ACTH-stimulation testing for relative adrenal insufficiency in critically ill patients (Box 1). The most common clinical feature associated with this condition, although very nonspecific in the critically ill, is hypotension refractory to fluid resuscitation and requiring vasopressors.8,36 Such patients frequently have hyperdynamic circulation, and the development of central nervous system dysfunction is common.8,19,36 Causes of adrenal insufficiency in critically ill patients are listed in Box 2.
Treatment with glucocorticoids during critical illness
The use of glucocorticoids during critical illness has been advocated mainly for two indications: acute respiratory distress syndrome and septic shock syndrome. Approaches to therapy have, however, undergone a major evolution in the past few decades, although on the basis of various rationales. From the 1960s until early 1980s, very high doses of corticosteroids were used to block the inflammatory cascade that perpetuates acute respiratory distress syndrome and septic shock syndrome. Daily doses of 30 mg/kg methylprednisolone were widely administered. Despite some promising reports from animal studies and small but methodologically flawed clinical studies, larger, multicenter, randomized, controlled trials37,38 and meta-analyses39,40 could not confirm beneficial effects in sepsis. Glucocorticoid therapy during acute respiratory distress syndrome fared similarly, as high-dose, short-course therapies aimed at inhibiting the inflammatory response during the early phase of illness yielded no improvements in survival.41,42
Moderate-dose glucocorticoid strategies are now being assessed. Two paradigms have led to this testing. Firstly, arterial resistance vessels are thought to become hyporesponsive to catecholamines during severe sepsis, yet improve with moderate-dose corticosteroids.43 Secondly, the concept of relative adrenal insufficiency was based on the observation that patients in septic shock have a blunted cortisol response to stimulation with ACTH, despite high baseline serum cortisol levels.22,27 Hence, substitution with so-called moderate-stress or physiological-stress doses of 200–300 mg hydrocortisone daily during septic shock or 1–2 mg/kg methylprednisolone daily in acute respiratory distress syndrome was hypothesized to be beneficial.
Acute respiratory distress syndrome
Meduri et al.44 sought to confirm whether long-term glucocorticoid treatment during acute respiratory distress syndrome could reduce mortality. In this study, 24 patients with severe acute respiratory distress syndrome in whom lung injury score had not improved by day 7 were randomly assigned placebo or 2 mg/kg methylprednisolone daily for 2 weeks, with doses gradually tapered over at least another 2 weeks. Reductions were seen in the lung injury score and multiple organ dysfunction scores in the methylprednisolone group. Mortality while in intensive care was strikingly decreased from 62% to 0%, and hospital mortality from 62% to 12%. Aspects of the study methods have, however, caused concern, as the protocol contained a crossover provision for patients who did not respond after 10 days of therapy. Hence, in the placebo group four of eight patients were switched to methylprednisolone, albeit in a blinded fashion. In addition, the study was designed as a sequential clinical trial, in which sample size was not calculated in advance and data were examined in a series of interim analyses, and the study was stopped when there was sufficient evidence that the treatments differed significantly.
To confirm these results, a multicenter, placebo-controlled, double-blind trial was performed by the National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome Clinical Trials Network.45 The primary end point in this study was mortality at 60 days, with the number of ventilator-free days and organ-failure-free days being secondary end points. All patients with chronic respiratory disease and those taking corticosteroids were excluded, leaving 180 patients to be randomly assigned placebo or 2 mg/kg methylprednisolone daily for up to 25 days. Methylprednisolone increased the number of days free from ventilator use and organ failure before day 28 compared with placebo, but mortality was not lowered at day 60 (placebo 28.6% [95% CI 20.8–38.6%] versus methylprednisolone 29.2% [95% CI 20.8–39.4%]). The short-term benefits in improved lung function achieved with methylprednisolone appeared to be offset by increased return to assisted breathing. Moreover, methylprednisolone treatment increased mortality risk when started at least 14 days after the onset of acute respiratory distress syndrome and augmented the incidence of myopathy and neuropathy. Of note, the study suffered from slow recruitment—spanning a period of 7 years—during which time intensive-care practice might have evolved considerably.
