
Abstract
GATA zinc finger domain-containing 2B (GATAD2B) is a subunit of the methyl-CpG-binding protein-1 complex (MECP1), which deacetylates methylated nucleosomes and regresses transcriptional activity. Recently, GATAD2B has been elucidated as a candidate gene in patients with intellectual disability (ID). In this study, we identified two novel heterozygous frameshift mutations of GATAD2B in two unrelated ID cases through next-generation sequencing (NGS). Both of the mutations c.80_81insGATGT and c.552_555delGAAA cause truncated proteins that might be detrimental to neurodevelopment. We performed western blotting and observed a reduction in the target protein compared with normal controls. This is the first report of GATAD2B in Chinese ID patients. Our findings will broaden the spectrum of GATAD2B mutations and facilitate genetic diagnosis and counseling.
Similar content being viewed by others


Introduction
Intellectual disability (ID) is a genetically heterogeneous disease representing a series of neurological dysfunctions, with a prevalence estimated at 1–3%.1 Patients usually suffer from conceptual, social and practical dysfunction at an early stage, generally before the age of 18.2 With the burgeoning studies on ID heredity in recent decades, the role of genetic factors in ID has been made clearer than ever before.3
GATAD2B is located on chromosome 1q21.3 and the coding region translates into a protein named p66β. Recent studies indicate that GATAD2B plays a significant role in ID patients and functional studies in Drosophila illustrate the importance for normal synapse development.4
Here we describe two Chinese cases exhibiting obvious intellectual disability with novel de novo GATAD2B frameshift mutations identified by next-generation sequencing (NGS). Protein immunoblotting results illustrated the changes in quantity of the target protein in the patients. Our findings support previous research, adding GATAD2B to the list of ID-associated genes.
Materials and methods
Patients
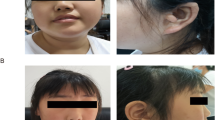
Individual 1 (Figures 1a and b), a 4-year-old boy, came to consult our geneticist for ID. His facial features included ocular hypertelorism, antimongoloid slant of palpebral fissure and flat nasal bridge. He could understand simple instructions although language developmental delay was obvious because he could only speak very few words. He could not walk stably. Gesell developmental schedules revealed that the developmental quotients of gross motor (DQ score 41), fine motor (DQ score 75), language (DQ score 19), adaptability (DQ score 57) and individuality (DQ score 46). Other family members did not suffer from neurological disorders. Recently, we paid a visit to the patient, who is now 11 years old. He was 151 cm tall with a normal physique, but still could only speak two or three words. He could walk but not run normally. He studied in a special education school and presented no obvious social problems.

Frontal and lateral photos of individual 1 and individual 2 with de novo GATAD2B mutations. Facial images of individual 1 (a, b) at 11 years old and individual 2 (c–e) at 12 years old. Note the common facial characteristics of small palpebral fissure, ocular hypertelorism and flat nasal bridge. A full color version of this figure is available at the Journal of Human Genetics journal online.
Individual 2 (Figures 1c–e) came to consult our geneticist when she was 9 years old. She was the first child of a non-consanguineous couple and was born at full-term after a normal pregnancy. Her facial dysmorphism included small palpebral fissure, ocular hypertelorism, a flat, low nasal bridge and dental misalignment. She could sit alone at 10 months, stand alone at 1 year and walk alone at 2 years old. The patient could not clearly speak a single word and showed a tendency toward hyperactivity. Intellectual test at 9 years showed DQ scores for each item far below the normal level (data not available). The patient was from a sporadic pedigree and had two unaffected siblings. Recently, we paid a visit to the patient, who is now 12 years old. She was 148 cm tall and still could not speak a word. She enjoyed running and playing alone. She could eat independently but could not wash her face.
Written informed consent and clinical information were acquired from the parents, and the research was approved by the local institutional review board.
