
Abstract
Withania somnifera or Ashwagandha is a medicinal herb of Ayurveda. Though the extract and purified molecules, withanolides, from this plant have been shown to have different pharmacological activities, their effect on bone formation has not been studied. Here, we show that one of the withanolide, withaferin A (WFA) acts as a proteasomal inhibitor (PI) and binds to specific catalytic β subunit of the 20S proteasome. It exerts positive effect on osteoblast by increasing osteoblast proliferation and differentiation. WFA increased expression of osteoblast-specific transcription factor and mineralizing genes, promoted osteoblast survival and suppressed inflammatory cytokines. In osteoclast, WFA treatment decreased osteoclast number directly by decreasing expression of tartarate-resistant acid phosphatase and receptor activator of nuclear factor kappa-B (RANK) and indirectly by decreasing osteoprotegrin/RANK ligand ratio. Our data show that in vitro treatment of WFA to calvarial osteoblast cells decreased expression of E3 ubiquitin ligase, Smad ubiquitin regulatory factor 2 (Smurf2), preventing degradation of Runt-related transcription factor 2 (RunX2) and relevant Smad proteins, which are phosphorylated by bone morphogenetic protein 2. Increased Smurf2 expression due to exogenous treatment of tumor necrosis factor α (TNFα) to primary osteoblast cells was decreased by WFA treatment. This was corroborated by using small interfering RNA against Smurf2. Further, WFA also blocked nuclear factor kappa-B (NF-kB) signaling as assessed by tumor necrosis factor stimulated nuclear translocation of p65-subunit of NF-kB. Overall data show that in vitro proteasome inhibition by WFA simultaneously promoted osteoblastogenesis by stabilizing RunX2 and suppressed osteoclast differentiation, by inhibiting osteoclastogenesis. Oral administration of WFA to osteopenic ovariectomized mice increased osteoprogenitor cells in the bone marrow and increased expression of osteogenic genes. WFA supplementation improved trabecular micro-architecture of the long bones, increased biomechanical strength parameters of the vertebra and femur, decreased bone turnover markers (osteocalcin and TNFα) and expression of skeletal osteoclastogenic genes. It also increased new bone formation and expression of osteogenic genes in the femur bone as compared with vehicle groups (Sham) and ovariectomy (OVx), Bortezomib (known PI), injectible parathyroid hormone and alendronate (FDA approved drugs). WFA promoted the process of cortical bone regeneration at drill-holes site in the femur mid-diaphysis region and cortical gap was bridged with woven bone within 11 days of both estrogen sufficient and deficient (ovariectomized, Ovx) mice. Together our data suggest that WFA stimulates bone formation by abrogating proteasomal machinery and provides knowledge base for its clinical evaluation as a bone anabolic agent.
Similar content being viewed by others


Main
Bone mass is dependent on the balance between bone formation by osteoblasts and bone resorption by osteoclasts.1, 2, 3 The differentiation and function of these cells is regulated by a number of regulatory factors including bone morphogenetic proteins (BMPs), key signal transduction molecules and critical transcription factors including Runt-related transcription factor 2 (RunX2).4 Out of these factors, signaling by BMP is important in bone formation, which involves the ubiquitin–proteasome pathway. In this pathway, Smad ubiquitin regulatory factor 1 (Smurf1), which is E3 ubiquitin ligase, directly interacts with RunX2 and induces its degradation in a ubiquitin- and proteasome-dependent manner.5 Conversely, BMP2 protects RunX2 from Smurf-catalyzed proteolysis by stimulating RunX2 acetylation, which occurs by Smad-dependent mechanism.6 Studies show that under patho-physiological conditions the inflammatory cytokine tumor necrosis factor α (TNFα) increases Smurf1 expression in osteoblasts.7 This results in degradation of Smad1 and RunX2, and subsequently impaired osteoblastogenesis.5 Experiments in transgenic mice support the above findings as Smurf1 knockout mice are born normal but exhibit age-dependent increases in bone mass.8 At the same time, transgenic mice overexpressing Smurf1 in osteoblasts have impaired bone formation during postnatal life.7, 8, 9, 10, 11 Reports also provide evidence for interaction of Smad ubiquitin regulatory factor 2 (Smurf2) with BMP-activated Smad1. Smurf2 that is a Smurf1-related member of hect domain E3 ligase, defines substrate specificity by recognizing the target protein and then degradation by 26 S proteasome.12, 13 The proteasome is also directly involved in osteoclastogenesis by directly controlling important signaling pathway like nuclear factor kappa-B (NF-kB) pathway.14
Clinical and experimental research revealed that inhibition of proteasome activity is critical for the activation of osteogenic transcription factors and bone metabolism.15 Further, preclinical studies on synthetic proteasomal inhibitor’s (PI’s) like bortezomib (Bzb; anti-myloma agent) show that it promotes osteoblastogenesis and suppresses osteoclastogenesis by regulating the ubiquitin–proteasome-dependent degradation of proteins.16, 17, 18 However, no natural PI is reported for enhanced osteogenic effect.
Withania somnifera is one of the most ancient herb used as medicine and dietary supplement.1920 In Ayurveda, the leaves and roots of Withania are prescribed to cure inflammation-related disorders.21 This plant has been studied extensively for its biologically active constituents, steroidal lactones and withanolides.22 The health benefits of Ashwagandha are supported by clinical trials in case of inflammation, immune modulation and reducing arthritis pain. The most abundant constituent is withaferin A (WFA), a highly oxygenated withanolide, which is the major biologically active constituent.23, 24 Despite these studies on pharmacological activities of WFA, their mechanism of action is not fully understood.
We demonstrate that WFA induces osteoblast differentiation by inhibiting the proteasomal machinery decreasing Smurf2 expression and degradation of RunX2 protein. This was validated using small interfering RNA (siRNA) against Smurf2 and TNFα that increases Smurf2 expression. To mimic post-menopausal condition, we carried out in vivo experiments, by ovariectomizing mice that is an equivalent animal model for post-menopausal osteoporosis.25, 26 Overall, our findings point towards a novel molecular mechanism for WFA on bone. Based on these preclinical data, we provide a rationale for the use of WFA, a natural PI, in the treatment of post-menopausal osteoporosis.
Results
Effect of WFA on osteoblast survival, proliferation and mineralization
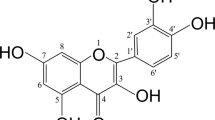
Figure 1a shows chemical structure of WFA (MW=470.6). Mice calvarial osteoblasts (MCOs) were tested with increasing concentrations (1 pM to 1 μM) of WFA (Figure 1b). WFA concentration dependently stimulated osteoblast proliferation as assessed by direct cell count (Figure 1bI) and 5-bromo-2-deoxyuridine (BrdU) incorporation assay (Figure 1bII). Changes in cell count were calculated compared with control group set as 100. As 10 nM concentration of WFA maximally stimulated MCOs’ proliferation, this concentration was used in remaining experiments. MCOs were treated with WFA and Bzb (1 pM to 1 μM) and stimulation of alkaline phosphatase (ALP) activity was studied. Dose of 10 nM was most effective in stimulating ALP activity (P<0.01) as compared with control (Figure 1d). At 10 nM ALP activity of WFA was approximately threefold higher as compared with Bzb (P<0.001). However, reduced proliferation observed at 1 μM may be due to steric hindrance of WFA in osteoblasts.

WFA has osteogenic effect in vitro. (a) Structure of WFA. (b) Effect of WFA on primary osteoblast proliferation. Primary osteoblasts were cultured in increasing concentrations of WFA and Bzb for 24 h and harvested for cell proliferation direct cell count for cell proliferation assay (I) and BrdU incorporation cell proliferation assay (II). The results were expressed as relative cell growth in percentage, which was compared with control group. We set the control group as 100. Values represents mean±S.E. of three independent experiments (n=3). *P<0.05, **P<0.01 when compared with control. (c) WFA treatment of MCOs for 48 h in osteoblast differentiation medium significantly increased ALP production compared with control. Values represent mean±S.E. of three independent experiments (n=3). **P<0.01, ***P<0.001 compared with control vehicle group. (d) Photomicrographs show that treatment of MCOs with WFA in osteoblast differentiation medium significantly increased mineralized nodules compared with control, as assessed by alizarin Red-S staining. (e) Quantitation of mineralization (Alizarin Red-S stain) data are shown as OD at 405 nm. Values represent mean±S.E. of three independent experiments (n=3). ***P<0.001 compared with control vehicle group. (f) WFA (10 nM) treatment of MCOs increased expression of osteogenic genes. Figure shows increased expression of RunX2, OCN and ColI with WFA treatment. Expression was normalized to GAPDH internal control. Values represent mean±S.E. of three independent experiments (n=3). *P<0.05, ***P<0.001 compared with vehicle control group. (g) WFA exerts anti-apoptotic effects in osteoblasts. Using Becton Dickinson FACS and FL-H channel (annexin-V) and FL2-H channel (PI) data show that WFA treatment inhibited apoptosis of osteoblast cells. Shown are representative dot plots. (h) Quantification of flow cytometry data are shown as percent of total cells. Values represents mean ±S.E. of three independent experiments (n=3). *P<0.05 compared with control for early apoptosis and ##P<0.01 compared with control for late apoptosis
WFA (10 nM) treatment stimulated osteoblast mineralization (Figure 1e) (P<0.001) compared with control and Bzb (P<0.001) (Figure 1e). Further, WFA increased expression of osteogenic genes (Figure 1f) with increased expression of transcription factor associated with osteoblast differentiation. Of the studied genes, RunX2 (2.5-fold, P<0.001), collagen type I (ColI, 1.5-fold, P<0.05) that is progression marker of mineralization, and osteocalcin (OCN), 3.6-fold, P<0.001) showed increase in expression. Expression of OCN by WFA was approximately twofold more than with Bzb.
