
Abstract
The strain SCSIO 01127, isolated from the South China Sea sediment, was identified as a member of Streptomyces by the 16S rDNA sequence analysis. Two new spirotetronate antibiotics lobophorins E (1) and F (2), along with two known analogs lobophorins A (3) and B (4), were isolated from Streptomyces sp. SCSIO 01127. Their structures were elucidated on the basis of detailed IR, NMR and MS spectroscopic analyses. The new compound lobophorin F (2) showed antibacterial activities against Staphylococcus aureus ATCC 29213 and Enterococcus faecalis ATCC 29212 with MIC values of 8 μg ml−1 for both the strains, better than that of lobophorin B (4). Lobophorin F (2) also displayed better cytotoxic activities than lobophorin B (4), with IC50 of 6.82, 2.93 and 3.16 μM against SF-268, MCF-7 and NCI-H460, respectively.
Similar content being viewed by others
Introduction
In recent years, more and more novel genera and species of marine actinomycetes have been isolated from sediments of the South China Sea,1, 2, 3, 4, 5 suggesting South China Sea to be a rich source for exploring new marine actinomycete taxa. In the past two decades, marine actinomycetes are emerging as the most prolific new sources for novel drug discovery and have produced lots of secondary metabolites exhibiting excellent biological activities.6, 7, 8 Recently, Marinactinospora thermotolerans SCSIO 00606, a newly identified novel genus from the South China Sea, was found to produce new γ-pyrones, marinactinones A-C, which showed moderate antitumor activity and weak DNA topoisomerase II-inhibiting activity.9 This discovery inspires our particular interests in exploring the numerous actinobacterial strains collected from the South China Sea for their potential in producing novel compounds. During our continuous searching efforts, a deep-sea actinobacterial strain SCSIO 01127 caught our attention, for that the organic extract of the fermentation broth of this strain showed excellent antibacterial activities. Here we report the preliminary characterization of the strain SCSIO 01127 as a member of the Streptomyces genus by 16s rDNA analysis, the isolation and structural elucidation of four spirotetronate antibiotics. Two compounds were identified to be novel lobophorin analogues, designated lobophorins E (1) and F (2), and the other two were found to be lobophorins A (3) and B (4).10 Interestingly, lobophorin F (2) exhibited better cytotoxic activities against three tumor cell lines (SF-268, MCF-7 and NCI-H460) and better antibacterial activities than lobophorin B (4).
Materials and Methods
General experimental procedures
Materials for column chromatography were silica gel (100–200 mesh; 300–400 mesh; Jiangyou Silica gel development, Yantai, PR China), Sephadex LH-20 (40–70 μm; Amersham Pharmacia Biotech AB, Uppsala, Sweden), and YMC*GEL ODS-A (12 nm S-50 μm; YMC Company, Kyoto, Japan). TLC(0.1–0.2 mm or 0.3–0.4 mm) was conducted with pre-coated glass plates (silica gel GF254, 10–40 nm, Jiangyou Silica gel development). Medium-pressure liquid chromatography was performed on automatic flash chromatography (EZ Purifier III, Leisure Science, Shanghai, China) with monitor wavelength of 210 nm and collector wavelength of 263 nm. Mass spectral data were obtained on a quadrupole-time-of-flight MS (Waters, Milford, MA, USA) for high-resolution FAB-MS. The optical rotation was recorded on a 341 Polarimeter (Perkin Elmer, Norwalk, CT, USA). UV spectra were obtained during detection of the monomer compounds on a Varian ProStar 210 HPLC (Varian Australia Pty., Ltd., Mulgrave, VIC, Australia). IR spectra were obtained in KBr with a FT-IR EQUINOX 55 Bruker (Bruker Optik GmbH, Ettlingen, Germany). 1H, 13C NMR and 2D NMR spectra were recorded on a Bruker AV-500 NMR spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) with tetramethylsilane (δ 0 p.p.m.) as the internal standard. 1H NMR data were reported as follows: chemical shift (multiplicity: singlet (s), doublet (d), triplet (t), and multiplet (m); coupling constants: Hz); 13C NMR data were reported as follows: chemical shift (quaternary carbon (s), methine (d), methylene (t) and methyl (q)). Deuterated NMR solvents were purchased from Cambridge Isotopes (Andover, MA, USA).