Next-generation sequencing
Peripheral blood samples were obtained for genomic DNA extraction. In this study, we used molecular inversion probes designed to capture the coding regions of ID-associated genes. The library was purified, quantified and sequenced on an Illumination Hiseq 2000 sequencer, which generated paired-end 150-bp reads matched through the distinctive barcode. The sequence reads were aligned to the reference human genome (hg19) and the position information of the sequence was obtained according to the specific barcode on the probe. After variants were annotated, all variants present in dbSNP (MAF⩾0.005), the NHLBI exome sequencing project exome variant server (NHLBI-ESP 6500) and the 1000 Genomes Project were removed. Candidate variants were validated by Sanger sequencing.
Western blot analysis
Target protein was extracted from immortalized lymphoblast cells of patients and healthy controls. Western blotting was performed to analyze the protein according to standard methodology. The protein product was separated by 8% SDS-PAGE and transferred onto a PVDF membrane. The membrane was incubated overnight at 4 °C with rabbit anti-human GATAD2B polyclonal antibody (Origene, Rockville, MD, USA) and mouse polyclonal anti-β-actin antibody (Sigma, St Louis, MO, USA), followed by detection with goat anti-rabbit/mouse immunoglobulin G antibody (Jackson, West Grove, PA, USA).
Results and Discussion
With the burgeoning studies on genetic testing in recent decades, NGS can be regarded as a milestone on the way to efficient and precise genetic diagnosis due to its decreased cost and increased throughput.5 We used molecular inversion probes (MIPs) to screen the coding region of ID-associated genes, which requires lower per-sample cost and sample input requirements.6, 7
Two mutations in GATAD2B were identified in the two unrelated ID patients: c.80_81insGATGT in case 1 (Figure 2a) and c.552_555delGAAA in case 2 (Figure 2b). The mutation c.80_81insGATGT locates upstream of conserved region 1 (CR1) domain, which will result in a polypeptide that includes 28 normal amino acids (p.L28Mfs*18). Another mutation c.552_555delGAAA is in the CR1 region (Figure 2c) and can lead to the truncated protein p.K184Nfs*2. Sanger sequencing confirmed that mutations in the two cases occurred de novo in their respective families. Western blotting demonstrated that GATAD2B expression was significantly higher in immortal lymphoblast cells of healthy controls than in the mutated samples with the same level of β-actin (Figure 3). As the truncated peptide in case 1 just included 28 normal amino acids, which was too short to be recognized by the antibody, it resulted in a weaker band than healthy controls. The resulting aberrant mRNA in case 2 was predicted to yield truncated GATAD2B proteins while the truncated proteins were not detected in triplicate experiments, we speculate that the truncated proteins were likely to be degraded, explaining the lower intensity of the corresponding band versus healthy controls.

(a) Sanger sequencing analysis of case 1 showed a de novo frameshift mutation in c.80_81insGATGT (p.L28Mfs*18). (b) Sanger sequencing analysis of case 2 showed a de novo frameshift mutation in c.552_555delGAAA (p.K184Nfs*2). (c) Schematic presentation of GATAD2B protein. The arrows above the diagram indicate the positions of the two mutations identified in this study and the arrows below show the genotype of patients in the literature mentioned before. CR1, conserved region 1; CR2, conserved region 2.

Western blotting of total proteins extracted from peripheral blood leukocytes. Lanes 1 and 2: leukocytes from healthy controls; lane 3: leukocytes from case 1; lane 4: leukocytes from case 2 probed with an anti-GATAD2B antibody against the N-terminal amino acids. The blots were probed with an anti-actin antibody as a control for protein loading.
Considering the two individuals together with four previous patients associated with GATAD2B mutations in the literature, there appear to be some obvious overlapping characteristics.4 Facial features like hypertelorism and narrow palpebral fissures were noticed in all of the reported patients in the literature mentioned before, and all of the four patients displayed very limited language competence, which are the same to our two cases. In the literature, all of the four patients showed motor ability development delay, one of them (p.N195Kfs*30) showed motor skills impairment at 6 years old and another one (p.Q190Afs*34) showed hyperactivity at the age of 12. Similarly, in this report, the two patients both learnt to walk without support later than normal children, motor skills impairment was recognized in case 1 (p.L28Mfs*18) as he could not run stably, while the patient of case 2 (p.K184Nfs*2) exhibited hyperactive and autistic tendency.