Apoptosis was induced in MCOs by serum deprivation (0.5% FBS containing medium), effect of WFA was analyzed by fluorescence-activated cell sorting (FACS) following annexin-V/PI staining (Figure 1g). MCOs cultured in 10% FBS contained 4% total apoptotic cells compared with ∼8% under apoptotic (0.5% FBS) conditions (Figure 1h). WFA when added to serum-deprived MCOs, only 3% of the cells were found to be apoptotic (Figure 1h) as compared with Bzb.
Effect of WFA on proteasome
WFA’s proteasomal effect on 20 S subunit in calvarial osteoblasts shows that WFA acts as a natural PI (Figure 2a). WFA inhibited proteasomal 20S β subunit activity within 3 h with maximum inhibition at 6 h (∼40–50%) as compared with Bzb (Figure 2a). Lactacystin (negative control) significantly inhibited the 20 S proteasomal activity. Consistent with proteasome inhibition, treatment with WFA and Bzb increased accumulation of ubiquitinated proteins at 6 h as observed by western blot (Figure 2b).

Effect of WFA on osteoblast signaling. (a) Proteasomal effect of WFA in calvarial osteoblast cells. 20S proteasome activities were measured 1, 3, 6 h after treatment with WFA. Values represent mean±S.E. of three (n=3) independent experiments. ***P<0.001, *P<0.001 compared with control. (b) Lower panel shows that 6 h of treatment with WFA induces ubiquitination of proteins through western blot. Protein ladders were detected by anti-ubiquitin antibody. (c) qPCR data of the osteogenic genes after WFA treatment. WFA targets BMP2 signaling by inducing mRNA levels of BMP2 responsive genes in osteoblast cells after 24 h of treatment. Data represent mean±S.E. of three independent experiments (n=3). *P<0.05, **P<0.01 ***P<0.001 when compared with control. (d) Western blot analysis of various osteogenic genes BMP2, Smad1 and its phosphorylation state in the presence of WFA. Expression of Smurf1, Smurf2 a negative regulator of osteogenesis is also shown. All blots were normalized with β-actin. (e) Densitometric analyses of western blot (d) showing fold change. Fold increase was calculated relative to control vehicle-treated cells. The values are the mean±S.E. of three experiments. ***P<0.001 versus vehicle-treated group. (f) Interaction of Smurf2 with Smad1 and RunX2 in the presence of WFA. Immuno-precipitation (IP) assays were performed using anti-Smurf antibody followed by western blot using anti-Smad1 and anti-RunX2 antibodies. (g) WFA prevents RunX2 ubiquitination and proteosomal degradation. Primary osteoblast cells were treated with 5 ng/ml TNFα for 24 h in the presence or absence of WFA, and endogenous RunX2 was immunoprecipated by anti- RunX2 antibody. Ubiquitinated RunX2 (Ub-RunX2) protein ladders were detected by anti-ubiquitin antibody (upper panel). After stripping the antibody, total un-ubiquitinated RunX2 protein levels were determined by anti- RunX2 antibody (lower panel). MW, molecular mass. (h) Western blot of RunX2 and Smurf2 in the presence of various treatments. All blots were normalized with housekeeping gene β-actin. (i) Densitometric analyses of western blot (h) showing fold change. Fold increase was calculated relative to control vehicle-treated cells. The values are the mean±S.E. of three experiments. *P<0.05, **P<0.01 ***P<0.001 versus vehicle -treated group. (j) Relative expression of Smurf2 SiRNA. Osteoblast cells were treated with Smurf2 siRNAs and mRNA levels were measured by real-time RT-PCR. The values are the mean±S.E. of three experiments. **P<0.01versus the vehicle-treated group. (k) Smurf2 knockdown reduced degradation of RunX2. Expression of RunX2 and Smurf2 was determined by western blot analysis after the osteoblast cells were treated with Smurf2 siRNA in the presence and absence of WFA and Bzb. (l) Densitometric analyses of western blot (k) showing fold change. Fold increase was calculated relative to control vehicle-treated cells. The values are the mean±S.E. of three experiments. *P<0.05, **P<0.01 ***P<0.001 versus vehicle (vehicle-treated) group. (m) Smurf2 siRNA blocks TNF-induced RunX2 degradation. Osteoblast cells were treated with Smurf2 siRNA and then treated with (5 ng/ml) TNF for 24 h. Smurf2 and RunX2 expression was determined by western blot analysis. (n) TNF-induced ubiquitination of RunX2 is reversed by Smurf2 knockdown. Endogenous Smurf2 was knocked down in primary osteoblast cells then treated with 5 ng/ml TNFα for 24 h and endogenous RunX2 was immunoprecipated by anti-RunX2 antibody. Ubiquitinated RunX2 (Ub-RunX2) protein ladders were detected by anti-ubiquitin antibody (upper panel). After stripping the antibody, total un-ubiquitinated RunX2 protein levels were determined by anti-RunX2 antibody (lower panel). MW, molecular mass. (o) Smurf2 knockdown prevents RunX2 ubiquitination and proteosomal degradation in presence of WFA. Primary osteoblast cells were treated with WFA for 24 h, and endogenous RunX2 immunoprecipated by anti- RunX2 antibody. Ubiquitinated RunX2 (Ub-RunX2) protein ladders were detected by anti-ubiquitin antibody. After stripping the antibody, total un-ubiquitinated RunX2 protein levels were determined by anti-RunX2 antibody (lower panel). (p) Smurf2 knockdown increased ALP activity in the presence of WFA and TNFα. WFA treatment of MCOs for 48 h in osteoblast differentiation medium significantly increased ALP production compared with control that was abrogated in the presence of TNFα. Treatment with Smurf2 siRNA in the presence of TNFα reversed the effect. Values represent mean±S.E. of three independent experiments (n=3). *P<0.05, ***P<0.001 compared with control vehicle group. (q) Smurf2 knockdown increased expression of osteogenic genes. mRNA expression of BMP2 and RunX2 was assessed by real-time PCR. The values are the mean±S.E. of three experiments. *P<0.05, ***P<0.001 versus control (vehicle) group
WFA’s effect on osteoblast differentiation was examined in primary osteoblasts in vitro; pre-treatment with WFA significantly increased expression of BMP2 (∼5.2-fold; P<0.001). At the same time, Smurf1 and Smurf2 expression decreased (P<0.05), (P<0.01), respectively (Figure 2c). Although data were consistent with Bzb, WFA decreased expression of Smurf2 significantly. Transcript data corroborated with protein data showing increased expression of BMP2 and phosphorylation of Smad (Figure 2d). Overall, WFA acts like PI, induces BMP2 responsive genes to prevent degradation of Smad that interacts with Smurf2 to increase bone formation by increasing expression of RunX2.4, 11
Effect of WFA on Smurf E3 ubiquitin ligase
Western blot with anti-Smad1 and anti-RunX2 antibody shows that expression of Smurf2 decreased in presence of WFA or Bzb with concomitant increase in expression of RunX2 and Smad1 (Figure 2f). Significant reduction in Smurf2 expression as compared with Smurf1 suggests that Smad/RunX2 exhibit greater affinity for Smurf2 in the presence of WFA/Bzb, as Smad/RunX2 are pulled down with reduced amounts of Smurf2 exhibiting greater affinity for Smurf2 protein.
To see if WFA prevents RunX2 ubiquitination, osteoblasts were treated with TNFα in the presence and absence of WFA and endogenous RunX2 was immuno-precipitated with anti- RunX2 antibody followed by western blot using anti-ubiquitin antibody. WFA treatment revealed reduced ubiquitinated RunX2 (Figure 2g). Increased ubiquitinated proteins in the presence of TNFα, indicates that TNFα induces ubiquitination of RunX2 protein leading to its rapid breakdown through proteasomal degradation.6 TNFα inhibits bone formation by upregulating Smurf and degradation of RunX2;7 however, WFA treatment decreased Smurf2 expression. To authenticate this, TNFα was used to increase Smurf levels (Figure 2h) and then see expression of Smurf2 in presence of WFA or Bzb. Osteoblasts treated with TNFα (5 ng/ml), WFA or Bzb increased expression of RunX2, in the presence of WFA or Bzb with reduced expression in the presence of TNFα (Figure 2h). Figure 2i shows ∼20% reduction in Smurf2 in the presence of WFA plus TNFα as compared with TNFα alone. WFA or Bzb significantly reduced Smurf1 and Smurf2; however, with TNFα the reduction in Smurf2 was more pronounced as compared with Smurf1 (Figure 2i). This suggests that WFA or Bzb seems to be regulated by Smurf2.
Smurf2 knockdown reduced degradation of RunX2
Reduction in Smurf2-dependent RunX2 degradation by WFA was explained using siRNA specific for Smurf2 in osteoblasts. Smurf2 siRNA reduced Smurf2 mRNA levels in vehicle cells to ∼50% (Figure 2j). Reduced endogenous Smurf2 due to Smurf2 siRNA led to approximately twofold increase in RunX2 expression (Figure 2k). Likewise, WFA and Bzb in the presence of SiSmurf2 increased RunX2 expression (approximately twofold) as shown in Figure 2l.
To see if TNFα regulates Smurf2 expression, osteoblasts were treated with TNFα (5 ng/ml) for 24 h, then Smurf2 expression was examined (Figure 2m). TNFα increased Smurf2 expression with abrogation of RunX2 expression. Blocking endogenous Smurf2 with Si in presence of TNFα led to reversal in TNFα-mediated response for Smurf2 and RunX2 (Figure 2m). Figure 2n shows increased ubiquitinated RunX2 complexes in TNF-treated cells. SiSmurf2 treatment revealed small amounts of ubiquitinated RunX2 with increased RunX2 expression. Assessment of SiSmurf2 on osteoblast function in the presence and absence of WFA and TNFα shows that Smurf2 knockdown increased ALP activity and expression of BMP2 and RunX2 that further increased to approximately twofold in the presence of WFA (Figures 2p and q) establishing that blocking endogenous Smurf2 increases osteoblast differentiation. This increase was more as compared with the effect by WFA alone. TNFα attenuated ALP activity that was compensated in the presence of SiSmurf2 (Figure 2p). Expression of RunX2 and BMP2 substantiated the ALP data (Figure 2q).
