
Abstract
Background:
The objectives of this study were to describe the impact of trial enrollment at diagnosis on event-free and overall survival in paediatric acute lymphoblastic leukaemic (ALL) using a population-based approach.
Methods:
We conducted a retrospective cohort study that included children newly diagnosed with ALL between 1 and 14 years of age. The data source was the Cancer in Young People in Canada (CYP-C) national paediatric cancer population-based database. We conducted univariate and multiple Cox proportional hazards models.
Results:
There were 2569 children with ALL; 1408 (54.8%) were enrolled on a clinical trial at initial diagnosis. Event-free survival at 5 years was 89.8%±0.9 vs 84.1%±1.2. (P<0.0001) for those enrolled and not enrolled on a clinical trial, respectively. Overall survival at 5 years was higher for those enrolled (94.1%±0.7) vs not enrolled (90.5%±1.0; P=0.001). In a model that adjusted for demographic, leukaemic and socioeconomic factors, enrollment on trials was significantly associated with better event-free survival (hazard ratio (HR) 0.67, 95% confidence interval (CI) 0.47–0.95; P=0.023), but not overall survival (HR 0.69, 95% CI 0.44–1.08; P=0.102).
Conclusions:
Event-free survival was significantly better in children with ALL enrolled on a clinical trial. Future research should identify barriers to clinical trial enrollment for children with ALL.
Similar content being viewed by others