As timing of methylprednisolone therapy seems to play a determining role in outcome, a third, important, randomized, double-blind, placebo-controlled trial was set up to study the effect of this drug (1 mg/kg daily) in 91 patients with early severe acute respiratory distress syndrome.46 Among these, 66% had concomitant sepsis, and a quarter had adrenal insufficiency at enrollment. All patients were enrolled within 3 days of the acute respiratory distress syndrome being diagnosed, and treatment lasted up to 28 days. Methylprednisolone therapy reduced the lung injury score, doubled the proportion of patients breathing without assistance, and halved intensive-care-unit mortality from 42.9% in the placebo group to 20.6% in the study group. A confounding factor in this study, however, was the use of open-label methylprednisolone treatment for patients whose lung injury had not improved by days 7–9.
In all three studies methylprednisolone did not increase the frequency of infectious complications; in fact, in the trials by Meduri et al.,44,46 incidence of infection was surveyed weekly by bronchoalveolar lavage.
On the basis of trials with mortality as the primary end point, the use of glucocorticoids, even at a low dose of 1 mg/kg methylprednisolone daily, cannot be systematically recommended in nonresolving acute respiratory distress syndrome, although a benefit on pulmonary function is quite likely. The controversy surrounding the use of corticosteroids in this syndrome is further highlighted by the contrasting findings of two meta-analyses, which included different trials and were limited by clinical and moderate statistical heterogeneity.47,48
Septic shock syndrome
The use of glucocorticoids during septic shock has also been promoted. The incentive has been the supplementation of relative adrenal insufficiency rather than the inhibition of the inflammatory response.
In the late 1990s several reports showed that administration of supraphysiologic doses of hydrocortisone reversed septic shock. Bollaert et al.49 and Briegel et al.50 demonstrated in 41 and 40 patients, respectively, that those receiving 300 mg hydrocortisone daily had earlier shock reversal and substantially shorter need for vasopressor support than those receiving placebo. These small trials led to the seminal multicenter study by Annane and co-workers,26 in which 300 patients with septic shock who were receiving vasopressor therapy were randomly assigned placebo or 200 mg hydrocortisone plus 50 µg fludrocortisone. This study's major strength was the use of 28-day mortality as end point. Additionally, relative adrenal insufficiency was tested with the short ACTH test. Hydrocortisone administration was associated with a reduction in the duration of vasopressor therapy and in mortality (10% absolute risk reduction) in patients who were unable to produce an adequate total cortisol response to ACTH.
The latter finding points to some complexity in the interpretation of the results of the Annane et al. trial. The effect of steroid supplementation on withdrawal of vasopressor therapy was significant in the entire study population (i.e. both responders and nonresponders to ACTH), yet the mortality reduction in the steroid supplementation group was present only in the patients who did not show an adequate cortisol response to ACTH. A confounding factor might have been introduced by the use of etomidate (a drug used as a narcotic for tracheal intubation but that is known to suppress adrenal function) in 72 (24%) patients, of whom 68 were classified as nonresponders to ACTH. Although the use of etomidate is an important feature of clinical practice, and patients who received this drug might benefit from treatment with hydrocortisone, these results do not allow generalization of such therapeutic effect of hydrocortisone replacement therapy to patients not receiving the drug.
There are some other methodological concerns relating to this pioneer study, such as the use of one-sided statistical testing, which basically excludes possible harmful effects of steroid administration from the analysis, the use of the Cox's proportional hazards model, which corrects for baseline differences between the randomized groups, and a power calculation that assumed 95% mortality in the nonresponder placebo subgroup. In hindsight, a more realistic sample size calculation would have shown that for a baseline mortality of 63%, 400 patients in each group of a parallel study, with two independent groups, would be needed to show an absolute risk reduction of 10% with a statistical power of 80%. These values are important for repeat studies on this topic.
