
Abstract
Lysosomal regulation is a poorly understood mechanism that is central to degradation and recycling processes. Here we report that LAMTOR1 (late endosomal/lysosomal adaptor, MAPK and mTOR activator 1) downregulation affects lysosomal activation, through mechanisms that are not solely due to mTORC1 inhibition. LAMTOR1 depletion strongly increases lysosomal structures that display a scattered intracellular positioning. Despite their altered positioning, those dispersed structures remain overall functional: (i) the trafficking and maturation of the lysosomal enzyme cathepsin B is not altered; (ii) the autophagic flux, ending up in the degradation of autophagic substrate inside lysosomes, is stimulated. Consequently, LAMTOR1-depleted cells face an aberrant lysosomal catabolism that produces excessive reactive oxygen species (ROS). ROS accumulation in turn triggers p53-dependent cell cycle arrest and apoptosis. Both mTORC1 activity and the stimulated autophagy are not necessary to this lysosomal cell death pathway. Thus, LAMTOR1 expression affects the tuning of lysosomal activation that can lead to p53-dependent apoptosis through excessive catabolism.
Similar content being viewed by others

Main
Lysosomes serve as the important intracellular function of compartmentalizing pH-dependent degradation. They receive extracellular components via endocytosis and intracellular material through autophagy or the biosynthetic pathway.1, 2 These catabolic functions are essential regulators of cellular homeostasis, and lysosomal dysfunction is implicated in various pathological processes including neurodegenerative diseases.3 Being part of a highly dynamic endocytic system that implicates constant exchange of incoming and outgoing membranes and multiple fusion events, cellular mechanisms regulating lysosomal maturation and activity remain incompletely understood.2 Growing evidence indicates that lysosome positioning in cells can modulate lysosomal function.4, 5, 6 For instance, their clustering in the perinuclear area facilitates autophagy,6 probably because of the spatially-restricted increased concentration of lysosomes available for fusion with autophagosomes.
Recently, we and others have simultaneously identified LAMTOR1 (late endosomal/lysosomal adaptor MAPK and mTOR activator 1), a novel adaptor of the lysosomal membrane.5, 7, 8, 9 This lipidated protein anchors on the surface of lysosomes the protein complex that drives amino acid-dependent mTORC1 activation.9 The activation status of mTORC1 depends not only on whether it is bound to lysosomes, but also on lysosomal positioning at the cell periphery.6 LAMTOR1 also serves as an essential anchor for the p14-MP1–MEK–ERK pathway in late endosomes/lysosomes.5 Its loss results in redistribution of lysosomes throughout the cytoplasm, which is associated with alterations in the processing of lysosomes and impaired cycling of rab11-positive recycling endosomes.5, 10 These defects are attributed to the downregulation of both late endosomal-associated MEK–ERK activity5 and mTORC1 activity.10 Noticeably, this altered endosome dynamics likely contributes to the perturbation in cellular free cholesterol homeostasis associated with LAMTOR1 depletion.8, 10 Lastly, LAMTOR1 may also regulate actin remodeling by activating RhoA and RhoC through its binding to p27kip1.7, 11
In this study, we show that the endolysosomal dysfunctions induced by LAMTOR1 depletion encompass an unexpected excessive catabolism that triggers a lysosomal reactive oxygen species (ROS)–p53 cell death pathway. These findings may help to better understand the pathologies associated with lysosomal dysfunction such as certain neurodegenerative diseases in which a lysosmal activity enhancement is often observed.
Results
LAMTOR1 depletion induces p53-mediated cell cycle arrest and apoptosis, independently of mTORC1 regulation
During the course of previous studies,8 we found that LAMTOR1 depletion via short interfering RNA (siRNA)-mediated knockdown led, within 72 h, to reduced SHEP cell numbers (Figure 1a) associated with apparent cell death. Cell cycle analysis of LAMTOR1-depleted cells, using pulse 5′-bromo-2-deoxyuridine (BrdU) labeling, showed a lower percentage of cells entering into S phase compared with control siRNA (Figure 1b), confirming a decreased cell cycle entry. Flow cytometric analysis of annexin V binding showed an increase in cellular staining that could be prevented by the pan-caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-(O-methyl)-fluoromethylketone (zVAD), but not by the z-phe-ala fluoromethylketone (zFA) inhibitor control (Figure 1c), indicating that LAMTOR1-depleted SHEP cells were concomitantly undergoing apoptosis. Given that SHEP cells are wild type for p53 and that p53 is an important cell cycle and cell death regulator,12 it was reasoned that the observed cellular response might be due to p53 activation. Both an increase in p53 expression and in p21 expression, a cyclin-dependent kinase inhibitor whose gene is transcriptionally activated by p53, could be readily observed in LAMTOR1-depleted SHEP cells (Figure 1d). LAMTOR1 silencing-induced biological outcomes are observed irrespective of the use of two independent siRNA A or B (as exemplified in Figure 1d), or when using of a pool of siRNA A and B, which further reduces off-target silencing due to competition among the siRNAs13 (Supplementary Figure S1). To confirm that LAMTOR1 depletion-induced apoptosis is elicited through p53, we compared an isogenic set of human colon adenocarcinoma cell lines (HCT116) differing only in their p53 status.14 The parental HCT116 line, containing wild-type p53 (p53+/+) exhibited a marked increase in annexin V staining upon LAMTOR1 depletion (Figure 2a). However, the p53-deficient derivative (p53−/−) was much less sensitive to LAMTOR1 depletion. Consistently, both an increase in p53 expression and in p21 expression was detected in the parental HCT116 line, but not in its p53−/− counterpart (Figure 2b). Therefore, LAMTOR1 depletion induces p53-dependent cell cycle inhibition and apoptosis.

