
Abstract
Purpose
To determine the prevalence of cystic macular oedema (CME) in patients with choroideremia (CHM) by using spectral-domain optical coherence tomography (SD-OCT).
Methods
A total 16 patients affected with CHM were enroled in the study. All patients underwent a complete eye examination. SD-OCT was performed using an OPKO spectral-domain OCT/SLO instrument.
Results
The average age of the study patients was 44.0±16.0 years (range, 13–63 years). Out of the 16 patients with CHM, 10 patients (62.5%) showed a degree of CME on SD-OCT testing in at least one eye, and 8 patients (50%) showed CME in both eyes.
Conclusions
Because of its notable prevalence, it would seem prudent to screen CHM patients by SD-OCT for the possible presence of CME and to identify those amenable to future treatment strategies for their macular oedema.
Similar content being viewed by others
Introduction
Choroideremia (CHM) is an X-linked progressive chorioretinal dystrophy.1 Affected males develop early onset night blindness, restriction of the peripheral visual field, and a decrease in central visual acuity, often leading to legal blindness in the advanced stages of the disease. Fundus examination shows a slowly progressive atrophy of the choroid and retinal pigment epithelium (RPE). Heterozygous female carriers are usually asymptomatic; however some can show patchy areas of chorioretinal degeneration and an abnormal electroretinogram.2, 3, 4 Fully affected females have also been previously described.4, 5
Choroideremia is caused by mutations in the CHM gene, encoding the Rab escort protein-1. The Rab escort protein-1 facilitates posttranslational modification of Rab proteins, which regulate intracellular trafficking in the RPE and photoreceptors, and are likely involved in the removal of outer segment disc membranes by the RPE.6, 7, 8
A recent study by Affortit-Demoge et al,9 by using time-domain OCT testing, showed a positive correlation between the retinal thickness and choriocapillaris preservation in CHM. Jacobson et al10 showed the presence of retinal thickening and normal retinal laminar structures in early stages of the disease with subsequent retinal thinning and disorganization with disease progression.
Cystic macular oedema (CME) is a common pathological finding of the macula that is the result of cystic accumulation of extracellular intraretinal fluid predominately in the outer plexiform and inner nuclear layers of the retina as a result of breakdown in the blood-retinal barrier.11
To our knowledge, there are no previous reports that demonstrate the presence of cystic macular lesions in patients with CHM, although it has been described in gyrate choroidal atrophy.12, 13 This paper aims to investigate the prevalence of CME in patients with CHM by using spectral-domain optical coherence tomography (SD-OCT).
Materials and methods
A total of 30 eyes of 16 patients with a diagnosis of CHM were included in this prospective investigational study including 3 patients with identified CHM gene mutations (Table 1). The study was conducted in the Department of Ophthalmology at the University of Illinois at Chicago; approval was obtained from an institutional review board at the University of Illinois and the investigation was conducted in accordance with tenets of the Declaration of Helsinki. An informed consent was obtained from all subjects after the study protocol and procedures were explained to each participant.
The diagnosis of CHM was based on the patients’ history of nyctalopia, variable degrees of peripheral field restriction, and the characteristic fundus findings (including choroidal and RPE degenerative changes throughout the posterior pole and mid-peripheral retina). Most patients (N=13) underwent full-field electroretinogram testing by either of two procedures previously described.14, 15
Inclusion criteria were patients with a diagnosis of CHM without an ocular opacity that might hinder OCT scan acquisition. Exclusion criteria were patients with diabetic retinopathy, uveitis, retinal vascular occlusion, previous history of intraocular surgery, or poor OCT image quality. Twenty-eight patients previously seen by one of the authors (GAF) were contacted by telephone and asked to participate in the study based on their previous diagnosis of CHM. A total of 16 patients agreed to participate in the present study.
All patients had a complete eye examination that included best-corrected visual acuity using an early treatment diabetic retinopathy study chart (The Lighthouse, Long Island City, NY, USA), slit-lamp biomicroscopy and intraocular pressure testing using a Goldmann applanation tonometer. Both pupils were dilated with 1% tropicamide and 2.5% phenylephrine. Detailed fundus examinations were performed with a +78 diopter lens by stereobiomicroscopy, and by both direct and indirect ophthalmoscopy.