To settle the controversy and evaluate the efficacy and safety of hydrocortisone therapy in a broad population of patients with septic shock, the Corticosteroid Therapy of Septic Shock (CORTICUS) study was performed.51 In this multicenter, randomized, double-blind, placebo-controlled trial patients received either placebo or 200 mg hydrocortisone daily for 5 days followed by a tapering period. The primary end point was mortality at 28 days in patients with inadequate total cortisol responses to ACTH. This trial confirmed that hydrocortisone treatment shortens the duration of time to shock reversal in the entire population of patients with septic shock (hydrocortisone 3.3 days [95% CI 2.9–3.9 days] versus placebo 5.8 days [95% CI 5.2–6.9 days]) as well as in responders to ACTH (hydrocortisone 2.8 days [95% CI 2.1–3.3 days] versus placebo 5.8 days [95% CI 5.2–6.9 days]). However, no mortality benefit could be shown at 28 days in nonresponders (hydrocortisone 39.2% [95% CI 30.5–47.9%] versus placebo 36.1% [95% CI 26.9–45.3%]) or the entire population (hydrocortisone 34.3% [95% CI 28.3–40.2%] versus placebo 31.5% [95% CI 25.6–37.3%]). Moreover, hydrocortisone supplementation increased the incidence of septic shock relapse, hyperglycemia and hypernatremia.
Two main factors could explain the negative findings in the above study. Firstly, because of slow recruitment the trial was stopped after only 500 of the planned 800 patients had been enrolled. Secondly, baseline mortality in the placebo subgroup of patients not responding to ACTH was much lower in the CORTICUS trial than in the landmark study by Annane et al.26 (36% versus 63%), which suggests a less-severely ill patient population. The combination of these factors resulted in an underpowered the study, which substantially weakens any negative results. The trial profile diagram was not published, thus preventing judgment on possible selection bias. In addition, the short ACTH test seemed not to satisfactorily predict which patients would benefit from hydrocortisone supplementation, despite being useful in the prediction of mortality.52
Other clinical conditions
An important study contributing to the debate on the benefit of using corticosteroids is the Corticosteroid Randomisation After Significant Head Injury (CRASH) trial.53 After numerous small studies hinted an advantage of corticosteroid administration in head trauma management, this large (n = 10,008), international, randomized, placebo-controlled trial showed that high-dose methylprednisolone increased the risk of death after considerable head injury. Other well-controlled studies suggested that corticosteroids prevent atrial fibrillation after cardiac surgery54 and laryngeal edema after extubation.55 The long term safety profile for these indications, however, remains unclear.
Eye to the future
As the clinical trials on the use of glucocorticoids in the acute respiratory distress syndrome and the septic shock syndrome remain inconclusive, further research on HPA axis function in critical illness is necessary. Ideally, the biological effects of steroids during critical illness should be assessed by measuring, for instance, glucocorticoid receptor activation, as total cortisol is rather nonspecific and free cortisol levels too hard to quantify directly. Furthermore, the behavior of the HPA axis in the chronic phase of critical illness, hallmarked by dysfunctional pituitary release of ACTH, deserves more attention. Stimulation of the HPA axis by upstream substitution with CRH or ACTH might, for example, be a better method to bring about the right level of HPA activity, but the clinical benefit of this approach is yet to be proven.56 Additionally, the interaction of the HPA axis with the somatotropic and thyrotropic axes and reactivation of the axes with releasing factors (growth-hormone-releasing hormone, growth-hormone-releasing peptide 2 and TSH-releasing hormone)1,2 would be an interesting area of research. As a counter-regulatory hormone, cortisol antagonizes the effects of insulin and aggravates hyperglycemia. As strict blood glucose control with intensive insulin therapy has been shown to decrease morbidity and mortality during critical illness,57,58 theoretically both strategies should be adopted in combination.
If the fundamentals of the concept of glucocorticoid supplementation are not better understood, drafting future guidelines for clinicians will remain a challenge. An absolute reduction in the risk of death in the intention-to-treat population seems unlikely to surpass the 10% mark (intensive insulin therapy 3–4% and drotrecogin α 6% absolute risk reduction);57,58,59 therefore, a study sufficiently powered to detect a difference in a clinically hard end point, such as mortality, would require a very large number of patients.
Conclusions
Undoubtedly, the discussion of whether glucocorticoids have a pivotal role in the management of inflammatory syndromes in critical illness will not be settled for some time. The major culprit for this ongoing debate is the great divide between the promising results in the original, small and thought-provoking studies that launched the concept and the less favorable data from the subsequent multicenter trials, which tried to evaluate these earlier findings in broader, real-life populations. Design-related features, such as slow recruitment of patient populations that differ from those in the original trials and too-small sample sizes, have compromised possible conclusions from these studies and hampered generalizability. The heterogeneity of the case-mix and lower risk of death is also not helpful in harnessing the statistical power of these studies.