LAMTOR-1 depletion induces an increase in both p53 and p21 expression. SHEP cells were transfected with scrambled or LAMTOR1 (B) siRNAs, and viable cells were counted (a) or stained with BrdU and PI for cell cycle analysis (b), 72 h after transfection. Values are means+S.E.M. of three independent experiments. **P<0.01 using Mann–Whitney test. (c) FACS analysis of Annexin V/PI staining of SHEP cells transfected with the indicated siRNAs as above and treated or not with zFA (20 μM) or zVAD (20 μM) for 72 h; n=3. **P<0.01 using Mann–Whitney test. (d) SHEP cells transfected with scrambled or LAMTOR1 (A or B) siRNAs were analyzed for expression levels of P53, P21, LAMTOR1 and actin proteins by western blotting; n=5

LAMTOR-1 depletion induces p53-mediated apoptosis. (a) Representative FACS analysis of Annexin V/PI staining of HCT116 cells transfected with scrambled or LAMTOR1 (B) siRNAs for 36 h; n=3. **P<0.01 using Mann–Whitney test. (b) HCT116 cells transfected with scrambled or LAMTOR1 (B) siRNAs for 36 h were analyzed for expression levels of P53, P21, LAMTOR1 and actin proteins by western blotting; n=4. (c) MCF10-A cells were transfected with scrambled shRNA plasmid (shCtrl) or shRNA plasmids targeted against Rictor or Raptor. The corresponding protein expression levels are shown in the left panel. These cells further transfected with scrambled or LAMTOR1 (A) siRNAs for 72 h were analyzed for expression levels of P53, P21, LAMTOR1 and actin proteins (right panel); n=3
LAMTOR1 depletion abrogates mTORC1 activation.9 To test whether p53 activation is linked to dysfunctional mTORC1 regulation, we investigated the effect of LAMTOR1 depletion in the non-tumorigenic human epithelial cell line MCF10-A in which the endogenous raptor (mTORC1) or rictor (mTORC2) have been knocked down using specific shRNA.15 These cells have significantly reduced levels (>90% reduction) of each of the targeted proteins (Figure 2c, left panel). In the different shRNA expressing cells, LAMTOR1 depletion could still increase p53 and p21 expressions (Figure 2c, right panel). Thus, mTORC1 inhibition due to LAMTOR1 depletion is not necessary to trigger p53 activation.
LAMTOR1 depletion causes lysosomal activation
As an initial step to characterize the mechanism by which LAMTOR1 depletion induces p53 activation, we used electron microscopy to assess intracellular perturbations at the ultrastructural level. Compared with control siRNA-treated cells, LAMTOR1 depletion induced numerous intracellular vesicles of different sizes and shapes (Figure 3a). Many of these vesicles met standard morphologic criteria for autophagosomes, autolysosomes and lysosomes.16, 17 The increase in autophagic and lysosomal structures upon LAMTOR1 depletion is consistent with an abrogated mTORC1 activation that stimulates the autophagic process.18, 19 Intriguingly, however, the increased lysosomal structures were scattered throughout the cytoplasm, instead of being clustered in the perinuclear area to facilitate autophagosome–lysosome fusion.6 Therefore, the autophagic process might not be fully functional in these cells. In addition, LAMTOR1 depletion can affect the processing and the final maturation of lysosomes.5, 10 Thus, we evaluated the extent of these lysosomal dysfunctions that might be linked with p53 activation.