Isolation, characterization and fermentation of the strain SCSIO 01127
The strain SCSIO 01127 was isolated from a sediment sample (E 111°54.693′, N 08°56.003′) at the depth of 1350 m in the South China Sea from an open voyage in July 2007, after incubation at 28 °C for 2 weeks on Gauze's Medium No. 1 (soluble starch 2%, KNO3 0.1%, K2HPO4 0.05%, MgSO4·7H2O 0.05% and FeSO4·7H2O 0.01% at pH 7.4), and was deposited in the type culture collection of Center for Marine Microbiology, Research Network of Applied Microbiology, South China Sea Institute of Oceanology and Chinese Academy of Sciences, Guangzhou, China. Genomic DNA isolation, PCR amplification of 16S rDNA, sequence comparison and phylogenetic tree construction of the strain SCSIO 01127 were performed as described previously.1 A single colony of SCSIO 01127 was inoculated into 50 ml of seed medium (soybean meal 0.3%, yeast extract 0.3%, trehalose 1%, proline 0.1%, beef extract 0.3%, glycerol 0.6%, K2HPO4 0.03%, MgSO4·7H2O 0.05%, FeSO4·7H2O 0.01%, CaCO3 0.2% and sea salt 3% at pH 7.4 before sterilization) in 250-ml Erlenmeyer flasks, and was cultured on a rotary shaker at 200 r.p.m., 28 °C for 2 days. A total of 5 ml of seed cultures were transferred into 50 ml production medium (the same as the seed medium) in 250 ml Erlenmeyer flasks, and were cultured on a rotary shaker at 200 r.p.m., 28 °C for 5 days.
Isolation of lobophorins from SCSIO 01127
The 4.5-l fermentation broth was extracted four times with 17 l butanone to afford residue A after removal of the solvent. The mycelia cake was extracted three times with 4 l acetone. After removing acetone, the residue was re-extracted by 4 l butanone to afford residue B upon removal of the solvent. Residues A and B were combined as the crude extract for further isolation. The crude extract was subjected to silica gel column chromatography (300–400 mesh, 150 g), and eluted with CHCl3/CH3OH (100:0–5:1) to give three fractions (Fr.1–Fr.3). Fr.1 was subjected to Sephadex LH-20, and eluted with CHCl3/CH3OH (1:1), to give two sub-fractions (Fr.1-1 and Fr.1-2). Fr.1-1 was first purified by preparative TLC on a silica gel plate (20 × 20 cm) developed with CHCl3/CH3OH (30:1), and then by semi-preparative HPLC on a Varian Star Workstation, using a reverse-phase column C18 (YMC*GEL ODS-A, 120A S-5 μm, 250 × 10 mm) with UV detection at 263 nm, to afford compound 2 (11.5 mg). The following HPLC program was used: solvent A, 10% acetonitrile in water; solvent B, 90% acetonitrile in water; 40% B to 100% B (linear gradient, 0–9 min), 100% B (9–17 min), 100% B to 40% B (17–18 min), 40% B (18–26 min); the flow rate was 2.5 ml min−1. Fr.1-2 was purified by semi-preparative C18 reverse-phase medium-pressure liquid chromatography (YMC*GEL ODS-A, 12 nm S-50 μm, 30 × 2.5 cm), with a linear gradient elution (CH3OH/H2O 10–90%, 15 ml min−1, 160 min). The desired fractions were further purified by semi-preparative HPLC to get compound 1 (2.5 mg), under the above mentioned conditions with a different developing program: 40% B to 100% B (linear gradient, 0–11 min), 100% B (11–16 min), 100% B to 40% B (16–17 min) and 40% B (17–23 min).