Effect of WFA on osteoclast
Whether, WFA alters osteoclast function, MCOs were treated with WFA and mRNA expression of osteoprotegrin (OPG) and receptor activator of nuclear factor kappa-B (RANK) ligand (RANKL) produced by osteoblasts were determined which stimulate formation of tartarate-resistant acid phosphatase (TRAP)-positive cells and thus osteoclast function.27 Data are expressed as ratio of RANKL/OPG, where decreased ratio (approximately threefold at day 9) represents induction of osteoclastogenesis (Figure 3a). Direct effect of WFA on osteoclasts shows that treatment to BMSC’s abrogated formation of multinucleated osteoclasts that resorb bone versus mononuclearcells. Multinucleated TRAP-positive cells reduced by ∼30–40% (P<0.01) as compared with mononuclear cells in the presence of WFA or Bzb (Figure 3b) followed by significantly reduced mRNA expression of TRAP and RANK (Figure 3c). WFA directly inhibits osteoclast differentiation, and indirectly osteoblast-generated osteoclastogenesis.

Effect of WFA on osteoclastogenesis indirectly through osteoblasts. (a) WFA increases mRNA levels of OPG but decreases mRNA levels of RANKL in primary osteoblasts. Ratio of RANKL: OPG were determined and quantified with qPCR and normalized with GAPDH. Values represents mean±S.E. ***P<0.001 and **P<0.01 of three independent experiments(n=3) when compared with vehicle-treated cells. (b) WFA treatment to OVx mice reduces osteoclastogenesis and increases osteoprogenitor cells in bone marrow. BMCs from mice of various experimental groups were seeded into 48-well plates and osteoclast differentiation was induced as described. Cells were stained for TRAP activity for osteoclast formation. Figure shows quantitative representation of TRAP +ive mononuclear and multinuclear cells. (c) WFA increases mRNA levels of OPG but decreases mRNA levels of TRAP and RANK in osteoclast culture. BMCs were isolated from 4- to 6-week-old mice. After overnight culture cells were cultured for 6 days in the presence of MCSF and RANKL. mRNA levels of TRAP and RANK related to osteoclastogenesis was determined by qPCR from the total RNA made from BMC’s. Data represent mean±S.E.M.; n=3. **P<0.01 and ***P<0.001 compared with vehicle-treated cells. (d) WFA treatment decreases expression of inflammatory cytokines in osteoblasts. qPCR data show mRNA levels IL-6, MCP-1. Values represents mean±S.E. **P<0.01 and ***P<0.001 of three independent experiments (n=3) when compared with control. (e) WFA inhibits TNFα-induced NF-kB nuclear translocation in osteoblast cells. Representative photomicrograph of sub cellular localization of p65 was determined by immuno fluorescence (magnification × 40) under control and treatment conditions from three independent experiments (n=3). (f) WFA treatment abolished the nuclear translocation of the NF-kB in osteoblasts. Osteoblasts were treated with TNFα and WFA. Cytoplasmic or nuclear extracts were prepared and NF-kB protein levels were detected by immunoblotting. TNFα enhanced the nuclear translocation of NF-kB, WFA abolished it. Histone H3 was used as a loading control for nuclear extract, β-actin was used as loading controls for cytoplasmic fraction. (g) This figure shows the densitometric analysis of NF-kB from three independent blots. Values represents mean±S.E. of three independent experiments (n=3), ***P<0.001, **P<0.01 when compared with control
Figure 3d shows that whereas TNFα robustly increased mRNA expression of MCP-1 and IL-6 (osteoclastogenic cytokines) in osteoblasts, presence of WFA/Bzb significantly reduced TNFα-stimulated expression. NF-KB p65-subunit labeling by immunofluorescence shows characteristic cytoplasmic and perinuclear localization in unstimulated osteoblasts (Figure 3e). Whereas TNFα resulted in intense nuclear labeling of p65-subunit, WFA/Bzb inhibited the nuclear labeling (Figure 3e). Nuclear translocation of p65-subunit by TNFα was quantified by western blot normalized with histone 3 antibody for nuclear extract (Figure 3f). TNFα increased nuclear translocation of p65-subunit by ∼twofold (Figure 3g). WFA/Bzb alone or in the presence of TNFα decreased TNFα-induced p65 nuclear translocation lower than the control. Altogether, WFA’s proteasomal effect on osteoblast and production of osteoclastic factors in osteogenic cells indirectly impacts osteoclastogenesis.
Pharmacokinetic studies of WFA after oral administration
WFA at an oral dose of 10 mg/kg resulted in maximum plasma levels of 8.41±1.4 μg/ml after 3 h. The AUC0–24 and t1/2 of WFA was found to be 55.01±8.4 μg h/ml was 7.1±1.2 h, respectively. The Vd and MRT was found to be 0.043 l and 6.52 h, respectively, as shown in Figure 4.

Plasma concentration time profile of WFA after oral administration at the dose of 10 mg/kg. The data are represented as mean±S.D. of three individual experiments
Effect of WFA on bone regeneration
In vivo regeneration of bone to monitor local repair process was done using μ-CT in Sham and ovariectomy (OVx) groups from days 11 to 21 after creation of the hole.28, 29, 30, 31 Intensity of calcein labeling in the drill hole shows that WFA at 5 and 10 mg/kg/day increased mineral deposition (Figures 5a and b). Modest mineralized calluses appeared at defect and intra-medulla region of OVx and Sham groups. By day 11, extent of callus mineralization increased, in the intra-medulla region. Post drilling 11th day, WFA treatment at 5 mg/kg/day dose led to predominantly occupied mineralized callus, and defect region was partially bridged, however, the effect was more significant at 10 mg/kg/day with more mineralized callus within intra-medulla region to be remodeled to completely bridge the defect (Figure 5c). Six weeks post-surgery OVx mice had significantly lower mineral deposition (50% less) than sham+vehicle group. However, Ovx+WFA at 5 and 10 mg/kg/day had the ability for mineral deposition compared with OVx+vehicle group.

WFA promotes bone regeneration in the drill-hole site in sham-operated (ovary intact) and OVx mice. (a) Representative confocal images (magnification= × 100) of calcein labeling shown in the drill-hole site of various groups 0, 11 and 21 days after injury without and with WFA treatment(5 and 10 mg/kg/day). (b) Data show the quantification of the mean intensity of calcein labeling. Values represent mean±S.E. **P<0.01, ***P<0.001 compared with Sham vehicle. #P<0.05, ##P<0.01, ###P<0.001 compared with OVx vehicle group. Inter-dose comparison shows that cP<0.05 when 10 mg/kg/day dose was compared with 5 mg/kg/day dose. *P<0.05 when OVx+V compared with Sham+V. (c) Representative μCT images from the center of the bony hole (defect region) on days 0, 11 and 21 in the sham and OVx groups. (d and e) Graph represents μCT analysis in the defect and intra-medulla regions of sham and sham+ WFA-treatment group. Data show BV/TV (%) at 5 and 10 mg/kg/day dose in both the regions.Values represent mean±S.E. **P<0.01, ***P<0.001 compared with Sham vehicle. ##P<0.01compared with OVx vehicle. Inter-dose comparison shows aP<0.05 when 10 mg/kg/day dose is compared with 5 mg/kg/day dose in sham group on day 11. (f and g) Graph represents μCT analysis in the defect and intra-medulla regions of OVx and OVx+ WFA treatment groups. Values represent Mean±S.E. *P<0.05, ***P<.001 compared with Sham vehicle. #P<0.05 and ##P<0.01 compared with OVx vehicle. Inter-dose comparison shows aP<0.05 when 10 mg/kg/day dose is compared with 5 mg/kg/day dose in sham group on day 11. (h–j) Effect of WFA on osteogenic genes after drill-hole injury. mRNA profile of RunX2, BMP2 and OCN extracted from callus of drill site of bone. Values represent mean±S.E. of three independent experiments n=3. **P<0.01, ***P<0.001 compared with Sham vehicle. #P<0.05, ##P<0.01, ###P<0.001 compared with OVx vehicle. Inter-dose comparison shows aP<0.05 when 10 mg/kg/day dose is compared with 5 mg/kg/day dose in sham group on day 11. bP<0.05 when 10 mg/kg/day dose is compared with 5 mg/kg/day in sham group on day 21. cP<0.05 when 10 mg/kg/daydose iseffective than 5 mg/kg/day in OVx group on day 11. dP<0.05 when 10 mg/kg/day dose is effective than 5 mg/kg/day in OVx group at day 21for RunX2 gene
Ovx group displayed lower BV/TV compared with Sham, from day 11 to 21 in the defect region; however, this was significantly increased with WFA treatment at 10 mg/kg/day opposed to 5 mg/kg/day group (Figures 5d–f). In both groups, BV/TV in the intra-medulla space moderately increased with time, peaked day 11, and returned to baseline on day 21. Inter-dose comparison in the intra-medulla region was not significant in the sham group, however, in the OVx group dose of 10 mg/kg/day was significantly better. (Figures 5e–g). Data were corroborated by qPCR. After drill-hole injury RunX2, BMP2 and OCN mRNA expression were significantly higher in Sham than OVx group at day 11 and 21 (Figures 5h–j). WFA treatment (5 and 10 mg/kg/day) increased expression of these genes with robust change at 10 mg/kg/day in Sham +WFA versus OVx +WFA group.