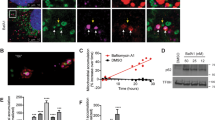
LAMTOR1 depletion induces lysosomal expansion. (a) Electron microscopy of SHEP cells transfected with scrambled siRNA (top panels), or with LAMTOR1 siRNA B (middle and bottom panels). Scale bars are indicated. Autophagosomes (white arrows), autolysosomes (black arrows) and lysosomes (arrow heads). (b) Confocal images of representative cathepsin D immuno-staining in SHEP cells transfected with scrambled or LAMTOR1 (B) siRNAs. Scale bar= 10 μm. (c) SHEP cells were transfected with scrambled or LAMTOR1 (B) siRNAs for the indicated times and the expression levels of P53, P21, mature cathepsin D, LAMP-1 and LAMTOR1 proteins were analyzed by western blotting; n=3. (d) SHEP cells transfected with scrambled or LAMTOR1 (B) siRNAs for 72h, were further treated or not during the last 6 h with 20 μM CQ to inhibit lysosomal activity. Expression levels of LC3-I, LC3-II, P62, LAMTOR1 and actin proteins were analyzed by western blotting; n=4
First, we assessed the maturation and levels of the major lysosomal enzyme cathepsin D. Cathepsin D is transported from the Golgi through late endosomes to lysosomes.20 Its mature chains (34 and 14 kDa) are produced by the cleavage of the intermediate chain (48 kDa) upon exposure to the acidic environment in lysosomes. Importantly, any change in its mature forms reflects altered late endocytic trafficking or perturbed lysosomal maturation. Consistent with previous report,5 LAMTOR1 depletion disrupted accumulation of cathepsin D-positive lysosomal structures in the perinuclear region and dispersed them throughout the cytoplasm (Figure 3b, see also Supplementary Figure S4). Despite this dispersion, LAMTOR1 depletion increased the level of mature cathepsin 34 kDa chain (Figure 3c, the antibody used does not recognize the 14 kDa chain) without increasing its intermediate or immature forms (Figure 4b). The increase in mature cathepsin D was not specific to SHEP cells as it was recapitulated in the p53+/+ HCT116 line (Supplementary Figure S2B). Thus, the altered spatial organization of lysosomes does not significantly impair lysosomal processing. Together with the augmented number of lysosomal structures, the increased levels of both mature cathepsin D and lysosome-associated membrane protein-1 (LAMP-1) (Figure 3c) are consistent with an expansion of the lysosomal system.

The increased lysosomal activity generates ROS. (a) Representative FACS histograms of H2DCF-DA analysis of scrambled or LAMTOR1 (B) siRNA-transfected SHEP cells for 72 h, treated or not with 2.5 μM of CQ; n=5. (b) LAMTOR1 (B) siRNA-treated SHEP cells were treated with indicated concentrations of CQ for 72 h. The maturation process of cathepsin D was analyzed by western blotting. Pro, pro-cathepsin D; Int, intermediate form of cathespsin D; Mat, mature form of cathespsin D; n=2 (c) Untransfected or transfected SHEP cells with the indicated siRNAs (LAMTOR1 siRNA B) for 72 h, were analyzed for expression levels of cathepsin D, LC3-I, LC3-II, p62, Beclin1, LAMTOR1, and actin by western blotting n=2. (d) Representative FACS histograms of H2DCF-DA analysis of SHEP cells transfected with the indicated siRNAs for 72 h; n=5
Second, to evaluate the proper function of lysosomes, we assessed autophagic flux, that is, the dynamic process of autophagosome synthesis, their fusion with lysosomes, and the degradation of autophagic substrates inside the lysosome. We analyzed the conversion and turnover of the autophagic marker microtubule-associated protein light chain 3 (LC3) and the degradation of the autophagic-specific substrate p62.21 LC3-II is a lipidated form of LC3-I that inserts into autophagic membranes. P62 is selectively incorporated into autophagosomes through direct binding to LC3-II. Both markers are eventually degraded by lysosomal hydrolases following fusion of autophagosomes with lysosomes. Compared with control siRNA-treated cells, LAMTOR1-depleted cells displayed increased levels of LC3-I and to a lesser extent of LC3-II, and decreased levels of p62 (Figure 3d). The lysosomotropic agent chloroquine (CQ) is an alkylamine that accumulates in the lysosome and neutralizes the lysosomal acidity by its weak base property,22 thereby reducing lysosomal activity. In the presence of CQ, the amounts of both LC3-II and p62 were substantially increased. Consistent with the current understanding of LC3 and p62 flux,21 these results indicate that despite a scattered lysosomal positioning, the autophagic flux is not substantially altered during LAMTOR1 depletion.
Therefore, despite an altered lysosome positioning, we present three lines of evidence (electron microscopic, lysosomal hydrolase and autophagic analysis) that consistently support the notion that LAMTOR1 depletion results in expansion of an overall functional lysosomal system.