SD-OCT examinations
All subjects included in the study underwent SD-OCT measurements by using an OPKO spectral-domain OCT/SLO instrument (OPKO Instrumentations, Miami, FL, USA), which is a combination OCT and confocal scanning ophthalmoscope designed to image the retinal layers as well as provide a confocal fundus image. Both the confocal fundus image and the OCT image are produced simultaneously from the same super-luminescent diode light source through the same optics, and therefore, provide pixel-to-pixel correspondence. The system provides up to 27 000 A-scans per second with a 5 μm axial resolution.
For the scan acquisition, both the three-dimensional Retinal Topography and Line Scan (B-scan) exam protocols were used, which can suitably detect cystic macular lesions. The Line Scan mode allows the capture of cross-sectional B-scan OCT images of the vitreoretinal, retinal, and chorioretinal structures. A red scanning line on the confocal scanning ophthalmoscope image represents the exact location of the cross-sectional OCT image. We used the ‘Max Frame Count’ of 64 frames that is defined as the maximum sequentially captured frames of OCT and confocal scanning ophthalmoscope images, which are captured and displayed as individual frames. The three-dimensional Retinal Topography mode covers an area of 8.5 × 8.5 mm with a 2.0 mm depth.
The onboard software calculates average retinal thickness measured in each of nine early treatment diabetic retinopathy study-like zones.16 Retinal thickness measurements were used to calculate the central 1-mm subfield. The mean±SD macular thickness from the central 1-mm subfield and from each of the four sectors of the inner circle (between 1- and 3-mm in diameter) was calculated. The macular thickness values were compared with normative data provided by the manufacturer and which were retrieved from 225 eyes of 119 normative control subjects (mean age of 47.8±16.3 years; Table 2). A paired Student's t-test was used to compare mean values between patients and control subjects to statistically analyse mean retinal thickness differences. A P-value <0.05 was considered to be significant. The number of macular cysts based on SD-OCT examinations were classified as mild, if the number of cysts were from 4–9 and microcystic in appearance, moderate, if the number of microcysts were from 10–15 or one or more macrocystic lesions were observed, and severe, if the number of microcysts were >15 or if there were diffuse and confluent cysts in any single scan image.
Results
Our study cohort included 30 eyes of 16 patients with CHM with 2 eyes excluded because of the presence of an old traumatic retinal detachment in one patient and poor fixation that made the acquisition of a reliable scan difficult in a second patient. The average age of the patients was 44.0±16.0 years (range, 13–63 years). All patients were males with the exception of one female patient, who was diagnosed with CHM based on characteristic clinical fundus appearance, electroretinogram changes, and a genetic molecular mutation. The study cohort was comprised of 15 Caucasian (93.8%) and 1 Hispanic (6.2%) patient. Full-field electroretinogram, which was performed on 13 patients, showed a non-detectable rod function for the isolated dark-adapted response, in 5 patients a non-detectable cone function, and in 8 patients’ cone function that was reduced 20–71% below the lower limits of normal for the single flash light-adapted response.
The average best-corrected visual acuity for all patients was 0.54±0.72 logMAR (range, 0.08–2.80), which is equivalent to 20/20−2– light perception on a Snellen acuity chart.
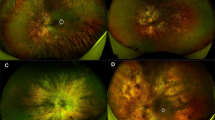
Figure 1 shows the characteristic fundus changes among three different patients (age range from 13 to 63 years), at three different stages of disease severity, where there were either less severe choroidal and RPE atrophic changes (panel a), or more severe fundus changes with diffuse atrophy of the choroid and RPE, and evidence of pigmentary changes in the posterior pole and mid-peripheral retina (panels b and c).

Fundus photographs of right eyes in a 13-year-old (a), 44-year-old (b), and 63-year-old (c) choroideremia affected males that show areas of choroidal and RPE atrophy at an earlier stage (a), and more extensive atrophy of the choroid and RPE noted at later stages of the disease (b and c).