Effect of WFA on bone formation in osteopenic mice
WFA’s pharmacokinetics was studied in osteoporotic mice after its oral administration in gum acacia. Particle size of WFA in dispersion was 1.023±0.28 μm with poly dispersity index 0.423 of particles. Zeta potential and viscosity was 40.7±8.06 mV and 2.86 Pas, respectively (data not shown). Post treatment, BMC’s harvested from bones of OVx+vehicle group had decreased mineralized nodules. OVx mice treated with WFA at 10 mg/kg/day Bzb or injectible parathyroid hormone (iPTH) had nodule formation that was comparable with sham group (Figures 6a and b); however, WFA, at lower dose, and alendronate (Ald) did not significantly induce mineralization.

WFA has anabolic effect in osteopenic bones. (a and b). Oral supplementation of WFA to OVx mice increases mineralized nodule formation in BMCs as assessed by alizarin red-S staining. Values represent Mean±S.E. of three independent experiments n=3. *P<0.05, **P<0.01 as compared with OVx vehicle group. (c and d). WFA supplementation increases mineral apposition rate and bone formation rate in OVx mice. Dynamic histomorphometric parameters (MAR and BFR) at the femur mid-diaphysis was calculated from the double fluorochrome labeling experiments in various groups.Values represent Mean±S.E. of three independent experiments n=3. ***P<0.001 compared with OVx vehicle group. aP<0.001 when 10 mg/kg/day dose compared with 1 mg/kg/day. dP<0.001 when 10 mg/kg/day dose compared with 5 mg/kg/day. (e) WFA supplementation restores the trabecular micro-architecture of the femur epiphysis. Representative μCT images of the femur epiphysis of various experimental groups. (f–i) μCT analysis of various trabecular parameters of the femur epiphysis, including BV/TV, Tb.No, Tb.Th and SMI are presented. All values are expressed as mean±S.E.M. *P<0.05, **P<0.01, ***P<0.001 aP<0.001 when 10 mg/kg/day dose compared with 1 mg/kg/day. dP<0.001 when 10 mg/kg/day dose compared with 5 mg/kg/day dose, fP<0.05 when 10 mg/kg/day dose compared with 5 mg/kg/day dose, gP<0.05 when 10 mg/kg/day dose compared with Ald 3 mg/kg/day dose, iP<0.05 when 10 mg/kg/day dose compared with Bzb 0.3 mg/kg/day dose WFA supplementation has a significant restorative effect on the trabecular micro-architecture of L5 vertebrae. Figure shows representative μCT images of various treated group. (k–n) Trabecular parameters of L5 vertebrae of various treatment groups. All values are expressed as mean±S.E.M. *P<0.05, **P<0.01, ***P<0.001 compared with OVx group, bP<0.01 when 10 mg/kg/day dose compared with 1 mg/kg/day dose, iP<0.05 when 10 mg/kg/day dose compared with Bzb 0.3 mg/kg/day dose
Dynamic histomorphometric data of cortical bone (Figures 6c and d) show that OVx group had significantly lower periosteal MAR and BFR compared with sham group. Comparison of WFA (10 mg/kg/day) or Ald, Bzb and iPTH groups with sham revealed no significant difference in MAR and BFR.
Effect of WFA on bone micro-architecture and biomechanical -strength
3D μCT shows deterioration of bone owing to destruction of the trabecular architecture as observed in OVx+vehicle group compared with well-developed trabeculae in sham+vehicle group (Figure 6e). Trabecular response to WFA treatment to ovariectomized rats was quantified in tibia proximal (Supplementary Table 2), femur epiphysial (Figures 6f–i) and lumbar vertebral L-5 region (Figures 6k–n), and values compared with positive controls. Femoral data show that compared with sham group, the OVx+vehicle group exhibited significantly reduced BV/TV, trabecular number (Tb.No), trabecular thickness (Tb.Th) and increased structure mode index (SMI). Post WFA treatment BV/TV, Tb. No was at par to sham group at the dose of 10 mg/kg/day. Likewise, SMI increased due to OVx and decreased with WFA, PTH and Bzb treatment but was comparable to Sham group. Inter-dose comparison with sham group revealed that out of the three doses of 1, 5 and 10 mg/kg/day WFA, 10 mg/kg/day was most effective. Vertebral data show the OVx+vehicle group had significantly reduced BV/TV, Tb.No, Tb.Th and increased SMI. WFA treatment at 10 mg/kg/day was comparable in BV/TV, Tb.No, Tb.Th and SMI with Sham group. Bone formed by WFA at 10 mg/kg/day was comparable with the osteogenic agent iPTH.
Biomechanical strength data suggest that OVx+Vehicle group had significantly reduced energy and stiffness, whereas WFA treatment increased these parameters. Whereas OVx group required less compressive energy for breaking LV5 compared with the sham group (Table 1), WFA treatment at all three doses for 60 days required more compressive energy than the OVx group with 10 mg/kg/day being the most efficient dose.
Effect of WFA on bone turnover marker
Serum OCN was elevated in the OVx+vehicle group compared with sham+vehicle group Figure 7(a). WFA-treated OVx mice reduced these bone turnover markers. These data suggest that WFA inhibits bone turnover that is characteristically elevated under estrogen deficiency.

WFA inhibits bone turnover and has direct effect on osteoblast-specific genes in femurs of OVx mice. (a) Serum OCN levels as measured at the end of experiment from various treatment groups. Data show that WFA inhibits bone turnover in OVx mice. All values are expressed as mean±S.E.M. *P<0.05, **P<0.01, ***P<0.001 compared with OVx group. aP<0.001 when 10 mg/kg/day dose was compared with 1 mg/kg/day dose, eP<0.01 when 10 mg/kg/day dose compared with 5 mg/kg/day dose. (b) Circulating TNFα levels from serum of various groups were measured by ELISA at the end of the experiment period. All values are expressed as mean±S.E. of three independent experiments (n=3). **P<0.01 compared with OVx group. (c) Direct effect of WFA at the end of experiment on long bones (femur). Data show qPCR of osteogenic genes RunX2, ColI and OCN in RNA isolated from bone. All values are expressed as mean±S.E. of three independent experiments (n=3). *P<0.05, **P<0.01, ***P<0.001 compared with OVx group. aP<0.001 when 10 mg/kg/day dose was compared with 1 mg/kg/day dose, bP<0.01 when 10 mg/kg/day dose compared with 1 mg/kg/day dose, dP<0.001 when 10 mg/kg/day dose compared with 5 mg/kg/day dose, eP<0.01 when 10 mg/kg/day dose compared with 5 mg/kg/day dose, gP<0.05 when 10 mg/kg/day dose compared with Ald 3 mg/kg/day dose, hP<0.01 when 10 mg/kg/day dose compared with PTH mg/kg/day dose, iP<0.05 when 10 mg/kg/day dose compared with Bzb 0.3 mg/kg/day dose for RunX2 gene expression. (d) Effect of WFA on mRNA expression of Smurf2 in femur at the end of experimental period. All values are expressed as mean±S.E.M. of three independent experiments (n=3). *P<0.05, **P<0.01, ***P<0.001 when compared with OVx group, bP<0.01 when 10 mg/kg/day dose was compared with 1 mg/kg/day dose, iP<0.05 when 10 mg/kg/day dose compared with Bzb 0.3 mg/kg/day dose
In vivo effect of WFA on TNFα
Serum TNFα levels studied in vivo show significantly increased levels of TNFα in OVx+vehicle group at the end of study, which significantly decreased with of WFA treatment (all three doses). Data were comparable to standard control groups Ald, Bzb and iPTH (Figure 7b).
Effect of WFA on osteogenic genes in femurs of OVx mice
Increased expression of osteogenic genes was observed by qPCR in OVx+WFA (1, 5 and 10 mg) compared with OVx+vehicle group, suggesting that WFA increased osteoblast number and/or function in bones (Figure 7c). The 10 mg/kg/day dose showed robust changes as compared with other two doses. Smurf2 was significantly elevated in OVx+vehicle group (P<0.001). Treatment with WFA decreased Smurf2 expression dose dependently (P<0.001). Maximum reduction was observed with Ald that was better than WFA, Bzb or iPTH. Therefore, WFA may increase osteoblast function in vivo by promoting RunX2 expression and simultaneous downregulation of Smurf E3 ligase (Figure 7d).