Lysosomal activation generates ROS
Lysosomal catabolism is recognized as a potent source of ROS.23, 24 Using the fluorescent ROS indicator dichlorodihydrofluorescein diacetate (H2DCF-DA), we next assessed whether the stimulated lysosomal activation could increase the intracellular levels of ROS. We found that LAMTOR1 depletion evoked an increased ROS production as compared with control siRNA (Figure 4a). To examine whether this increased ROS production was due to a stimulated lysosomal catabolism, we used the lysosomal activity inhibitor CQ. As a parameter of lysosomal degradative capacity, we examined the pH-sensitive cleavage of cathepsin D. Concentrations above 2.5 μM CQ completely blocked cathepsin D cleavage, allowing only the detection of the pro-cathepsin D intermediate chains (Figure 4b). As previously reported,25 these CQ concentrations also induced significant cell death (data not shown). In contrast, cellular treatment with 2.5 μM CQ for 72 h, significantly reduced the appearance of the 34 kDa mature chain of cathepsin D (Figure 4b), without affecting the viability of SHEP cells (Figure 5a). In these cells that display a reduced lysosomal degradative capacity, LAMTOR1 depletion could no longer trigger ROS production (Figure 4a). A reproducible, albeit less robust increase in ROS production was also demonstrated in p53+/+ HCT116 line upon LAMTORT1 depletion (Supplementary Figure S2C). This ROS production was also prevented by low doses of CQ (Supplementary Figure S2B and S2C). Thus, these results strongly support a functional link between an increased lysosomal activity and ROS production.

ROS are causative in LAMTOR-1 depletion-induced cell death. (a) Representative FACS analysis of Annexin V/PI-stained cells. SHEP cells transfected with scrambled or LAMTOR1 (A) siRNAs were treated or not with 2.5 μM of CQ for 72 h; n=3. **P<0.01 using Mann–Whitney test. (b) SHEP cells transfected with the indicated scrambled or LAMTOR1 (B) siRNAs, were treated or not with anti-oxydant TROLOX at the indicated concentrations, for 72 h. The expression levels of P53, P21, cathepsin D, LAMTOR1 and actin were measured by western blotting; n=3. (c) SHEP cells transfected with scrambled or LAMTOR1 (B) siRNAs were treated or not with TROLOX (500 nM) for 72 h, and viable cells were counted. Values are mean+S.E.M. of three independent experiments. **P<0.01 using Mann–Whitney test
We then evaluated the contribution of the autophagic flux, which increases lysosomal catabolism, to ROS production. Beclin1 and autophagy-related gene 5 (ATG5) are an important autophagic components.21 We found that Beclin1 siRNA efficiently down-regulated Beclin1 protein expression, and consequently limited the increase in LC3-I and LC3-II and the decrease in p62, upon LAMTOR1 silencing (Figure 4c). However, under this condition of impaired autophagic flux, both ROS production (Figure 4d) and the increase in cathepsin D (Figure 4c) remained unaffected. It is of note that the reduced cellular viability of LAMTOR1-depleted cells was also not affected when autophagic flux was impaired (cells numberx105: Scrambled, 8.5±0.69; siBeclin, 7.8±0.49; siLAMTOR1, 4.2±0.55; siLAMTOR+siBeclin, 3.9±0.39. Values are means±S.E.M.). Similar results were obtained upon ATG5 silencing (Supplementary Figure S3). Thus, the stimulated autophagic flux does not contribute significantly to ROS production.
ROS are causative in LAMTOR1 depletion-induced apoptosis
Lysosomal ROS generation can cause lysosomal membrane permeabilization (LMP), whereby lysosomal cathepsins are released from the lysosomal lumen to the cytosol and can trigger cell death.26, 27 However, the absence of diffuse cathepsin D staining in the cytosol, a translocation that is widely used to monitor lysosomal membrane permeabilization,26 suggested a conserved integrity of lysosomal membranes (see Figure 3b and Supplementary Figure S4 for a time course study). Even though we cannot exclude the presence of LMP at levels below the detection limit of the assay, this nonetheless contrasts with the readily detectable cytoplasmic cathepsin D staining upon positive control for partial LMP (Supplementary Figure S4).
To ascertain the causal relationship between lysosomal ROS production and p53 activation, we treated cells with 2.5 μM CQ that inhibits ROS production, as depicted in Figure 4a. This CQ concentration substantially reduced LAMTOR1 depletion-induced apoptosis (Figure 5a), and p53+/+ HCT116 cell death (Supplementary Figure S2D). Thus, the inhibition of ROS production correlates with reduced apoptosis. Then, we investigated the effect of the widely used radical-scavenging vitamine E derivative 6-hydroxy-2,5,7,8-tetramethyl-chroman-2-carboxylic acid (Trolox, Sigma, Saint Quentin Fallavier, France).28 Trolox prevented both p53 and p21 increase following LAMTOR1 depletion (Figure 5b, and Supplementary Figure S2E). Furthermore, Trolox treatment could partially protected SHEP cells from LAMTOR1 depletion-induced death (Figure 5c). Therefore, these data indicate that ROS production mediates, at least in part, p53-induced apoptosis.
Discussion
In this study, we show that LAMTOR1 depletion leads to p53-dependent apoptosis due to a deregulated lysosomal catabolism and ROS production.