Of the 16 patients (30 eyes) without cystic-appearing macular lesions on fundus examination, 18 eyes (60%) of 10 patients (62.5%) showed a degree of CME on SD-OCT testing in at least one eye, and 8 patients (50%) showed CME in both eyes. The average age in the group of patients with CME on SD-OCT testing was 39.0±18.4 years. Six patients (37.5%) did not show CME on SD-OCT testing in either eye with an average age of 52.3±5.7 years.
Among the group of patients with CME (N=10), 10 eyes (55.6%) of seven patients (70%) showed mild CME, 4 eyes (22.2%) of four patients (40%) showed moderate CME, and 4 eyes (22.2%) of two patients (20%) showed severe CME in at least one eye.
The sizes and shapes of the macular cysts were variable from localized microcystic (11 eyes) or macrocystic changes (4 eyes), to diffuse cysts (3 eyes). The locations of the macular cysts were mostly noted at the level of outer retinal layers in the outer plexiform and inner nuclear layers and extrafoveally at 10–15 degrees. However, central foveal cysts were occasionally observed as well (five eyes of three patients; Figures 2a and b).

Horizontal SD-OCT scans of the left eyes (a and b) and both eyes (c) of three affected males with choroideremia (ages, 62-, 44-, and 63-year-old, respectively), which show evidence of cystic macular oedema in the fovea and parafoveal regions (arrows). Black arrow head (c) shows a rosette-like retinal structure, whereas the white arrow head (b) shows retinal nerve fibre layer thickening temporal to optic nerve (ON). Patient (a) shows evidence of an epiretinal retinal membrane (between the two white lines).
Besides the presence of CME in 62.5% of our study patients by a qualitative method of assessment, the SD-OCT testing showed the presence of normal central macular structure and thickness (N=2 patients, ages 16 and 42 years), a central macular thickening (N=7 patients, mean age of 44.4 years, range from 23 to 55 years), and a central macular thinning was also noted (N=7 patients, mean age of 52.4 years, range from 42 to 63 years). In most of our study patients (N=13), the inner segment/outer segment junction of the photoreceptors was either disrupted/disorganized or lost, even in the macula, from the degenerative process. Only three patients (younger group of patients, age range from 13 to 23 years) showed an intact inner segment/outer segment junction of the photoreceptors within the macula.
Of interest, six patients (37.5%) showed the presence of rosette-like retinal structures at the level of RPE, mostly extrafoveally, with disorganization and loss of the RPE (Figure 2c).
In the entire study cohort, the mean±SD of the macular thickness in the central foveal subfield combined with all sectors of the inner circle are displayed in Table 2, which are compared with the normative data provided by the manufacturer. No statistically significant differences in the central foveal subfield thickness were detected (P=0.18), whereas the mean retinal thickness in all of the four sectors of the inner circle (between 1- and 3-mm in diameter) was selectively thinner than normal, which was highly statistically significant (Table 2). The central foveal subfield thickness in patients who had CME in at least one eye was 327±62 μm, which was significantly thicker statistically when compared with normative data (P<0.0001; Figure 3).

Confocal scanning ophthalmoscopic images (a and c) and retinal thickness measured in early treatment diabetic retinopathy study-like zones (b and d) of the left eyes in two affected males with choroideremia (ages, 62- and 63-year-old, respectively), which show characteristic fundus changes and the presence of central foveal and parafoveal retinal thickening.
Discussion
OCT is a sensitive tool for detecting the presence of CME in various retinal diseases, which may be observed independent of leakage on fluorescein angiography.17, 18 Improvements in OCT technology have recently been introduced, including three-dimensional, high-resolution, and high-speed OCT that uses spectral-domain detection to provide increased resolution.
In our current study on patients with CHM, we report the presence of cystic macular changes of variable degrees on SD-OCT with an overall prevalence of 62.5% in at least one eye (10 patients) and 50% in both eyes (8 patients). CME has been previously reported in different hereditary retinal diseases such retinitis pigmentosa and Usher syndrome.19, 20, 21 CME has also been described in patients with gyrate atrophy, another hereditary choroidal dystrophy.12, 13, 22 Feldman et al22 reported a patient with an epiretinal membrane and CME associated with gyrate atrophy of the choroid and retina. Vannas-Sulonen,23 from a series of 21 patients with gyrate atrophy, observed that 9 (43%) had macular involvement, 1 (5%) with bilateral cystoid macular oedema confirmed by fundus fluorescein angiography.