Discussion
Menopause contributes to bone loss in osteoporosis.32, 33, 34 Under these conditions, osteoblast-mediated bone formation cannot compensate for accelerated osteoclastic bone resorption, suggesting direct inhibitory effect on osteoblasts35 and an increase in adipocytes.36, 37, 38 Emerging data indicate that the PIs may have a critical role in the regulation of osteoblast proliferation and differentiation.5 PIs come in two different types synthetic and natural, and each has been known to induce different biological effects in different cell types.39, 40 In light of the systemic toxicities of the known PI’s, we provide an evidence for a naturally occurring PI WFA from leaves of W. somnifera. Of the three withanolides, WFA showed best activity (Supplementary Figure S2). Stimulatory effect of WFA in osteoblast was observed in our system at concentrations as low as 10 nM, with no effect at higher dose. This behavior was not related to toxic effect of WFA, because it did not induce apoptosis or inhibit proliferation of either osteoblast progenitors or osteoblasts at concentration ranging between 1 pM and 1 μM. Higher dose of WFA (1 μM) may be imparting steric hindrance to the osteoblasts that may have attenuated proliferation at this concentration. Our findings provide the first report to show that WFA promotes osteoblast function by direct involvement of the ubiquitin–proteasome pathway in BMP signaling and controlling the ubiquitination status of RunX2 protein. WFA decreased expression of Smurf1 and Smurf2 although decrease in Smurf2 was more significant as compared with Smurf1 that was modestly modulated, may be due to differences in response threshold between Smurf1 and Smurf2. The effect of WFA on signaling, function and gene expression of osteoblasts leading to possible anabolic action is shown in Figure 8. We have several lines of evidence implicating Smurfs in the regulation of WFA-induced RunX2 expression and thus bone formation. TNFα was taken as a positive control as increased TNFα level is known to mediate osteoblast inhibition.41, 42, 43 Gilbert et al.44 have shown that increased TNFα levels reduce nuclear RunX2 protein in osteoblasts by 90% and transcription by 40–50%. We tested this hypothesis and show that TNFα treatment to calvarial osteoblast cells inhibited osteoblast differentiation because of increased expression of Smurf2 and simultaneously decreased expression of RunX2 protein. Treatment with WFA attenuated TNFα-induced Smurf2 expression, and increased RunX2 protein. This was confirmed by blocking endogenous Smurf2 levels by siRNA against Smurf2. Overall, proteasome inhibition by WFA simultaneously increased osteoblastogenesis and reduced osteoclast number in two ways. WFA indirectly lowered the inflammatory cytokines secreted by the osteoblast cells and the RANKL/OPG ratio. WFA’s direct effect involves the inhibition of the p65 nuclear translocation of NF-kB subunit whereas, activation of NF-kB has been found to inhibit BMP2-induced osteoblast differentiation by inhibiting Smad DNA binding.45 Smurf2 mediated protein degradation in TNFα-induced osteoblast inhibition was tested and found that proteasome is an in vivo target of WFA. Bones of ovariectomized mice exhibited elevated TNFα levels in serum, increased Smurf2 and decreased RunX2 mRNA expression.

Schematic diagram outlining the potential molecular targets and in vivo effect of WFA leading to bone anabolic effect in osteogenic cells and osteoclast precursors induced by proteasome inhibition. BMP signaling induced by WFA prevents degradation of Smad receptors. The transcription factor Runx2, which is induced by BMP2, is further stabilized by preventing completion of proteasomal degradation by E3 ubiquitin ligase, Smurf2. WFA decreases tumor necrosis factor α (TNFα), induced NF-Kb and Smurf2 expression resulting in increased Smad and Runx2 levels. In vivo proteasome inhibition simultaneously increases osteoblastogenesis by stabilizing RunX2 and reduces osteoclast numbers directly by inhibiting RANKL: OPG ratio. Thus, osteoclast differentiation is suppressed by inability of RANKL to bind with RANK. This results in reduced number of TRAP-positive cells. Simultaneous induction of osteoblastogenesis and suppression of osteoclastogenesis results in increased bone mass. Our in vivo data demonstrate that WFA exerted an osteogenic effect in osteopenic OVx mice and accelerated the bone-healing process after bone marrow injury of long bones (See text for details
WFA treatment induced expression of collagen I (ColI) and OCN restored bone in OVx rats and enhanced bone growth and healing. Dynamic measures of bone formation at the femur diaphysis and vertebrae demonstrated that WFA increased MAR and BFR. Absorption pattern of WFA dispersion suggests that the mono-disperse population of particles in gum acacia along with highly negative zeta potential facilitated absorption of WFA. Peak plasma concentration of WFA (55.01± μg/h/ml) reached within 3 h with high half-life of 7 h could have resulted in positive impact on bone. Data also suggest that significant lowering of S-OCN levels and preservation of trabecular micro-architecture by WFA in OVx vehicle group may have significantly contributed to increased bone strength. Restoration of bone by WFA was beyond the suppressive effect of Ald on bone although comparable to iPTH and Bzb.
The effect of WFA on temporal and spatial changes of bone-healing in drill-hole defect was studied between sham-operated and OVx-induced groups.28, 29, 30, 31Fluorochrome labeling demonstrated that intramembranous and endochondral ossification in osteoporotic bone healing was impaired in osteogenesis and remodeling. Micro-CT reconstruction consistently demonstrated less new bone formation in both defect and intra-medulla region (day 11) in OVx mice as compared with Sham mice. During the late phase of bone healing (by day 21), the newly formed bone in the defect region was fully remodeled into cortical like structure with WFA treatment compared with Sham and osteopenic mice, whereas remodeling was incomplete in the OVx mice. This indicated that remodeling capability from woven bone to lamellar bone was impaired in the OVx versus sham mice as evidenced by lower expression of osteogenic genes in the callus. Impaired healing in OVx mice was rescued rapidly with WFA treatment that augmented the process of bone healing.
In conclusion, the study demonstrates increased bone mass results from proteasome inhibition by WFA. WFA stimulates osteoblast growth and differentiation, accelerates bone healing following injury and exerts an anabolic effect on osteoporotic bone. Given the lack of uterine estrogenicity (Supplementary Figure S1) and intrinsic bioavailability of WFA, these investigations provide important findings pertaining to WFA-induced bone anabolic effect at 10 mg/kg/day. Data suggest that WFA can reach an optimum therapeutic concentration in vivo that can be anabolic in nature. The present study supports the usefulness of natural PI WFA, making it an attractive alternative anabolic candidate as a new treatment of post-menopausal osteoporosis as reviewed in review.46, 47
Materials and Methods
Reagents and chemicals
Cell culture medium and supplements were purchased from Invitrogen (Carlsbad, CA, USA). All fine chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA). Human PTH (1–34) was purchased from Calbiochem (EMD Millipore Corporation, Billerica, MA, USA) and alendronate sodium trihydrate (Ald) were purchased from Sigma-Aldrich. The Bzb used was purchased from Santa Cruz (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Dimethyl sulfoxide (DMSO) from Calbiochem used as vehicle to dissolve WFA and Bzb. Final concentration of DMSO was below 0.01% that is below the toxic range to the cells.28
MCO culture
For each experiment, 1- to 2-day-old MCOs were used to harvest 20–35 calvaria at room temperature. Briefly, individual calvaria were surgically isolated from the skull, sutures were segregated and adherent tissue material was cleaned by gentle scrapping. As described previously,48 the pooled calvarias were kept for repeated digestion (15 min/digestion) with 0.05% trypsin and 0.1% collagenase P to release cells. First digestion was discarded and cell was collected from next four digestions. These cells were cultured in a modified essential medium (a-MEM) containing 10% fetal calf serum (FCS) and 1% penicillin/streptomycin (complete growth medium). Cultures of MCOs were allowed to reach 80% confluence for the experiments.28
Cell proliferation assay
We assayed cell proliferation by using direct cell count and BrdU incorporation assay. MCOs at 80% confluence (FBS) were exposed with various concentrations (100 pM to 1 μM) of WFA and Bzb for 24 h. Direct cell count followed growth of the primary osteoblast cells in regular media for 24 h and growth arrest with serum-free media for another 24 h, the cell numbers were assessed by directly counting the number of cells with a hemocytometer under a light microscope. Percent growth of the cells was calculated as (cell number in treatment group/cell number in control vehicle group). For BrdU incorporation assay, primary osteoblast cells were seeded on chamber slides. After growth in regular media for 24 h and growth arrest with serum-free media for another 24 h, the media was changed to regular media, and BrdU (10 ng/ml; Sigma, St. Louis, MO, USA) was added to the media. Then the cells were incubated for 24 h. After overnight incubation, cells were treated with or without WFA and Bzb at concentration 10 nM for 24 h. The cells were washed, fixated and then stained with the BrdU antibody by using a BrdU staining kit (Calbiochem, Darmstadt, Germany) following the manufacturer’s instruction. BrdU-positive nuclei number and total nucleated cells were counted. Cell proliferation has been represented as a percentage of BrdU-positive nuclei over total nucleated cells.Counts were performed in duplicate and repeated in three cultures.
20S proteasome activity assay
Cells (2.0–4.0 × 103) were plated in each well of a six-well plate and treated with either WFA or Bzb at different time interval (1, 3 and 6 h) at 10 nM concentration. Lysate at different time interval was collected using the manufacture’s protocol, (Proteasome Activity Assay Kit, Chemicon’s). The assay is based on detection of the fluorophore 7-amino-4-methylcoumarin (AMC) after cleavage from the labeled substrate LLVY-AMC. Free AMC fluorescence can be quantified using a 380/460 nm filter set in a fluorometer. Purified 20S proteosome subunit has been used as a positive control and a proteasome inhibitor, Lactacystin, is included as a test inhibitor (control) for screening purpose.