The downregulation of LAMTOR1 protein results in a strong increase in lysosomal structures and in their dispersion throughout the cytoplasm. The subcellular distribution of lysosomes depends on their being transported along microtubules.29, 30 However, Takahashi et al.10 reported that vesicle movement is not affected in LAMTOR−/− mouse embryonic fibroblasts cells and that dispersion is more likely due to defects in lysosomal maturation. Regardless of the explanation for how lysosomes become scattered, the alteration in lysosome positioning can be associated with certain dysfunctions.4, 5, 6, 10 Interestingly, we found that these scattered lysosomal structures remain nonetheless overall functional. The assessment of cathepsin D maturation reveals no major defect in late endocytic trafficking or perturbed lysosomal maturation. Furthermore, we observed an increase in autophagic flux, indicating that the fusion of autophagosomes with lysosomes and the subsequent degradation of autophagic cargo are not substantially altered. Consistent with these findings, it has been reported that although depletion of Huntingtin or Dynein disperses the endosomes and lysosomes through the cytoplasm, only minor defects in intracellular trafficking are observed.31 Thus, LAMTOR1-depleted cells cope with a lysosomal enhancement.
How LAMTOR1 regulates lysosomal activity is unclear at present. The altered positioning of lysosomes on its own may not be sufficient to trigger an aberrant lysosomal catabolism. Indeed, the downregulation of mTORC1 activity has been implicated in lysosomal scattering,10 but we have shown that mTORC1 inhibition is not sufficient to trigger p53 activation. Furthermore, we have found that the silencing of Huntingtin, which compromised intracellular vesicular trafficking, did not lead to cell death in SHEP cells (data not shown). A lysosomal enhancement is often observed in neurodegenerative diseases where it is sought to represent a compensatory response to promote the clearance of toxic protein accumulation.32 It is possible that the increased macropinocytosis associated with LAMTOR1 depletion8 together with the altered lysosomal positioning, results in an accumulation of endocytic material that triggers such a compensatory response.
In order to investigate whether LAMTOR1's expression is altered in some neurodegenerative diseases, we analyzed the European bioinformatics institute (EBI) center databases, which contained studies (E-GEOD-3790) (E-AFMX-6) showing that LAMTOR1′s expression is clearly and significantly decreased in Huntington's disease (t-test=−3, P=0.035).33 Furthermore, another study published in the EBI database (E-GEOD-7307) shows that LAMTOR1′s expression in Parkinson's disease decreases significantly (t-test=−7.1, P=1.71 × 10–10). These findings, together with our results, support a potential role for LAMTOR1 in the apoptotic process of some neurons induced in certain neurodegenerative disease. It is of note that enhancement of lysosomal activity has been proposed as a treatment strategy against certain neurodegenerative diseases.32 Our results call for caution in developing such potential treatment as, at least in certain contexts exemplified by LAMTOR1 depletion, lysosomal enhancement can lead to cell death.
Even though macroautophagy proceeded normally, we have nonetheless investigated whether mitophagy was compromised upon LAMTOR1 silencing. As shown in Supplementary Figure S5, mitophagy was unaffected, and therefore did not contribute to ROS production. In addition to mitochondria, lysosomes are now recognized as independent intracellular sources of ROS.23, 24 The current view is that ROS are produced by the specific activity of lysosomes such as the redox action of iron, through the catalysis of Fenton-type reactions. The use of low doses of the lysosomal inhibitor CQ to limit lysosomal catabolism revealed that the expansion of functional lysosomes generates excessive ROS production. Noticeably, this increased ROS production does not require the implementation of autophagy, indicating that the autophagic catabolism does not contribute significantly to ROS production in this system. In turn, ROS that are known to regulate p53 function through multiple mechanisms,34 lead to p53-dependent apoptosis. It is well described that ROS can disrupt the integrity of the lysosomes, and that this LMP then triggers caspase-mediated apoptosis or cathepsin-mediated necrotic cell death.26, 27 However, we did not find strong evidence for LMP in our studies. Whether there is some LMP that is below the detection limit of our assays remains formally possible. Nevertheless, because LMP was readily detectable in positive controls, these results suggested that lysosmal membranes are more stable upon LAMTOR1 depletion. In support of this, it has been reported that lysosomal membrane stability increases upon cholesterol accumulation,35 and we and others have demonstrated that LAMTOR1 depletion resulted in increased intracellular levels of cholesterol.8, 10
Thus, LAMTOR1 downregulation affects the tuning of lysosomal activation, which can trigger a ROS-p53-dependent apoptosis. We anticipate that these findings will be of importance to better understand the pathologies associated with lysosomal dysfunction.