The presence of macular oedema in a high percentage of our CHM patients is consistent with malfunction of the blood-retinal barrier, with diffusion of fluids to within the retina.
Previous studies10, 24 that used time-domain OCT testing on CHM patient (N=1) and carriers (N=21) did not show any evidence of CME. However, they noted the presence of retinal thickening, which was similar in our study cohort. We similarly observed that the central foveal thickness in the group of CME patients was thicker (327±62 μm) when compared with normative data (235±24 μm). This finding could relate to the macular oedema itself or may be related to retinal gliosis as has been previously described by MacDonald et al.25
One of the interesting findings we observed in our patients (N=6) was the presence of rosette-like retinal structures at the level of outer retinal layers observed on SD-OCT testing. This finding was similar to a previous report by Rodrigues et al,26 who described that these rosette-like structures in CHM may be due to a defect in outer segment phagocytosis. A more recent report by MacDonald et al,25 described these rosette-like structures as formed by abnormal photoreceptors.
Our study showed that CHM patients can have CME on SD-OCT exam. This finding has potential significance for future treatment trials in such patients where patient selection might be influenced by the degree of macular oedema. The use of high-speed, high-resolution tools such as SD-OCT may be useful to measure macular thickness during patient selection and for monitoring patients with CHM in future therapeutic trials.

References
McCulloch C, McCulloch RJP . A hereditary and clinical study of choroideremia. Trans Am Acad Ophthalmol Otolaryngol 1948; 52: 160–190.
Renner AB, Kellner U, Cropp E, Preising MN, MacDonald IM, van den Hurk JA et al. Choroideremia: variability of clinical and electrophysiological characteristics and first report of a negative electroretinogram. Ophthalmology 2006; 113: 2066–2073.
Potter MJ, Wong E, Szabo SM, McTaggart KE . Clinical findings in a carrier of a new mutation in the choroideremia gene. Ophthalmology 2004; 111: 1905–1909.
Majid MA, Horsborough B, Gray RH . Unusal macular findings in known choroideremia carrier. Eye (Lond) 1998; 12: 740–741.
Fraser GR, Friedmann AI . Choroideremia in a female. Br Med J 1968; 2: 732–734.
van den Hurk JA, van de Pol TJ, Molloy CM, Brunsmann F, Rüther K, Zrenner E et al. Detection and characterization of point mutations in the choroideremia candidate gene by PCR-SSCP analysis and direct DNA sequencing. Am J Hum Genet 1992; 50 (6): 1195–1202.
Cremers FP, van de Pol DJ, van Kerkhoff LP, Wieringa B, Ropers HH . Cloning of a gene that is rearranged in patients with choroideraemia. Nature 1990; 347: 674–677.
Seabra MC, Brown MS, Goldstein JL . Retinal degeneration in choroideremia: deficiency of rab geranylgeranyl transferase. Science 1993; 259: 377–381.
Affortit-Demoge A, Querques G, Angulo-Bocco C, D'Athis P, Soubrane G, Souied EH . Optical coherence tomography features of x-linked choroideremia. Retinal Cases Brief Rep 2009; 3 (2): 180–182.
Jacobson SG, Cideciyan AV, Sumaroka A, Aleman TS, Schwartz SB, Windsor EA et al. Remodeling of the human retina in choroideremia: rab escort protein 1 REP-1) mutations. Invest Ophthalmol Vis Sci 2006; 47 (9): 4113–4120.
Quinn CJ . Cystoid macular edema. Optom Clin 1996; 5 (1): 111–130.
Oliveira TL, Andrade RE, Muccioli C, Sallum J, Belfort Jr R . Cystoid macular edema in gyrate atrophy of the choroid and retina: a fluorescein angiography and optical coherence tomography evaluation. Am J Ophthalmol 2005; 140 (1): 147–149.
Vasconcelos-Santos DV, Magalhães EP, Nehemy MB . Macular edema associated with gyrate atrophy managed with intravitreal triamcinolone: a case report. Arq Bras Oftalmol 2007; 70 (5): 858–861.