Quantitative real-time PCR (qPCR)
Cells were homogenized using 1 ml of TRIzol reagent (Invitrogen), and total RNA was extracted according to the manufacturer’s protocol. cDNA was synthesized using RevertAid First Strand cDNA synthesis kit (no. K1621 Fermentas, Burlington, ON, Canada) from 1 μg of total RNA. Quantitative real-time PCR amplifications were performed in The LightCycler 480 Real-Time PCR System (Roche Diagnostics, Indianapolis, IN, USA) using LightCycler SYBR Green (Roche Diagnostics) according to the manufacturer’s instruction. The sequences of primer sets for Smurf1, Smurf2, RunX2, BMP2, ALP, OCN, ColI and GAPDH mRNAs, target sites on mRNAs and product sizes by PCR are shown in Supplementary Table S1. The quantity of each sample was normalized using the CT (threshold cycle) value obtained for the GAPDH mRNA amplifications.49
Osteoclast culture and real-time expression of TRAP and RANK
In vitro osteoclastogenesis was performed using a standard protocol.50, 51 Mouse bone marrow cells were flushed from femur using α-MEM. Cells were seeded in flask for overnight in Osteoclast medium (α-MEM, 10% FCS, antibiotic, EBSS, 10 ng/ml MCSF). After overnight incubation, non-adherent bone marrow cells were seeded in 48-well plates at a density of 2 00 000cells/well and cultured for 5–6 days in α-MEM containing 10% FCS, EBSS, 50 ng/ml RANKL, and 10 ng/ml MCSF and WFA, Bzb at different concentrations. Medium was replaced after every 48 hours; after 6 days of culture, cells were washed with vehicle and then fixed in 4% paraformaldehyde or use for RNA extraction using TRIZOL for analysis of TRAP, receptor activator of NF-kB (RANK) mRNA levels by real-time PCR. Fixed cells were followed by TRAP staining using the standardized protocol.52
Apoptosis assay
Mice calverial osteoblast were grown to 50–60% confluence, followed by serum withdrawal for 3 h and treatment with WFA (10 nM) and Bzb (10 nM) or without treatment for 24 h in a-MEM containing 0.5% FBS. Annexin-V/PI staining for FACS analysis was carried out using Calbiochem Annexin-V-FITC Apoptosis Detection Kit (Calbiochem) according to manufacturer’s instructions.28
ALP assay
For the measurement of ALP activity, MCOs at ∼80% confluence were trypsinized, and 2 × 103 cells/well were seeded onto 96-well plates. Cells were treated with WFA and Bzb at different concentration (1 pM, 100 pM, 10 nM, 1 μM) or vehicle for 48 h in α-MEM supplemented with 10% FBS, 10 mM β-glycerophosphate, 50 mg/ml of ascorbic acid, and 1% penicillin/streptomycin (osteoblast differentiation medium). At the end of incubation period, total ALP activity was measured using p-nitrophenylphosphate as substrate, and absorbance was read at 405 nm.28, 53
Mineralization assay
MCOs were seeded onto 12-well plates (25 000 cells/well) in osteoblast differentiation medium with or without WFA (10 nM) and Bzb (10 nM) for 21 days with a medium change every 48 h. At the end of the experiment, cells were washed with vehicle and fixed with paraformaldehyde (4%) in vehicle for 15 min. Alizarin red-S stain was used for staining mineralized nodules, followed by extraction of the stain using 10% acetic acid and 10% ammonium hydroxide for colorimetric quantification at 405 nm.48, 54
Western blot analysis
MCOs was grown to 80% confluency followed by exposure to compounds (WFA and Bzb) for different time periods. Cells were washed with cold phosphate-buffered saline (vehicle), and whole-cell lysates were prepared by the addition of lysis buffer from Sigma-Aldrich containing a protease inhibitor mixture from Sigma-Aldrich. Protein (30–50 μg) was loaded per lane and separated on a 10% polyacrylamide gel, followed by transfer to a PVDF membrane (Millipore, Billerica, MA, USA) by electroblotting. Membrane was blocked for nonspecific binding in 5% nonfat dry milk and followed by incubation with an primary antibody (abcam, Cambridge, MA, USA) at 4 °C overnight. Membranes were washed and were probed with a horseradish peroxidase-conjugated secondary antibody (abcam) and visualized by an enhanced chemi-luminescence system (GE Healthcare Life Sciences, Bangalore, India) according to the manufacturer’s instructions.55
Ubiquitination of RunX2
Primary osteoblast cells were incubated in medium containing 5 ng/ml of TNF for 24 h in the presence of vehicle or WFA. For the immuno-precipitation, cell lysate was incubated wit anti-RunX2 antibody overnight at 4 °C followed by the addition of protein G-agarose overnight at 4 °C. The immune precipitates were washed with 50 mM Tris-HCl buffer (pH 8.0), containing 150 mM NaCl, 1% Nonidet P-40, 0.05% deoxycholate and 0.1% SDS, resuspended in 1x reducing buffer, and subjected to western blot analysis with an anti-ubiquitin antibody. The same membrane was stripped and reprobed for RunX2.
RNA interference for Smurf2
RNA interference was used to downregulate the expression of Smurf2 in osteoblast cells. SMARTpool siRNA and SMARTpool reagents for Smurf2 and nonspecific control siRNA duplexes were designed and synthesized by Customer SMARTpool siRNA Design from Dharmacon (Lafayettee, CO, USA). Following are the target sequences of On-target plus SMARTpool siRNA for Smurf2. GCA AUG UCG UCA AGU GGU U, AAA CGA UAU CAC AGG UGU U, GCU CAU UAU UUG UGG ACU, GCA GAA GGG CUU UAA UGA A. For gene knockdown experiments, osteoblast cells were plated in 6 cm dish and cultured for 48 h α-MEM containing 10% FBS and antibiotics, cells were transfected with siRNAs (50 nM) using transfection reagent according to the manufacturer’s instructions. After another 48 h of culture, cells were recultured in α-MEM containing 10% FBS and antibiotics. The cells were then used for RNA extraction or western blot analysis.
Nuclear and cytosolic protein extraction experiment
Mice primary osteoblast were incubated in medium with or without Bzb (10 nM), WFA (10 nM) and TNFα (5 ng/ml) for 24 h. Nuclear and cytosolic protein was extracted by different lysis buffer using manufacturer’s protocol (CelLytic NuCLEAR Extraction Kit, Sigma-Aldrich). These extracted protein were immunoblotted with NF-kB (abcam) for nuclear translocation.
Immuno-precipitation experiment
Mice primary osteoblast were incubated in medium with or without Bzb (10 nM) and WFA (10 nM) for 24 h. For immuno-precipitation, cell lysate was collected using Catch and Release v2.0 Immuno-precipitation Kit (Millipore) according to manufacturer protocol. Smurf2 was immuno-precipitated with anti-smurf2 antibody raised in rabbit and incubated with anti-RunX2, anti-Smad1 antibody raised in mouse for western blotting. Similarly, in another set RunX2 was immuno-precipitated antibody raised in mouse and incubated with anti-ubiquitin antibody raised in rabbit for western blotting.
Immunocytochemistry
Mice primary osteoblast were incubated in medium with or without Bzb (10 nM) and WFA (10 nM) and were grown in Lab-Tek Chamber Slides (Nunc, Denmark) for 24 h. For immunocytochemistry, cells were fixed with 4% formaldehyde followed by permeabilization with 0.1% triton X-100 and incubation in primary antibody (NF-kB) for overnight. Alexa flour-488 Invitrogen used as secondary antibody. Fluorescence was captured using fluorescent microscope (Eclipse 80i, Nikon, Tokyo, Japan), with the aid of appropriate filter (excitation 495 nm and emission 519 nm).
Animal Studies
Drill-hole defect at the mid-diaphysis of the femur
Sixty adult Balb/c mice (18–20 g each) were taken and divided into six groups of three each for the study. Each group included 12 animals per group. First three groups of Sham (ovary intact) include (Sham+V, Sham +5 mg/kg/day of WFA, Sham+10 mg/kg/day of WFA) and the other three groups with OVx (bilateral OVx include OVx+V, OVx +5 mg/kg/day, OVx+10 mg/kg/day of WFA). Mice were left for 8 weeks for the OVx mice to develop osteopenia. After 8 weeks, drill-hole injury was created in both sham and OVx groups. In mid femur region, the front skin was incised straight and longitudinally 1 cm in length under anesthetic condition. After splitting the muscle, periosteum was stripped to expose the femoral bone surface. A drill-hole injury was made 1 cm above the knee joint by inserting a drill bit with a diameter of 0.1 mm in the anterior portion of the diaphysis of the bilateral femurs. Treatment of WFA started from the next day of injury and continued for 21 days. Each animal received intraperitoneal administration of fluorochrome calcein (20 mg/kg) 24 h before autopsy. After 21 days of treatment as described above, all mice were killed and autopsied to collect their femurs for the measurement of bone micro architectural parameters at the drill-hole site. Bones were embedded in acrylic material and 50-μm sections were made using an Isomet BoneCutter (Buehler, Lake Bluff, IL, USA) and photographs taken under confocal microscope LSM 510 Meta, Carl Zeiss, Maple Grove, MN, USA) aided with appropriate filters. The intensity of calcein binding, which is an indicator of the amount of new mineral deposition, was calculated using Carl Zeiss AM 4.2 image-analysis software.28, 29, 30, 31
Preparation of WFA dispersion in gum acacia and its characterization
WFA was purified from W. somnifera grown in CSIR-CIMAP. Leaf tissue was used to purify WFA as discussed by Sanghwan et al.19 in the US patent.
WFA dispersion was prepared by dispersing fine powder gum acacia in 1 : 1 ratio. The mean particle size and size distribution of WFA dispersion (% in gum acacia) was determined using Malvern Zetasizer NanoZS (Malvern, Worcestershire, UK) as reported previously.56 The sample was dispersed in milli Q water and sonicated at 10% amplitude for 30 s before data acquisition. Similarly, charge of WFA dispersion was determined by measuring zeta potential (ξ) using Malvern Zetasizer NanoZS (Malvern). Rheological parameters of WFA dispersion (% in gum acacia) were calculated and analyzed using Bohlin Visco 88 viscometer equipped with Bohlin software (Malvern). Cone and plate geometry (cp 5.4°/30) was employed with 0.05 mm gap. The samples were centrifuged free of bubbles, and an amount sufficient to fill the cone and plate gap completely was carefully scooped out and spread over the cone (∼1 ml). The ambient temperature was maintained at 25±1 °C.