Materials and Methods
Materials and reagents
Peroxidase-conjugated goat anti-mouse and anti-rabbit IgG, and Cy3-conjugated donkey anti-mouse IgG were from Jackson Immuno-Research (Suffolk, England, UK). The primary antibodies included anti-lamp-1 (H4A3, Developmental Studies Hybridoma Bank, Iowa City, IA, USA), anti-cathepsin D (Ab-2, Calbiochem, Saint Quentin en Yvelines, France), anti-p62 (SQSTM1, Enzo, Villeurbane, France), anti-LC3 (51–11, MBL International Corporation, Nanterre, France), anti-p53 (D0–7, DAKO), anti-Beclin (E-8, Santa-Cruz, Le perray en Yvelines, France), anti-ATG5 (D1G9, Cell Signaling, Montigny le Bretonneux, France), anti-p21 Waf1/Cip1 (DCS60, Cell Signaling), anti-Raptor (24C12, Cell Signaling), anti-Rictor (53A2, Cell Signaling), and anti-BrdU (3D4, fluorescein isothiocyanate (FITC)-conjugated, Pharmingen, Le pont de Claix, France). Anti-LAMTOR-1 (rabbit pAb) was developed in collaboration with Eurogenetec. The reagents included Trolox ((±)-6-Hydroxy-2,5,7,8-tetramethylchromane-2-carboxylic acid (Sigma)), CQ diphosphate salt (Sigma), Lumi-light western blotting substrate (Roche, Meylan, France), human Annexin V-FITC Kit (AbCys SA, Les Ulis Courtaboeuf, France), and propidium iodide (PI, Sigma).
Cells culture
SHEP cell line is a clone derivative of the neuroblastoma cell line SK-N-SH 36 that lacks most neuronal characteristics and has a wild-type p53.37 SHEP cells were grown in RPMI-1640 (Invitrogen, Saint Aubin, France) supplemented with 10% heat-inactivated fetal calf serum (Lonza, Basel, Switzerland) and antibiotics. Human colon carcinoma HCT116 p53+/+ and p53−/− cells were cultured in Macoy′s medium (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum. MCF10-A ShCtrl, ShRictor, and ShRaptor cells lines were cultured as described.38
SiRNA & ShRNA gene knockdown
Cells were transfected with nonspecific scrambled siRNA (siRNA#3, Dharmacon, Villebon sur Yvette, France), or LAMTOR1 siRNA A (5′-GGAGCUGGUUGUACAGUUU-3′), LAMTOR1 siRNA B (5′-CAGCCAGCAACAUCAUUGAUU-3′), or a pool of LAMTOR1 siRNA A and B, by Hiperfect transfection reagent (QIAGEN, Les Ulis, France) according to the manufacturer’s instructions. Experiments were repeated using alternatively siRNA A or B, or the A/B pool. ShCtrl (Addgene plasmid 1864), ShRaptor (Addgene plasmid 1857) and ShRictor (Addgene plasmid 1853) plasmids were obtained from Dr. DM Sabatini laboratory.15 MCF10-A cells expressing the different shRNAs were established according to Addgene’s instructions and as described.15
Apoptosis assays
In all, 2 × 105 cells were plated in six-well plates such that cells would be 50% confluent the following day at the time of transfections and/or treatments. For analysis, floating and adherent cells were collected and centrifuged 5 min at 1000, r.p.m. The cell pellet was re-suspended in Annexin V/PI labeling solution according to the manufacturer′s instructions, and analyzed by fluorescence-activated cell sorting (FACS).
ROS assay
ROS were monitored by treating cells with 10 μM H2.DCFDA (Molecular Probes, Saint Aubin, France) in complete medium, for 20 min. Cells were then harvested, washed twice with phosphate-buffered saline (PBS) containing 10% heat-inactivated fetal calf serum, re-suspended in 200 μl of PBS and analyzed by FACS.
Transmission electron microscopy
Transfected cells were fixed by adding 4% glutaraldehyde in cell culture medium for 15 min at 4°C. Fixation was completed with 4% glutaraldehyde–0.2 M Na-cacodylate/HCl buffer at pH 7.4 for 30 min at 4 °C and samples were washed three times with a 0.2 M Na-cacodylate/HCl buffer at pH 7.4 and 0.4 M saccharose solution for 30 min. Cells were then post-fixed in 2% OsO4–0.3 M Na-cacodylate/HCl buffer at pH 7.4 for 45 min at 4 °C, dehydrated in graded ethanol (5 min in 30%, 50 and 70% ethanol, 15 min in 95% ethanol and three times for 15 min in pure ethanol), and embedded in Epon. Ultrathin sections were cut with a RMX-MTL Ultramicrotome (VENTANA, Illkirch, France), contrasted with methanolic uranyl acetate and lead citrate, and then examined using a JEOL 1200 EX transmission electron microscope equipped with MEGAVIEW II SIS camera (Olympus SIS-ELOÏSE, Roissy, France) and AnalySIS software (Olympus SIS-ELOÏSE).
Immunofluorescence
Immunostaining was performed as described.8 Primary antibody used to label cathepsin D was revealed by incubation with Cy5-Conjugated anti-mouse secondary antibody. Fixed cells were examined under a Leica DMRB (Leica, Nanterre, France) for conventional immunofluorescence, or under a Leica TCS-SP confocal microscope (Leica) for confocal analysis and sections were constructed with Leica Confocal Software (Leica). Fluorescence signals were recorded using a 63X Plan-NEOFLUAR oil immersion objective (Leica).