The nucleosome remodeling and histone deacetylase (NuRD) complex was highly conserved during evolution.8 The transcriptional repressor p66β, a subunit of MeCP1 encoded by GATAD2B, is related to chromatin modification and transcriptional repression.9 P66β contains two highly conserved regions CR1 and CR2, and the main function of the protein is enhancing MBD2-mediated repression and targeting the chromatin through interactions with MBD2.9, 10 Studies have shown that the conserved regions CR1 and CR2 domains of p66β are essential for gene silencing.11, 12 Further studies about GATAD2B loss-of-function research demonstrates that GATAD2B is necessary for synaptic morphology and function in Drosophila neurons and neuronal knockdown of the orthologous gene can lead to adaptability dysfunction, which indicates its contribution to the ID phenotype.4
In summary, we describe ID in two unrelated individuals owing to GATAD2B frameshift mutations. Our research validates the importance of GATAD2B in neurodevelopment and supports the statement that ID could be incurred due to GATAD2B haploinsufficiency. Our findings broaden the spectrum of GATAD2B mutations and facilitate genetic diagnosis and counseling.
References
Srivastava, A. K. & Schwartz, C. E. Intellectual disability and autism spectrum disorders: causal genes and molecular mechanisms. Neurosci. Biobehav. Rev. 46 (Pt 2), 161–174 (2014).
Vissers, L. E., Gilissen, C. & Veltman, J. A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 17, 9–18 (2016).
de Ligt, J., Willemsen, M. H., van Bon, B. W., Kleefstra, T., Yntema, H. G., Kroes, T. et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 367, 1921–1929 (2012).
Willemsen, M. H., Nijhof, B., Fenckova, M., Nillesen, W. M., Bongers, E. M., Castells-Nobau, A. et al. GATAD2B loss-of-function mutations cause a recognisable syndrome with intellectual disability and are associated with learning deficits and synaptic undergrowth in Drosophila. J. Med. Genet. 50, 507–514 (2013).
Xue, Y., Ankala, A., Wilcox, W. R. & Hegde, M. R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet. Med. 17, 444–451 (2015).
Hiatt, J. B., Pritchard, C. C., Salipante, S. J., O'Roak, B. J. & Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 23, 843–854 (2013).
O'Roak, B. J., Vives, L., Fu, W., Egertson, J. D., Stanaway, I. B., Phelps, I. G. et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622 (2012).
Kloet, S. L., Baymaz, H. I., Makowski, M., Groenewold, V., Jansen, P. W., Berendsen, M. et al. Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J. 282, 1774–1785 (2015).
Feng, Q., Cao, R., Xia, L., Erdjument-Bromage, H., Tempst, P. & Zhang, Y. Identification and functional characterization of the p66/p68 components of the MeCP1 complex. Mol. Cell. Biol. 22, 536–546 (2002).
Brackertz, M., Boeke, J., Zhang, R. & Renkawitz, R. Two highly related p66 proteins comprise a new family of potent transcriptional repressors interacting with MBD2 and MBD3. J. Biol. Chem. 277, 40958–40966 (2002).
Allen, H. F., Wade, P. A. & Kutateladze, T. G. The NuRD architecture. Cell. Mol. Life Sci. 70, 3513–3524 (2013).
Lai, A. Y. & Wade, P. A. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer 11, 588–596 (2011).
Acknowledgements
We thank the patients and their families for their anticipation. This work was supported by National Key Basic Research Program of China (2012CB944600).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Luo, X., Zou, Y., Tan, B. et al. Novel GATAD2B loss-of-function mutations cause intellectual disability in two unrelated cases. J Hum Genet 62, 513–516 (2017). https://doi.org/10.1038/jhg.2016.164
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.164