Fr. 2 was subjected to Sephadex LH-20 chromatography, and eluted with CHCl3/CH3OH (1:1). The desired fractions were further purified by medium-pressure liquid chromatography (YMC*GEL ODS-A, 12 nm S-50 μm, 30 × 2.5 cm). After elution with a linear gradient (CH3OH/H2O 10–60%, 15 ml min−1, 160 min), compound 3 (56.2 mg) was obtained. Fr. 3 was first separated by Sephadex LH-20 chromatography with CHCl3/CH3OH (1:1) elution, and then by medium-pressure liquid chromatography (YMC*GEL ODS-A, 12 nm S-50 μm, 30 × 2.5 cm), with a linear gradient elution (CH3OH/H2O 15–95%, 15 ml min−1, 160 min), to afford compound 4 (210.6 mg).
Structure determination
Lobophorin E (1): White powder, [α]20D−20.0 (c=0.38, MeOH), UV (in CH3CN: H2O: trifluoroacetic acid) 214.9, 263.1 nm. IR (KBr) 3522, 3439, 2932, 1734, 1627, 1545 and 1454 cm−1. 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Table 1; HR-MS m/z 1169.5874 (calcd for C61H89N2O20, [M–H]− 1169.6009); ESI-MS m/z 1194.1 [M+Na]+, 1170.2 [M−H]−.
Lobophorin F (2): White powder, [α]20D−60.0 (c=0.76, MeOH), UV (in CH3CN: H2O: trifluoroacetic acid) 210.6, 264.1 nm. IR (KBr) 3440, 2934, 1737, 1629, 1545 and 1454 cm−1. 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Table 1; HR-MS m/z 1049.5160 (calcd for C54H78N2O17Na, [M+Na]+ 1049.5198); ESI-MS m/z 1050.0 [M+Na]+, 1026.0 [M−H]−.
Lobophorin A (3): White powder, ESI-MS: m/z 1157.8 [M+H]+, 1155.9 [M−H]−.
Lobophorin B (4): White powder, 1H NMR (500 MHz, CDCl3) and 13C NMR (125 MHz, CDCl3), see Table 1; ESI-MS m/z 1210.3 [M+Na]+, 1186.1 [M−H]−.
Biological activity
MIC values of lobophorins (1–4) were determined against three bacterial strains, including Staphylococcus aureus ATCC 29213, Enterococcus faecalis ATCC 29212 and Bacillus thuringiensis SCSIO BT01, according to previously described methods.11 Cytotoxicities of lobophorins F (2), A (3) and B (4) were assayed against three tumor cell lines, including MCF-7 cells (human breast adenocarcinoma cell line), NCI-H460 (human non-small cell lung cancer cell line) and SF-268 (human glioma cell line). Assays were performed as described previously.12
Results and Discussion
Taxonomy of the producing strain
The producing strain SCSIO 01127, with a colonial morphology characteristic for actinobacteria, was isolated from deep-sea sediment of the South China Sea. The 16S rDNA of SCSIO 01127 was PCR amplified, sequenced and submitted to GenBank (accession number is JF794566). The phylogenetic tree generated by a neighbor-joining method clearly revealed the evolutionary relationship of the strain SCSIO 01127 with a group of Streptomyces species (Figure 1). Blast search showed that the new isolate had the highest similarities to Streptomyces olivaceus NBRC 12805T (AB249920) (100%) and Streptomyces pactum NBRC 13433T (100%). Thus, this strain was designated as Streptomyces sp. SCSIO 01127.

Phylogenetic dendrogram of the strain SCSIO 01127 and its closest relatives reconstructed by the neighbor-joining method on the basis of 16S rDNA gene sequences.