Cell cycle assay
Treated cells were incubated in RPMI medium containing BrdU (1:1000) for 1h at 37 °C and 5% CO2. After washing with PBS, cells were fixed with cold Ethanol 70% at 4 °C for 20 min and incubated at room temperature sequentially with HCL3N (20 min), Borax (0,1 M, 3 min), and anti-BrdU-FITC (20 min). Then cells were re-suspended in Propidium Iodide buffer (RNAse buffer+PI 5%+RNAse 5%) and analyzed by FACS.
Proliferation assay
Cells were collected and re-suspended in PBS. Dead cells were stained by trypan blue 0,04%. The numbers of living cells were determined using the Countess Automated Cell Counter (Invitrogen).
Mitochondrial integrity
Treated cells were stained with MitoTraker deep red (50 nM, Invitrogen) for 30 min at 37 °C. After a wash with serum-free medium, cells were harvested, washed twice with PBS, re-suspended in PBS containing 1% heat-inactivated fetal calf serum and analyzed by FACS to assess levels of mitochondrial function and integrity.39
Western blotting analysis
Cells were lysed as described8 and proteins were separated on 10–15% SDS–PAGE. Immunoblotting was performed using primary antibodies dilutions according to the manufacturer′s instructions. Secondary antibody horseradish peroxidase conjugates were used at 1 : 10 000. Blots were developed using the lumi-light western blotting substrate.
Abbreviations
- ATG5:
-
autophagy-related gene 5
- BrdU:
-
5′-bromo-2-deoxyuridine
- CQ:
-
chloroquine
- ERK:
-
extracellular signal-regulated kinase
- FACS:
-
fluorescence-activated cell sorting
- FITC:
-
fluorescein isothiocyanate
- H2DCF-DA:
-
dichlorodihydrofluorescein diacetate
- LAMP-1:
-
lysosome-associated membrane protein-1
- LAMTOR1:
-
late endosomal/lysosomal adaptor MAPK and mTOR activator 1
- LC3:
-
microtubule-associated protein light chain 3
- LMP:
-
lysosomal membrane permeabilization
- MEK:
-
MAP or ERK kinase
- MP1:
-
mek partner 1
- mTOR:
-
mammalian target of rapamycin
- ROS:
-
reactive oxygen species
- PBS:
-
phosphate-buffered saline
- PI:
-
propidium iodide
- siRNA:
-
short interfering RNA
- Trolox:
-
6-hydroxy-2,5,7,8-tetramethyl-chroman-2-carboxylic acid
- zVAD:
-
carbobenzoxy-valyl-alanyl-aspartyl-(O-methyl)-fluoromethylketone
- zFA:
-
z-phe-ala fluoromethylketone
References
Luzio JP, Pryor PR, Bright NA . Lysosomes: fusion and function. Nat Rev Mol Cell Biol 2007; 8: 622–632.
Saftig P, Klumperman J . Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat Rev Mol Cell Biol 2009; 10: 623–635.
Zhang L, Sheng R, Qin Z . The lysosome and neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai) 2009; 41: 437–445.
Teis D, Taub N, Kurzbauer R, Hilber D, de Araujo ME, Erlacher M et al. p14-MP1-MEK1 signaling regulates endosomal traffic and cellular proliferation during tissue homeostasis. J Cell Biol 2006; 175: 861–868.
Nada S, Hondo A, Kasai A, Koike M, Saito K, Uchiyama Y et al. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J 2009; 28: 477–489.
Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 2011; 13: 453–460.
Hoshino D, Tomari T, Nagano M, Koshikawa N, Seiki M . A novel protein associated with membrane-type 1 matrix metalloproteinase binds p27(kip1) and regulates RhoA activation, actin remodeling, and matrigel invasion. J Biol Chem 2009; 284: 27315–27326.
Guillaumot P, Luquain C, Malek M, Huber AL, Brugiere S, Garin J et al. Pdro, a protein associated with late endosomes and lysosomes and implicated in cellular cholesterol homeostasis. PLoS One 2010; 5: e10977.
Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM . Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010; 141: 290–303.
Takahashi Y, Nada S, Mori S, Soma-Nagae T, Oneyama C, Okada M . The late endosome/lysosome-anchored p18-mTORC1 pathway controls terminal maturation of lysosomes. Biochem Biophys Res Commun 2012; 417: 1151–1157.
Hoshino D, Koshikawa N, Seiki M A . p27(kip1)-binding protein, p27RF-Rho, promotes cancer metastasis via activation of RhoA and RhoC. J Biol Chem 2011; 286: 3139–3148.
Pietsch EC, Sykes SM, McMahon SB, Murphy ME . The p53 family and programmed cell death. Oncogene 2008; 27: 6507–6521.