Fishman GA, Farber MD, Derlacki DJ . X-linked retinitis pigmentosa. Profile of clinical findings. Arch Ophthalmol 1988; 106: 369–375.
Peachey NS, Fishman GA, Derlacki DJ, Alexander KR . Rod and cone dysfunction in carriers of X-linked retinitis pigmentosa. Ophthalmology 1988; 95: 677–685.
Hee MR, Puliafito CA, Duker JS, Reichel E, Coker JG, Wilkins JR et al. Topography of diabetic macular edema with optical coherence tomography. Ophthalmology 1998; 105 (2): 360–370.
Hirakawa H, Iijima H, Gohdo T, Tsukahara S . Optical coherence tomography of cystoid macular edema associated with retinitis pigmentosa. Am J Ophthalmol 1999; 128 (2): 185–191.
Jittpoonkuson T, Garcia P, Rosen RB . Correlation between fluorescein angiography and spectral domain optical coherence tomography in the diagnosis of cystoid macular edema. Br J Ophthalmol 2009; 94 (9): 1197–1200.
Walia S, Fishman GA, Hajali M . Prevalence of cystic macular lesions in patients with Usher II syndrome. Eye (Lond) 2009; 23 (5): 1206–1209.
Adackapara CA, Sunness JS, Dibernardo CW, Melia BM, Dagnelie G . Prevalence of cystoid macular edema and stability in oct retinal thickness in eyes with retinitis pigmentosa during a 48-week lutein trial. Retina 2008; 28 (1): 103–110.
Hajali M, Fishman GA . The prevalence of cystoid macular oedema on optical coherence tomography in retinitis pigmentosa patients without cystic changes on fundus examination. Eye (Lond) 2009; 23 (4): 915–919.
Feldman RB, Mayo SS, Robertson DM, Jones JD, Rostvold JA . Epiretinal membranes and cystoid macular edema in gyrate atrophy of the choroid and retina. Retina 1989; 9: 139–142.
Vannas-Sulonen K . Progression of gyrate atrophy of the choroid and retina. A long-term follow-up by fluorescein angiography. Acta Ophthalmol (Copenh) 1987; 65: 101–109.
Mura M, Sereda C, Jablonski MM, MacDonald IM, Iannaccone A . Clinical and functional findings in choroideremia due to complete deletion of the CHM gene. Arch Ophthalmol 2007; 125 (8): 1107–1113.
MacDonald IM, Russell L, Chan CC . Choroideremia: new findings from ocular pathology and review of recent literature. Surv Ophthalmol 2009; 54 (3): 401–407.
Rodrigues MM, Ballintine EJ, Wiggert BN, Lee L, Fletcher RT, Chader GJ . Choroideremia: a clinical, electron microscopic, and biochemical report. Ophthalmology 1984; 91 (7): 873–883.
Acknowledgements
This study was supported by funds from the Foundation Fighting Blindness, Owings Mills, Maryland; Grant Healthcare Foundation, Lake Forest, Illinois; NIH core Grant EYO1792; and an unrestricted departmental grant from Research to Prevent Blindness. We thank Mr Martin Lindeman, COMT for his technical support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Genead, M., Fishman, G. Cystic macular oedema on spectral-domain optical coherence tomography in choroideremia patients without cystic changes on fundus examination. Eye 25, 84–90 (2011). https://doi.org/10.1038/eye.2010.157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2010.157
Keywords
This article is cited by
-
Cystoid macular oedema without leakage in fluorescein angiography: a literature review
Eye (2023)
-
Bilateral visual acuity decline in males with choroideremia: a pooled, cross-sectional meta-analysis
BMC Ophthalmology (2022)
-
Lamellar Hole-associated Epiretinal Proliferation in choroideremia: a case report
International Journal of Retina and Vitreous (2021)
-
Prospective deep phenotyping of choroideremia patients using multimodal structure-function approaches
Eye (2021)
-
Optical coherence tomography (OCT) features of cystoid spaces in choroideremia (CHM)
Graefe's Archive for Clinical and Experimental Ophthalmology (2019)