Structural elucidation
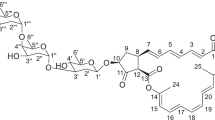
Compound 3 was determined to be lobophorin A by comparing the 1H and 13C NMR spectra data with those previously reported.10 Compound 4 was determined to be lobophorin B by comparing 1H, 13C, 1H-1H COSY and HMBC NMR spectral data with those reported in literature (Table 1).10 The molecular formula of compound 1 was established as C61H89N2O20 by HR-MS (m/z 1169.5874 [M−H]−, calcd 1169.6009). The IR spectrum of 1 showed absorption bands characteristic of hydroxyl (3522 and 3439 cm−1), methylene (2932 cm−1), carboxyl (1734 cm−1) and olefinic groups (1627, 1545 and 1454 cm−1). The 1H and 13C NMR spectra of 1 were almost identical to those of lobophorin B (4).10 The difference between 1 and 4 is that the oxygenated methylene (δH 4.15 (2H, m, H-32) and δC 64.7 (t, C-32)) in 4 was replaced by a methyl singlet (δH 1.80 (3H, s, Me-32) and δC 21.8 (q, Me-32)) in 1, indicating the absence of C-32-OH in 1 (Table 1). Consistently, the HMBC correlations from proton signals of the methyl singlet H3-32 to C-21, C-22 and C-23 confirmed the location of this methyl group at C-32 in 1. On the basis of these data, 1 was identified to be 32-deshydroxyl-lobophorin B, designated as lobophorin E (Figure 2).

Chemical structures of lobophorins 1–4.
According to the HR-MS analysis, the molecular formula of compound 2 (m/z 1049.5160 [M+Na]+, calcd 1049.5198) was established as C54H78N2O17. The IR spectrum of 2 showed absorption bands corresponding to hydroxyl (3440 cm−1), methylene (2934 cm−1), carboxyl (1737 cm−1) and olefinic groups (1629, 1545 and 1454 cm−1). In comparison of the 1H and 13C NMR spectra data of 2 with 1, the signals of 4-O-methyl-L-digitoxose moiety (sugar C, Figure 2) in 1 were absent in compound 2. Consistent with the loss of this sugar moiety in 2, 9.7 p.p.m. upfield shifting was observed for C-4B (δC 72.5) in the sugar B moiety (Figure 2). In accordance with this change occurring at C-4B, the COSY correlation from H-4B to H-3B and H-5B, and the HMBC correlation from H-6B to C-4B were observed (Figure 3). Based on these data, the structure of compound 2 was determined to be lobophorin F (Figure 2).

Selected key 1H-1H COSY and HMBC correlations of lobophorin F (2).
Biological activity
Antibacterial activities of lobophorins (1–4) were evaluated against S. aureus ATCC 29213, E. faecalis ATCC 29212 and B. thuringensis SCSIO BT01 (Table 2). All four compounds exhibited activities against B. thuringensis SCSIO BT01 with MIC values of 8, 2, 8 and 2 μg ml−1, respectively. Compounds 1, 3 and 4 showed no activities or weak activities against E. faecalis ATCC 29212 and S. aureus ATCC 29213 (Table 2). In contrast, compound 2 (lobophorin F) displayed antibacterial activities against these two strains with MIC values of 8 μg ml−1. Compounds 2–4 were also assayed for in vitro cytotoxic activities against three human tumor cell lines: SF-268, MCF-7 and NCI-H460. Compound 2 showed overall better cytotoxic activities against these three tumor cell lines than 4, (Table 2) with almost ninefold improvement against NCI-H460 (3.16 and 26.6 μM for 2 and 4, respectively). However, no cytotoxities were observed for compound 3 (lobophorin A) for all assayed three cell lines (>100 μM).