Kittler R, Surendranath V, Heninger AK, Slabicki M, Theis M, Putz G et al. Genome-wide resources of endoribonuclease-prepared short interfering RNAs for specific loss-of-function studies. Nat Methods 2007; 4: 337–344.
Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282: 1497–1501.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM . Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307: 1098–1101.
Liao G, Yao Y, Liu J, Yu Z, Cheung S, Xie A et al. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am J Pathol 2007; 171: 962–975.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005; 64: 113–122.
Noda T, Ohsumi Y . Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 1998; 273: 3963–3966.
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem 2009; 284: 8023–8032.
Benes P, Vetvicka V, Fusek M . Cathepsin D--many functions of one aspartic protease. Crit Rev Oncol Hematol 2008; 68: 12–28.
Mizushima N, Yoshimori T, Levine B . Methods in mammalian autophagy research. Cell 2010; 140: 313–326.
Seglen PO . Inhibitors of lysosomal function. Methods Enzymol 1983; 96: 737–764.
Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H et al. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem 2010; 285: 667–674.
Hamacher-Brady A, Stein HA, Turschner S, Toegel I, Mora R, Jennewein N et al. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalysed lysosomal reactive oxygen species production. J Biol Chem 2011; 286: 6587–6601.
Maclean KH, Dorsey FC, Cleveland JL, Kastan MB . Targeting lysosomal degradation induces p53-dependent cell death and prevents cancer in mouse models of lymphomagenesis. J Clin Invest 2008; 118: 79–88.
Kroemer G, Jaattela M . Lysosomes and autophagy in cell death control. Nat Rev Cancer 2005; 5: 886–897.
Kurz T, Terman A, Brunk UT . Autophagy ageing and apoptosis: the role of oxidative stress and lysosomal iron. Arch Biochem Biophys 2007; 462: 220–230.
McClain DE, Kalinich JF, Ramakrishnan N . Trolox inhibits apoptosis in irradiated MOLT-4 lymphocytes. FASEB J 1995; 9: 1345–1354.
Jahreiss L, Menzies FM, Rubinsztein DC . The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic 2008; 9: 574–587.
Kimura S, Noda T, Yoshimori T . Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct 2008; 33: 109–122.
Caviston JP, Zajac AL, Tokito M, Holzbaur EL . Huntingtin coordinates the dynein-mediated dynamic positioning of endosomes and lysosomes. Mol Biol Cell 2011; 22: 478–492.
Butler D, Nixon RA, Bahr BA . Potential compensatory responses through autophagic/lysosomal pathways in neurodegenerative diseases. Autophagy 2006; 2: 234–237.
Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet 2006; 15: 965–977.
Liu B, Chen Y, Clair DK . ROS and p53: a versatile partnership. Free Radic Biol Med 2008; 44: 1529–1535.
Appelqvist H, Nilsson C, Garner B, Brown AJ, Kagedal K, Ollinger K . Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am J Pathol 2011; 178: 629–639.
Ross RA, Spengler BA, Biedler JL . Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst 1983; 71: 741–747.
Xue C, Haber M, Flemming C, Marshall GM, Lock RB, MacKenzie KL et al. p53 determines multidrug sensitivity of childhood neuroblastoma. Cancer Res 2007; 67: 10351–10360.
Debnath J, Muthuswamy SK, Brugge JS . Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30: 256–268.
Kalbacova M, Vrbacky M, Drahota Z, Melkova Z . Comparison of the effect of mitochondrial inhibitors on mitochondrial membrane potential in two different cell lines using flow cytometry and spectrofluorometry. Cytometry A 2003; 52: 110–116.
Acknowledgements
We thank Bert Vogelstein for providing us with p53+/+ and p53−/− HCT116 cells. This work was supported by the Association pour la Recherche Contre le Cancer (ARC, SFI20101201945) and by the Ligue Contre le Cancer Comité du Rhône. VP is supported by a Marie Curie fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by P Salomoni
Supplementary Information accompanies the paper on Cell Death and Disease website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Malek, M., Guillaumot, P., Huber, AL. et al. LAMTOR1 depletion induces p53-dependent apoptosis via aberrant lysosomal activation. Cell Death Dis 3, e300 (2012). https://doi.org/10.1038/cddis.2012.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.39
Keywords
This article is cited by
-
The Ragulator complex: delving its multifunctional impact on metabolism and beyond
Inflammation and Regeneration (2023)
-
N-myristoyltransferase-1 is necessary for lysosomal degradation and mTORC1 activation in cancer cells
Scientific Reports (2020)
-
Varroa destructor parasitism has a greater effect on proteome changes than the deformed wing virus and activates TGF-β signaling pathways
Scientific Reports (2019)
-
Identification of host proteins interacting with the integrin-like A domain of Toxoplasma gondii micronemal protein MIC2 by yeast-two-hybrid screening
Parasites & Vectors (2014)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}