While awaiting larger, adequately powered and targeted studies, designed by trials consortia,60 physicians should adopt a common sense policy for steroid use in individual patients. This approach is supported by a consensus statement from the American College of Critical Care Medicine61 and by the Surviving Sepsis Campaign guidelines62 (Box 3). Clinical decision-making based on results from the ACTH test remains a huge challenge, as the most appropriate interpretation of such results in the critically ill is unclear and the test cannot accurately define a patient population that will benefit from hydrocortisone therapy. An interesting alternative approach recommends a three-level test: first, clinical suspicion of adrenal insufficiency; second, basal cortisol testing; and finally, if necessary, the ACTH test with peak cortisol levels as the end-point (Figure 1).15 Moreover, judging on the presently available clinical data, glucocorticoids cannot be generally recommended as a routine adjuvant therapy, neither in acute respiratory distress nor in septic shock. Glucocorticoids have, however, earned their spot among other rescue strategies in subgroups with the highest mortality risk.

Abbreviation: ACTH, adrenocorticotropic hormone.15
Review criteria
Publications discussed in this Review were identified by searching the PubMed database. Different combinations of the following search terms were used: “critical illness”, “sepsis”, “acute respiratory distress syndrome (ARDS)”, “trauma”, “surgery”, “burn”, “endocrinology”, “hypothalamic-pituitary axis”, “adrenal”, “hormone”, “steroid”, “glucocorticoid”, “cortisol”, “corticotropin (ACTH)”, “corticotropin-releasing hormone (CRH)” and “dehydroepiandrosterone (sulfate)”. A manual search of some references cited in these papers was also performed. All selected papers were English-language, full-text articles. Some references identified could not be included because of space restrictions.
References
Van den Berghe G et al. (1998) Acute and prolonged critical illness as different neuroendocrine paradigms. J Clin Endocrinol Metab 83: 1827–1834
Vanhorebeek I et al. (2006) Endocrine aspects of acute and prolonged critical illness. Nature Clin Pract Endocrinol Metab 2: 20–31
Venkatesh B et al. (2007) Evidence of altered cortisol metabolism in critically ill patients: a prospective study. Intensive Care Med 33: 1746–1753
White PC et al. (1997) 11β-Hydroxysteroid dehydrogenase and the syndrome of apparent mineralocorticoid excess. Endocr Rev 18: 135–156
Prigent H et al. (2004) Corticotherapy in sepsis. Crit Care 8: 122–129
Venkataraman S et al. (2007) The hypothalamic–pituitary–adrenal axis in critical illness. Rev Endocr Metab Disord 8: 365–373
Prigent H et al. (2004) Mechanisms of impaired adrenal function in sepsis and molecular actions of glucocorticoids. Crit Care 8: 243–252
Cooper MS and Stewart PM (2003) Corticosteroid insufficiency in acutely ill patients. N Engl J Med 348: 727–734
Rivier C and Vale W (1983) Modulation of stress-induced ACTH release by corticotropin-releasing factor, catecholamines and vasopressin. Nature 305: 325–327
Rook GAW (1999) Glucocorticoids and immune function. Baillieres Best Pract Res Clin Endocrinol Metab 13: 567–581
Marik PE and Zaloga GP (2002) Adrenal insufficiency in the critically ill: a new look at an old problem. Chest 122: 1784–1796
Cooper MS et al. (2001) Modulation of 11β-hydroxysteroid dehydrogenase isozymes by proinflammatory cytokines in osteoblasts: an autocrine switch from glucocorticoid inactivation to activation. J Bone Miner Res 16: 1037–1044
Beishuizen A et al. (2001) Patterns of corticosteroid-binding globulin and the free cortisol index during septic shock and multitrauma. Intensive Care Med 27: 1584–1591
Vanhorebeek I et al. (2006) Cortisol response to critical illness: effect of intensive insulin therapy. J Clin Endocrinol Metab 91: 3803–3813
Cooper MS and Stewart PM (2007) Adrenal insufficiency in critical illness. J Intensive Care Med 22: 348–362
Pemberton PA et al. (1988) Hormone binding globulins undergo serpin conformational change in inflammation. Nature 336: 257–258