The spirotetronate antibiotics comprise of a class of antibacterial and antitumor natural products, such as antlermicin,13 chlorothricin,14 kijanimicin,15 decatromicins,16 saccharocarcins,17 tetrocarcins18 and versipelostatins.19 A pair of spirotetranate antibiotics lobophorins A (3) and B (4), previously isolated from an alga-associated acintobacterium, were shown to exhibit anti-inflammatory but no antimicrobial activities.10 Very recently, two C-8 stereoisomers of 3 and 4, named lobophorins C and D, were isolated from a marine sponge-related Streptomyces strain and were shown to have selective cytotoxicities against different cell lines.20 Interestingly, our study reveals that the absence of the C-32 hydroxyl group in lobophorins E (1) and F (2), when compared with compound 4 (>128 μg ml−1), significantly enhances their antimicrobial properties against S. aureus ATCC 29213: >16-fold improvement with 2 (8 μg ml−1). In comparison with the MIC values of 1 and 2, and the cytotoxicities of 2 and 4, (Table 2) one could also infer that the presence of the terminal sugar moiety (4-O-L-digitoxose, sugar C, Figure 2) is disadvantageous for the antimicrobial and antitumor property. The cytotoxicity of 4 is significantly better than that of 3 (Table 2), indicating that the presence of the nitro-sugar moiety is critical. These data also support that the change of sugar moieties can define, alter or mask natural product bioactivity.21, 22 Finally, this study highlights the need of engineering the lobophorin biosynthetic pathway in Streptomyces sp. SCSIO 01127, to rationally design hydroxylation and glycosylations, for directed production of lobophorin F (2) or other novel analogs with improved bioactivities.
Accession codes
References
Tian, X. P. et al. Sciscionella marina gen. nov., sp. nov., a marine actinomycete isolated from a sediment in the northern South China Sea. Int. J. Syst. Evol. Microbiol. 59, 222–228 (2009).
Tian, X. P. et al. Streptomyces nanshensis sp. nov., isolated from the Nansha Islands in the South China Sea. Int. J. Syst. Evol. Microbiol. 59, 745–749 (2009).
Tian, X. P. et al. Marinactinospora thermotolerans gen. nov., sp. nov., a marine actinomycete isolated from a sediment in the northern South China Sea. Int. J. Syst. Evol. Microbiol. 59, 948–952 (2009).
Dai, H. Q. et al. Verrucosispora sediminis sp. nov., a cyclodipeptide-producing actinomycete from deep-sea sediment. Int. J. Syst. Evol. Microbiol. 60, 1807–1812 (2010).
Wang, J. et al. Prauserella marina sp. nov., isolated from ocean sediment of the South China Sea. Int. J. Syst. Evol. Microbiol. 60, 985–989 (2010).
Bull, A. T. & Stach, J. E. Marine actinobacteria: new opportunities for natural product search and discovery. Trends. Microbiol. 15, 491–499 (2007).
Goodfellow, M. & Fiedler, H. P. A guide to successful bioprospecting: informed by actinobacterial systematics. Antonie Van Leeuwenhoek 98, 119–142 (2010).
Olano, C., Mendez, C. & Salas, J. A. Antitumor compounds from marine actinomycetes. Mar. Drugs 7, 210–248 (2009).
Wang, F., Tian, X., Huang, C., Li, Q. & Zhang, S. Marinactinones A-C, new gamma-pyrones from marine actinomycete Marinactinospora thermotolerans SCSIO 00606. J. Antibiot. 64, 189–192 (2011).
Jiang, Z. D., Jensen, P. R. & Fenical, W. Lobophorins A and B, new antiinflammatory macrolides produced by a tropical marine bacterium. Bioorg. Med. Chem. Lett. 9, 2003–2006 (1999).
Xiao, Y. et al. Characterization of tiacumicin B biosynthetic gene cluster affording diversified tiacumicin analogues and revealing a tailoring di-halogenase. J. Am. Chem. Soc. 133, 1092–1105 (2011).
Wu, Z. C., Li, D. L., Chen, Y. C. & Zhang, W. M. A new isofuranonaphthalenone and benzopyrans from the endophytic fungus Nodulisporium sp. A4 from Aquilaria sinensis. Helv. Chim. Acta 93, 920–924 (2010).