Studies on OVx mice
Eighty adult Balb/c mice (18–20 g each) were randomly divided into eight equal groups as follows: sham-operated (ovary intact) vehicle (gum acacia in distilled water), OVx vehicle, OVx+40 μg/kg of iPTH (3 days/week), OVx+ 3 mg/kg/day of Aln, OVx +1 mg/kg/day of WFA, OVx +5 mg/kg/day of WFA, OVx+10 mg/kg/day of WFA, OVx +0.3 mg/kg/day of Bzb. The intraperitoneal dose for iPTH (40 μg/kg for 3 days/week) used in this study as known anabolic agent.57 The possible role of various treatment (WFA, Bzb) was assessed in estrogen-deficient bone loss model for 8 weeks post OVx. The above mentioned various treatments started 8 weeks after the surgery and continued for 8 weeks. For dynamic histomorphometry measurements, each animal was administered intraperitoneally with fluorochromes tetracycline (20 mg/kg) on day 30 (1 week) and calcein (20 mg/kg) on day 60 (8 weeks) after the commencement of various treatments. After 8 weeks of treatment, all mice were killed and autopsied to collect bones (i.e., tibias and femurs) for measurement of bone parameter. BMCs from tibias and femurs of vehicle- or WFA-treated mice were harvested, and mineralization was studied as described previously. BMCs were cultured in bone marrow differentiation medium to induce mineralization for 18 days. Mineralization was quantified as described.53 Alizarin red-S stain was used for staining mineralized nodules, followed by extraction of the stain for quantification. For pharmacokinetic analysis the blood samples were withdrawn at various time intervals (0, 0/25,. 0.5, 1, 2, 3, 4, 5, 8, 24 h) after oral administration of WFA at the dose of 10 mg/kg. The plasma samples were analyzed by HPLC according to reported method with slight modification.58
Microcomputed tomography (μCT)
Sky Scan 1076 μCT scanner (Sky Scan, Aartselaar, Belgium) was used to carryout μCT scanning of excised bones as described before.59 The bone samples were scanned at a resolution of 9 μm and reconstruction was done using the Sky Scan Nrecon software. The X-ray source was set at 50 kV and 200 mA, with a pixel size of 9 μm. A hundred projections were acquired over an angul arrange of 180°. CT analyzer (CTAn, Skyscan) software was used to draw ellipsoid contours in selected trabecular bone. Mean intercept length method was used to calculate trabecular bone volume, Tb.No and trabecular separation of the distal femoral epiphysis (covering secondary spongiosa and the secondary ossification center) and proximal tibialmetaphysis. Tb.Th and SMI was calculated according to the method of Hildebrand and Ruegsegger.60 3D parameters were based on analysis of a Marchingcubes-type model with a rendered surface. CT Vol software was used to create 3D model of the bones.59
Uterine histology and histomorphometry
The uterus of each mouse was weighed and then fixed in 4% paraformaldehyde. A middle segment of each uterus was dehydrated in ascending grades of isopropanol, cleared in xylene and sections of about 5 μm thickness was prepared. Haematoxylin and eosin staining was done of 5 μm transverse sections and representative images were captured. Leica Qwin-Semiautomatic Image-Analysis software (Leica Microsystems GmbH, Solms, Germany) was used to measure total uterine area, luminal area and luminal epithelial height (Supplementary Figure S1).
Bone turnover and inflammatory marker assay
Rat-MID OCN EIA (Immunodiagnostic, Boldon, UK), TNF-alpha (R&D, Minneapolis, MN, USA) performed using serum from treated and vehicle groups using manufacturer’s protocol.
Statistics analysis
Data are expressed as mean±S.E. The data obtained in experiments with multiple treatments were subjected to one-way ANOVA followed by Newman–Keuls multiple comparison test of significance using Graph Pad Prism 3.02 software. Qualitative observations have been represented following assessments made by three individual experiments.
Abbreviations
- Ald:
-
alendronate
- ALP:
-
alkaline phosphatase activity
- AUC:
-
area under the curve
- BFR:
-
bone formation rate
- BrdU:
-
5-Bromo-2′-deoxyuridine
- Bzb:
-
Bortezomib
- BMCs:
-
bone marrow cells
- BMP2:
-
bone morphogenetic protein 2
- BV/TV:
-
bone volume/tissue volume
- Col I:
-
collagen type I
- EBSS:
-
earle's balanced salt solution
- IL-6:
-
interleukin-6
- MAR:
-
mineral apposition rate
- MCO:
-
mouse calverial osteoblast
- μ-CT:
-
micro-computed tomography
- iPTH:
-
injectible parathyroid hormone
- MCP-1:
-
monocyte chemo-attractant protein-1
- MRT:
-
mean residence time
- NF-κB:
-
nuclear factor κ-B
- OCN:
-
osteocalcin
- OPG:
-
osteoprotegrin
- OVx:
-
ovariectomy
- PI:
-
proteasomal inhibitor
- PNPP:
-
p-nitrophenyl pyro-phosphate
- Q-PCR:
-
quantitative real time PCR
- RunX2:
-
runt related transcription factor 2
- RANKL:
-
receptor activator of NF-κB ligand
- RANK:
-
receptor activator of nuclear factor kappa-B
- Smurf2:
-
smad ubiquitin regulatory factor
- SMI:
-
structural model index
- TRAP:
-
tartarate resistant acid phosphatase
- Tb.No:
-
trabecular number
- Tb.Th:
-
trabecular thickness
- TNF-α:
-
tumor necrosis factor α
- Vd:
-
volume of distribution
- WFA:
-
withaferin A
References
Katagiri T, Takahashi N . Regulatory mechanisms of osteoblast and osteoclast differentiation. Oral Dis 2002; 8: 147–159.
Kawamura N, Kugimiya F, Oshima Y, Ohba S, Ikeda T, Saito T et al. Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS ONE 2007; 2: e1058.
Hie M, Tsukamoto I . Increased expression of the receptor for activation of NF-kappaB and decreased runt-related transcription factor 2 expression in bone of rats with streptozotocin-induced diabetes. Int J Mol Med 2010; 26: 611–618.
Canalis E, Economides AN, Gazzerro E . Bone morphogenetic proteins, their antagonists, and the skeleton. Endocr Rev 2003; 24: 218–235.
Garrett IR, Chen D, Gutierrez G, Zhao M, Escobedo A, Rossini G et al. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J Clin Invest 2003; 111: 1771–1782.
Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem 2006; 281: 16502–16511.
Kaneki H, Guo R, Chen D, Yao Z, Schwarz EM, Zhang YE et al. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem 2006; 281: 4326–4333.
Cao Y, Zhang L . A Smurf1 tale: function and regulation of an ubiquitin ligase in multiple cellular networks. Cell Mol Life Sci 2013; 70: 2305–2317.
Chen D, Zhao M, Mundy GR . Bone morphogenetic proteins. Growth Factors 2004; 22: 233–241.
Chen AL, Fang C, Liu C, Leslie MP, Chang E, Di Cesare PE . Expression of bone morphogenetic proteins, receptors, and tissue inhibitors in human fetal, adult, and osteoarthritic articular cartilage. J Orthop Res 2004; 22: 1188–1192.
Cao X, Chen D . The BMP signaling and in vivo bone formation. Gene 2005; 357: 1–8.
Zhang Y, Chang C, Gehling DJ, Hemmati-Brivanlou A, Derynck R . Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc Natl Acad Sci USA 2001; 98: 974–979.
Severe N, Dieudonne FX, Marie PJ . E3 ubiquitin ligase-mediated regulation of bone formation and tumorigenesis. Cell Death Dis 2013; 4: e463.
Wertz IE, Dixit VM . Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol 2010; 2: a003350.
Giuliani N, Morandi F, Tagliaferri S, Lazzaretti M, Bonomini S, Crugnola M et al. The proteasome inhibitor bortezomib affects osteoblast differentiation in vitro and in vivo in multiple myeloma patients. Blood 2007; 110: 334–338.
Ito Y, Fukushima H, Katagiri T, Seo Y, Hirata S, Zhang M et al. Lactacystin, a proteasome inhibitor, enhances BMP-induced osteoblastic differentiation by increasing active Smads. Biochem Biophys Res Commun 2011; 407: 225–229.
Shapovalov Y, Benavidez D, Zuch D, Eliseev RA . Proteasome inhibition with bortezomib suppresses growth and induces apoptosis in osteosarcoma. Int J Cancer 2010; 127: 67–76.
Murray SS, Tu KN, Young KL, Murray EJ . The effects of lovastatin on proteasome activities in highly purified rabbit 20S proteasome preparations and mouse MC3T3-E1 osteoblastic cells. Metabolism 2002; 51: 1153–1160.
Sangwan RS, Chaurasiya ND, Misra LN, al P, Uniyal CC, Sangwan NS et al. An improved processfor isolation of withaferin-A from plant materials and products. US Patent 2006; 7: 870.
Gupta P, Goel R, Pathak S, Srivastava A, Singh SP, Sangwan RS et al. De Novo Assembly, Functional Annotation and Comparative Analysis of Withania somnifera Leaf and Root Transcriptomes to Identify Putative Genes Involved in the Withanolides Biosynthesis. PLoS One 2013; 8: e62714.
Maitra R, Porter MA, Huang S, Gilmour BP . Inhibition of NFkappaB by the natural product Withaferin A in cellular models of Cystic Fibrosis inflammation. J Inflamm 2009; 6: 15.
Matsuda H, Murakami T, Kishi A, Yoshikawa M . Structures of withanosides I, II, III, IV, V, VI, and VII, new withanolide glycosides, from the roots of Indian Withania somnifera DUNAL. and inhibitory activity for tachyphylaxis to clonidine in isolated guinea-pig ileum. Bioorg Med Chem 2001; 9: 1499–1507.
Mohan R, Hammers HJ, Bargagna-Mohan P, Zhan XH, Herbstritt CJ, Ruiz A et al. Withaferin A is a potent inhibitor of angiogenesis. Angiogenesis 2004; 7: 115–122.
Yang H, Shi G, Dou QP . The tumor proteasome is a primary target for the natural anticancer compound Withaferin A isolated from "Indian winter cherry". Mol Pharmacol 2007; 71: 426–437.
Mohamed AM . Bone loss and bone size after menopause. N Engl J Med 2003; 349: 1671 author reply 1671.
Ahlborg HG, Johnell O, Turner CH, Rannevik G, Karlsson MK . Bone loss and bone size after menopause. N Engl J Med 2003; 349: 327–334.