Hammond GL et al. (1990) A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab 71: 34–39
Hamrahian AH et al. (2004) Measurements of serum free cortisol in critically ill patients. N Engl J Med 350: 1629–1638
Vermes I and Beishuizen A (2001) The hypothalamic-pituitary-adrenal response to critical illness. Best Pract Res Clin Endocrinol Metab 15: 495–511
Bornstein SR and Chrousos GP (1999) Clinical review 104: adrenocorticotropin (ACTH)- and non-ACTH-mediated regulation of the adrenal cortex: neural and immune inputs. J Clin Endocrinol Metab 84: 1729–1736
Rothwell PM and Lawler PG. (1995) Prediction of outcome in intensive care patients using endocrine parameters. Crit Care Med 23: 78–83
Annane D et al. (2000) A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. J Am Med Assoc 283: 1038–1045
Arlt W et al. (2006) Dissociation of serum dehydroepiandrosterone and dehydroepiandrosterone sulfate in septic shock. J Clin Endocrinol Metab 91: 2548–2554
Marx C et al. (2003) Adrenocortical hormones in survivors and nonsurvivors of severe sepsis: diverse time course of dehydroepiandrosterone, dehydroepiandrosterone sulfate, and cortisol. Crit Care Med 31: 1382–1388
Arafah BM (2006) Hypothalamic pituitary adrenal function during critical illness: limitations of current assessment methods. J Clin Endocrinol Metab 91: 3725–3745
Annane D et al. (2002) Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA 288: 862–871
Rothwell PM et al. (1991) Cortisol response to corticotropin and survival in septic shock. Lancet 337: 582–583
Siraux V et al. (2005) Relative adrenal insufficiency in patients with septic shock: comparison of low-dose and conventional corticotropin tests. Crit Care Med 33: 2479–2486
Widmer IE et al. (2005) Cortisol response in relation to the severity of stress and illness. J Clin Endocrinol Metab 90: 4579–4586
Dickstein G (2005) On the term “relative adrenal insufficiency”—or what do we really measure with the adrenal stimulation tests? J Clin Endocrinol Metab 90: 4973–4974
Annane D (2005) Low-dose adrenocorticotropic hormone test is not ready for routine adrenal function testing in the intensive care unit. Crit Care Med 33: 2688–2689
Venkatesh et al. (2005) Evaluation of random cortisol and the low dose corticotropin test as indicators of adrenal secretory capacity in critically ill patients: a prospective study. Anaesth Intensive Care 33: 201–209
Ho JT et al. (2006) Septic shock and sepsis: a comparison of total and free plasma cortisol levels. J Clin Endocrinol Metab, 91: 105–114
Bendel S et al. (2008) Free cortisol in sepsis and septic shock. Anesth Analg 106: 1813–1819
Cohen et al. (2006) Variability of cortisol assays can confound the diagnosis of adrenal insufficiency in the critically ill population. Intensive Care Med 32: 1901–1905
Marik PE (2007) Mechanisms and clinical consequences of critical illness associated adrenal insufficiency. Curr Opin Crit Care 13: 363–369
Veterans Administration Systemic Sepsis Cooperative Study Group (1987) Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. N Engl J Med 317: 659–665
Bone RC et al. (1987) A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med 317: 653–658
Lefering R and Neugebauer EAM (1995) Steroid controversy in sepsis and septic shock: a meta-analysis. Crit Care Med 23: 1294–1303
Cronin L et al. (1995) Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med 23: 1430–1439
Luce JM et al. (1988) Ineffectiveness of high-dose methylprednisolone in preventing parenchymal lung injury and improving mortality in patients with septic shock. Am Rev Respir Dis 138: 62–68
Bernard GR et al. (1987) High dose corticosteroids in patients with adult respiratory distress syndrome. N Engl J Med 317: 1565–1570
Altura BM and Altura BT (1974) Peripheral vascular actions of glucocorticoids and their relationship to protection in circulatory shock. J Pharmacol Exp Ther 190: 300–315
Meduri GU et al. (1998) Effect of prolonged methylprednisolone therapy in unresolving acute respiratory stress syndrome: a randomized controlled trial. JAMA 280: 159–165