Kobinata, K., Uramoto, M., Mizuno, T. & Isono, K. A new antibiotic antlermicin A. J. Antibiot. 33, 244–246 (1980).
Keller-Schierlein, W., Muntwyler, R., Pache, W. & Zähner, H. Metabolic products of microorganisms. Chlorothricin and deschlorothricin. Helv. Chim. Acta. 52, 127–142 (1969).
Mallams, A. K., Puar, M. S., Rossman, R. R., McPhail, A. T. & Macfarlane, R. D. Kijanimicin. 2. Structure and absolute stereochemistry of kijanimicin. J. Am. Chem. Soc. 103, 3940–3943 (1981).
Momose, I. et al. Decatromicins A and B, new antibiotics produced by Actinomadura sp. MK73-NF4. I. Taxonomy, isolation, physico-chemical properties and biological activities. J. Antibiot. 52, 781–786 (1999).
Hegde, V. R., Patel, M. G., Das, P. R., Pramanik, B. & Puar, M. S. A family of novel macrocyclic lactones, the saccharocarcins produced by Saccharothrix aerocolonigenes subsp. antibiotica. II. Physico-chemical properties and structure determination. J. Antibiot. 50, 126–134 (1997).
Tomita, F. et al. Novel antitumor antibiotics, tetrocarcins. J. Antibiot. 33, 668–670 (1980).
Park, H.- R., Furihata, K., Hayakawa, Y. & Shin-ya, K. Versipelostatin, a novel GRP78/Bip molecular chaperone down-regulator of microbial origin. Tetrahedron Lett. 43, 6941–6945 (2002).
Wei, R. B. et al. Lobophorin C and D, new Kijanimicin derivatives from a marine sponge-associated actinomycetal strain AZS17. Mar. Drugs 9, 359–368 (2011).
Griffith, B. R., Langenhan, J. M. & Thorson, J. S. ‘Sweetening’ natural products via glycorandomization. Curr. Opin. Biotechnol. 16, 622–630 (2005).
Zhang, C. et al. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science 313, 1291–1294 (2006).
Acknowledgements
This study was supported in part by the funds of the 973 program (2010CB833805), the Chinese Academy of Sciences for Key Topics in Innovation Engineering (KSCX2-YW-G-065, KZCX2-YW-JC202, LYQY200805 and KSCX2-EW-G-12), National Science Foundation for Young Scientists of China (41006089), Natural Science Funds of South China Sea Institute of Oceanology for Young Scholar (SQ200903) and Open Project Program of Key Laboratory of Marine Bio-resources Sustainable Utilization (LMB091013). CZ is a scholar of the ‘100 Talents Project’ of the Chinese Academy of Sciences (08SL111002). We are grateful to Ms Xiao, Ms Sun and Mr Li of the South China Sea Institute of Oceanology, CAS for recording NMR data, Mr Liu and Ms Jia of South China Botanical Garden, CAS for MS data and OR data and Ms Rui of Guangdong Pharmaceutical University for HR-MS data.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Niu, S., Li, S., Chen, Y. et al. Lobophorins E and F, new spirotetronate antibiotics from a South China Sea-derived Streptomyces sp. SCSIO 01127. J Antibiot 64, 711–716 (2011). https://doi.org/10.1038/ja.2011.78
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2011.78
Keywords
This article is cited by
-
Redefining bioactive small molecules from microbial metabolites as revolutionary anticancer agents
Cancer Gene Therapy (2024)
-
Promising bioactive compounds from the marine environment and their potential effects on various diseases
Journal of Genetic Engineering and Biotechnology (2022)
-
Genomic insight into a novel actinobacterium, Actinomadura rubrisoli sp. nov., reveals high potential for bioactive metabolites
Antonie van Leeuwenhoek (2021)
-
Streptomyces sp. VN1, a producer of diverse metabolites including non-natural furan-type anticancer compound
Scientific Reports (2020)
-
Antimicrobial compounds from marine actinomycetes
Archives of Pharmacal Research (2020)