Pacifici R . T cells and post menopausal osteoporosis in murine models. Arthritis Res Ther 2007; 9: 102.
Sharan K, Mishra JS, Swarnkar G, Siddiqui JA, Khan K, Kumari R et al. A novel quercetin analogue from a medicinal plant promotes peak bone mass achievement and bone healing after injury and exerts an anabolic effect on osteoporotic bone: the role of aryl hydrocarbon receptor as a mediator of osteogenic action. J Bone Miner Res 2011; 26: 2096–2111.
Nagashima M, Sakai A, Uchida S, Tanaka S, Tanaka M, Nakamura T . Bisphosphonate (YM529) delays the repair of cortical bone defect after drill-hole injury by reducing terminal differentiation of osteoblasts in the mouse femur. Bone 2005; 36: 502–511.
Tanaka K, Tanaka S, Sakai A, Ninomiya T, Arai Y, Nakamura T . Deficiency of vitamin A delays bone healing process in association with reduced BMP2 expression after drill-hole injury in mice. Bone 2010; 47: 1006–1012.
He YX, Zhang G, Pan XH, Liu Z, Zheng LZ, Chan CW et al. Impaired bone healing pattern in mice with ovariectomy-induced osteoporosis: A drill-hole defect model. Bone 2011; 48: 1388–1400.
Tremollieres FA, Pouilles JM, Ribot C . Withdrawal of hormone replacement therapy is associated with significant vertebral bone loss in postmenopausal women. Osteoporos Int 2001; 12: 385–390.
Weitzmann MN, Pacifici R . Estrogen deficiency and bone loss: an inflammatory tale. J Clin Invest 2006; 116: 1186–1194.
Takano-Yamamoto T, Rodan GA . Direct effects of 17 beta-estradiol on trabecular bone in ovariectomized rats. Proc Natl Acad Sci USA 1990; 87: 2172–2176.
Teitelbaum SL . Bone resorption by osteoclasts. Science 2000; 289: 1504–1508.
Cao JJ . Effects of obesity on bone metabolism. J Orthop Surg Res 2011; 6: 30.
Rosen CJ, Bouxsein ML . Mechanisms of disease: is osteoporosis the obesity of bone? Nat Clin Pract Rheumatol 2006; 2: 35–43.
Gautam J, Kushwaha P, Swarnkar G, Khedgikar V, Nagar GK, Singh D et al. EGb 761 promotes osteoblastogenesis, lowers bone marrow adipogenesis and atherosclerotic plaque formation. Phytomedicine 2012; 19: 1134–1142.
Grover A, Shandilya A, Bisaria VS, Sundar D . Probing the anticancer mechanism of prospective herbal drug Withaferin A on mammals: a case study on human and bovine proteasomes. BMC Genomics 2010; 11 (Suppl 4): S15.
Kitson RP, Lu M, Siman R, Goldfarb RH . Proteasome inhibitor and lymphocyte function: partial inhibition of cell-mediated cytotoxicity and implication that the lymphocyte proteasome may contain multiple chymotryptic domains. In Vivo 2000; 14: 265–268.
Furuya Y, Mori K, Ninomiya T, Tomimori Y, Tanaka S, Takahashi N et al. Increased bone mass in mice after single injection of anti-receptor activator of nuclear factor-kappaB ligand-neutralizing antibody: evidence for bone anabolic effect of parathyroid hormone in mice with few osteoclasts. J Biol Chem 2011; 286: 37023–37031.
Majewski PM, Thurston RD, Ramalingam R, Kiela PR, Ghishan FK . Cooperative role of NF-{kappa}B and poly(ADP-ribose) polymerase 1 (PARP-1) in the TNF-induced inhibition of PHEX expression in osteoblasts. J Biol Chem 2010; 285: 34828–34838.
Li YP, Stashenko P . Proinflammatory cytokines tumor necrosis factor-alpha and IL-6, but not IL-1, down-regulate the osteocalcin gene promoter. J Immunol 1992; 148: 788–794.
Gilbert L, He X, Farmer P, Rubin J, Drissi H, van Wijnen AJ et al. Expression of the osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A) is inhibited by tumor necrosis factor-alpha. J Biol Chem 2002; 277: 2695–2701.
Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C, Komori T et al. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol Cell Biol 2000; 20: 8783–8792.
Trivedi R, Goswami R, Chattopadhyay N . Investigational anabolic therapies for osteoporosis. Expert Opin Investig Drugs 2010; 19: 995–1005.
Trivedi R, Mithal A, Chattopadhyay N . Anabolics in osteoporosis: the emerging therapeutic tool. Curr Mol Med 2010; 10: 14–28.
Dixit P, Chillara R, Khedgikar V, Gautam J, Kushwaha P, Kumar A et al. Constituents of Dalbergia sissoo Roxb. leaves with osteogenic activity. Bioorg Med Chem Lett 2012; 22: 890–897.
Kumar A, Singh AK, Gautam AK, Chandra D, Singh D, Changkija B et al. Identification of kaempferol-regulated proteins in rat calvarial osteoblasts during mineralization by proteomics. Proteomics 2010; 10: 1730–1739.
Cappellen D, Luong-Nguyen NH, Bongiovanni S, Grenet O, Wanke C, Susa M . Transcriptional program of mouse osteoclast differentiation governed by the macrophage colony-stimulating factor and the ligand for the receptor activator of NFkappa B. J Biol Chem 2002; 277: 21971–21982.
Tyagi AM, Gautam AK, Kumar A, Srivastava K, Bhargavan B, Trivedi R et al. Medicarpin inhibits osteoclastogenesis and has nonestrogenic bone conserving effect in ovariectomized mice. Mol Cell Endocrinol 2010; 325: 101–109.
Ariyoshi W, Takahashi T, Kanno T, Ichimiya H, Shinmyouzu K, Takano H et al. Heparin inhibits osteoclastic differentiation and function. J Cell Biochem 2008; 103: 1707–1717.
Khedgikar V, Gautam J, Kushwaha P, Kumar A, Nagar GK, Dixit P et al. A standardized phytopreparation from an Indian medicinal plant (Dalbergia sissoo) has antiresorptive and bone-forming effects on a postmenopausal osteoporosis model of rat. Menopause 2012; 19: 1336–1346.
Siddiqui JA, Swarnkar G, Sharan K, Chakravarti B, Gautam AK, Rawat P et al. A naturally occurring rare analog of quercetin promotes peak bone mass achievement and exerts anabolic effect on osteoporotic bone. Osteoporos Int 2011; 22: 3013–3027.
Sashidhara KV, Kumar M, Khedgikar V, Kushwaha P, Modukuri RK, Kumar A et al. Discovery of coumarin-dihydropyridine hybrids as bone anabolic agents. J Med Chem 2013; 56: 109–122.
Kumar A, Gupta GK, Khedgikar V, Gautam J, Kushwaha P, Changkija B et al. In vivo efficacy studies of layer-by-layer nano-matrix bearing kaempferol for the conditions of osteoporosis: a study in ovariectomized rat model. Eur J Pharm Biopharm 2012; 82: 508–517.
Motyl KJ, McCauley LK, McCabe LR . Amelioration of type I diabetes-induced osteoporosis by parathyroid hormone is associated with improved osteoblast survival. J Cell Physiol 2012; 227: 1326–1334.
Gupta GK, Kumar A, Khedgikar V, Kushwaha P, Gautam J, Nagar GK et al. Osteogenic efficacy enhancement of kaempferol through an engineered layer-by-layer matrix: a study in ovariectomized rats. Nanomedicine 2013; 8: 757–771.
Trivedi R, Kumar A, Gupta V, Kumar S, Nagar GK, Romero JR et al. Effects of Egb 761 on bone mineral density, bone microstructure, and osteoblast function: Possible roles of quercetin and kaempferol. Mol Cell Endocrinol 2009; 302: 86–91.
Hildebrand T, Ruegsegger P . Quantification of Bone Microarchitecture with the Structure Model Index. Comput Methods Biomech Biomed Engin 1997; 1: 15–23.
Acknowledgements
Grant from CSIR (BSC0201) is acknowledged. PKT and NSS acknowledge Council of Scientific and Industrial Research, New Delhi, Govt. of India, for providing the financial support to carry out this work under NMITLI scheme. PRM acknowledge funding from BIOCERAM (ESC0103). VK, JG acknowledge University Grants Commission, New Delhi, Govt. of India for fellowship, PK acknowledge Council of Scientific and Industrial Research CSIR, New Delhi, Govt. of India for fellowship. This is CDRI communication number 156/2013/RT.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Federici
Supplementary Information accompanies this paper on Cell Death and Disease website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Khedgikar, V., Kushwaha, P., Gautam, J. et al. Withaferin A: a proteasomal inhibitor promotes healing after injury and exerts anabolic effect on osteoporotic bone. Cell Death Dis 4, e778 (2013). https://doi.org/10.1038/cddis.2013.294
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.294
Keywords
This article is cited by
-
Involvement of MYB family transcription factors, WsMYBL1 and WsMYBL2, in withanolide biosynthesis in Withania somnifera, a medicinal plant
Plant Cell, Tissue and Organ Culture (PCTOC) (2024)
-
Alternative Medical Therapy
Indian Journal of Orthopaedics (2023)
-
Growth kinetics and withanolide production in novel transformed roots of Withania somnifera and measurement of their antioxidant potential using chemiluminescence
Plant Cell, Tissue and Organ Culture (PCTOC) (2018)
-
Bortezomib prevents ovariectomy-induced osteoporosis in mice by inhibiting osteoclast differentiation
Journal of Bone and Mineral Metabolism (2018)
-
Withania somnifera as a Potential Anxiolytic and Anti-inflammatory Candidate Against Systemic Lipopolysaccharide-Induced Neuroinflammation
NeuroMolecular Medicine (2018)