The National Heart, Lung, and Blood Institute Acute Respiratory Distress Syndrome Clinical Trials Network (2006) Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med 354: 1671–1684
Meduri GU et al. (2007) Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest 131: 954–963
Meduri GU et al. (2008) Steroid treatment in ARDS: a critical appraisal of the ARDS network trial and the recent literature. Intensive Care Med 34: 61–69
Peter JV et al. (2008) Corticosteroids in the prevention and treatment of acute respiratory distress syndrome (ARDS) in adults: meta-analysis. BMJ 336: 1006–1009
Bollaert P-E et al. (1998) Reversal of late septic shock with supraphysiologic doses of hydrocortisone. Crit Care Med 26: 645–650
Briegel J et al. (1999) Stress doses of hydrocortisone reverse hyperdynamic septic shock: a prospective, randomized, double-blind, single-center study. Crit Care Med 27: 723–732
Sprung CL et al. for the Corticus Study Group (2008) Hydrocortisone therapy for patients with septic shock. N Engl J Med 358: 111–124
Lipiner-Friedman D et al. for the Corticus Study Group (2007) Adrenal function in sepsis: the retrospective Corticus cohort study. Crit Care Med 35: 1012–1018
Roberts I et al. for the CRASH trial collaborators (2004) Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): a randomized placebo-controlled trial. Lancet 364: 1321–1328
Halonen J et al. (2007) Corticosteroids for the prevention of atrial fibrillation after cardiac surgery: a randomized controlled trial. JAMA 297: 1562–1567
François B et al. for the Association des Réanimateur du Centre-Ouest (ARCO) (2007) 12-h treatment with methylprednisolone versus placebo for prevention of postextubation laryngeal oedema: a randomized double-blind trial. Lancet 369: 1083–1089
Reincke M et al. (1993) The hypothalamic-pituitary-adrenal axis in critical illness: response to dexamethasone and corticotropin-releasing hormone. J Clin Endocrinol Metab 77: 151–156
Van den Berghe G et al. (2001) Intensive insulin therapy in critically ill patients. N Engl J Med 345: 1359–1367
Van den Berghe G et al. (2006) Intensive insulin therapy in medical intensive care patients. N Engl J Med 354: 449–461
Bernard GR et al. for the Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344: 699–709
Finfer S (2008) Corticosteroids in septic shock. N Engl J Med 358: 188–190
Marik PE et al. (2008) Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 36: 1937–1949
Dellinger RP et al. (2008) Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med 36: 296–327
Acknowledgements
Our work cited in this Review was supported by research grants from Catholic University of Leuven, Leuven and the Fund for Scientific Research, Flanders, Belgium. I Vanhorebeek is a Postdoctoral Fellow of the Fund for Scientific Research. Charles P Vega, University of California, Irvine, CA, is the author of and is solely responsible for the content of the learning objectives, questions and answers of the Medscape-accredited continuing medical education activity associated with this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Mesotten, D., Vanhorebeek, I. & Van den Berghe, G. The altered adrenal axis and treatment with glucocorticoids during critical illness. Nat Rev Endocrinol 4, 496–505 (2008). https://doi.org/10.1038/ncpendmet0921
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncpendmet0921
This article is cited by
-
Persisting neuroendocrine abnormalities and their association with physical impairment 5 years after critical illness
Critical Care (2021)
-
Relation between Baseline Total Serum Cortisol Level and Outcome in Pediatric Intensive Care Unit
Scientific Reports (2019)
-
Clinical characteristics of adrenal crisis in adult population with and without predisposing chronic adrenal insufficiency: a retrospective cohort study
BMC Endocrine Disorders (2017)
-
Glucocorticoids as an Emerging Pharmacologic Agent for Cardiopulmonary Resuscitation
Cardiovascular Drugs and Therapy (2014)
-
Corticosteroids for severe sepsis: an evidence-based guide for physicians
Annals of Intensive Care (2011)