Main
There has been debate about the impact of enrollment on clinical trials and survival outcomes for children with cancer. Retrospective studies found that patients enrolled on clinical trials have better outcomes compared with patients not enrolled on clinical trials (Stiller and Eatock, 1994; Wagner et al, 1995). However, a systematic review determined that these studies were limited because of confounding and selection bias, and thus concluded that the impact of trial enrollment is not clear (Peppercorn et al, 2004). Further, a Cochrane review found that, in general, outcomes are similar between those who do and those who do not participate in randomised trials (Vist et al, 2008).
We recently used the Cancer in Young People in Canada (CYP-C) database, a national paediatric cancer population-based data source, to describe the proportion of all children with cancer enrolled on a clinical trial and to describe factors associated with enrollment. About one in four children with cancer were enrolled on a clinical trial (Pole et al, 2017). The most common reasons cited for non-enrollment were lack of an available trial and physician choice. Acute lymphoblastic leukaemic (ALL) is the most common paediatric cancer, accounting for approximately one-third of all cancers in children aged 0–14 years of age. We found that children with ALL had the highest rates of enrollment among paediatric cancer diagnoses (Pole et al, 2017).
Given this data, we next wanted to determine if enrollment on a clinical trial is associated with better survival for children with ALL. Therefore, the objectives of this analysis were to describe the impact of trial enrollment at diagnosis on event-free survival (EFS) and overall survival (OS) in paediatric ALL.
Materials and methods
Population of interest and sampling methods
We included children with newly diagnosed ALL with ICD-O M codes 9835, 9836 and 9837; these codes define patients with precursor lymphoblastic leukaemic NOS, B-cell and T-cell, respectively. Other inclusion criteria were 1–14 years of age, diagnosed between 01 January 2001 and 31 December 2012, and treated at one of the 17 paediatric oncology centres in Canada. We excluded patients in whom enrollment status was unknown, those diagnosed <1 year of age (infant ALL) and those with Burkitt’s leukaemic. We chose to exclude infant and Burkitt’s ALL as the treatment approach is fundamentally different compared with precursor lymphoblastic leukaemic.
Data source
We used the data from CYP-C, a population-based registry that aims to include all paediatric patients with cancer diagnosed between 0 and 14 years of age since 2001, who were diagnosed and treated at one of the 17 tertiary paediatric oncology centres in Canada. Two data collection approaches are used for CYP-C data. For the 5 centres in Ontario, the data are transferred to CYP-C from the Paediatric Oncology Group of Ontario (POGO) Networked Information System (POGONIS), which is a provincial population-based registry that includes similar data to CYP-C. For the 12 centres outside of Ontario, the data are entered directly into CYP-C. Elements captured by both databases include the following: (1) demographic variables, including sex, date of birth, postal code and race; (2) diagnostic details; (3) times to diagnosis and treatment; (4) treatment plan details, including enrollment on a therapeutic trial and whether the initial treatment plan was terminated early or completed as planned; and (5) outcomes such as relapse, second malignancy and death.
The reasons for non-enrollment have been consistently collected by CYP-C throughout the study period, whereas a standardised list has only recently been incorporated into POGONIS. Thus, description of reasons for non-enrollment was restricted to the 12 non-Ontario sites.
The CYP-C program achieves high-quality data through multiple approaches. A community of practice composed of each site’s data manager was established to maximise the data quality through monthly teleconference and annual face-to-face training combined with site audits. The data were provided for the purpose of this analysis on 19 September 2016.
Statistical plan
Event-free survival was defined as time from diagnosis to relapse or death from any cause, whichever occurred first. Those without an event were censored on the date of last follow-up. Overall survival was defined as time from diagnosis to death from any cause or date of last follow-up. Survival was described for those enrolled and not enrolled on a therapeutic trial at diagnosis using the Kaplan–Meier method and compared using the log rank test.
In order to evaluate potential confounders, Cox proportional hazards models were created. The following variables were examined: (1) demographic features: age at diagnosis (1–4, 5–9 and 10–14), sex, race, and diagnostic era (<2007 vs ⩾2007, the approximate mid-point); (2) Leukaemia features: initial white blood cell count (WBC) (⩾50 vs <50 × 109 l−1), central nervous system (CNS) status 1 (no blasts), 2 (presence <5 μl−1 WBCs and cytospin positive for blasts) or 3 (CSF >5 μl−1 WBCs and cytospin positive for blasts), immunophenotype (B-precursor vs T), and cytogenetic risk group; and (3) Socioeconomic factors: kilometers to the nearest tertiary care paediatric centre and neighborhood income quintile. We also stratified analyses by National Cancer Institute (NCI) standard (age <10 years and initial WBC <50 × 109l−1) and high (age ⩾10 years or initial WBC ⩾50 × 109 l−1) risk groups. Favourable cytogenetics were defined as trisomies 4 and 10 and t(12;21). Unfavourable cytogenetics were defined as hypodiploidy (<45 chromosomes), t(9;22), MLL (11q23) rearrangements, and RUNX1 (AML1) amplification.
We used postal codes at diagnosis to determine distance to the nearest tertiary care paediatric cancer centre and area-level socioeconomic status by linking to the census data. Full 6 digit postal codes were available for all provinces except for British Columbia, in which 3 digit postal codes were available. Using the Statistics Canada Postal Code Conversion File software (PCCF+, Version 4J), we linked the postal code to a 2001 census dissemination area. Dissemination areas are the smallest area unit defined by Statistics Canada and include between 400 and 700 persons. Using the 2001 census, we determined income quintiles that adjust for household size and regional differences (Borugian et al, 2005).
Adjusted associations between enrollment on trials and survival outcomes were described using hazard ratios (HR) with 95% confidence intervals (CIs). Models adjusted for all demographic, leukaemic and socioeconomic factors separately and then together. Statistical significance was defined as P-value<0.05. Statistical analysis was conducted using the SAS statistical program (SAS-PC, version 9.4; SAS Institute Inc, Cary, NC, USA).
Results
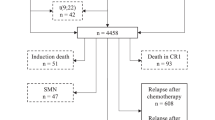
There were 2732 identified children with ALL in CYP-C, among whom 72 had unknown enrollment status, 76 were infants<1 year of age and 15 had Burkitt’s leukaemic, leaving 2569 patients available for analysis. Of the 2569 included patients, 1408 (54.8%) were enrolled on a clinical trial at diagnosis. For the 1408 enrolled on a clinical trial, 562 (39.9%) were enrolled on Children’s Oncology Group protocols, 336 (23.9%) were enrolled on Dana Farber Cancer Institute ALL Consortium protocols and the remainder were others or unknown. Conversely, for the 1161 not enrolled on a trial, 506 (43.6%) were treated according to Children’s Oncology Group protocols, 78 (6.7%) were treated according to Dana Farber Cancer Institute protocols and the remainder were others or unknown.
Table 1 shows the demographic, leukaemic and socioeconomic features of the study cohort by enrollment status. Those enrolled were more likely to be white, have an initial WBC<50 × 109l−1, CNS 2 status, B precursor immunophenotype and favourable cytogenetic features. Table 2 illustrates the reasons for non-enrollments for centres outside of Ontario and shows that the most common known reason for non-enrollment was lack of an available trial.
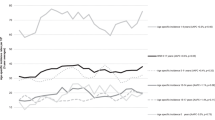
Event-free survival at 5 years was 89.8%±0.9 vs 84.1%±1.2, P<0.0001, for those enrolled and not enrolled on a therapeutic trial at diagnosis respectively (Figure 1). Overall survival at 5 years was 94.1%±0.7 vs 90.5%±1.0, P=0.001 (Figure 2). Table 3 shows univariate Cox proportional hazards models evaluating factors associated with survival. Enrollment on trials was significantly associated with better EFS (HR: 0.62, 95% CI: 0.49–0.78; P< 0.0001) and OS (HR: 0.61, 95% CI: 0.45–0.82; P=0.001). When stratified by NCI risk status and presented by those enrolled vs not enrolled, EFS was 91.0±1.0 vs 86.8±1.3 (P=0.006) for standard-risk patients and was 86.9±1.8 vs 78.7±2.3 (P=0.006) for high-risk patients, respectively. Overall survival was 96.2±0.7 vs 93.4±1.0 (P=0.014) for standard-risk patients and was 88.5±1.9 vs 84.1±2.3 (P=0.093) for high-risk patients.

Event-free survival for children with acute lymphoblastic leukaemic enrolled and not enrolled on a clinical trial.

Overall survival for children with acute lymphoblastic leukaemic enrolled and not enrolled on a clinical trial.
Table 4 illustrates adjusted analyses by demographic, leukaemic and socioeconomic factors and adjustment for all factors together. In the fully adjusted model, enrollment on trials was significantly associated with better EFS (HR: 0.67, 95% CI: 0.47–0.95; P=0.023) but not OS (HR: 0.69, 95% CI: 0.44–1.08; P=0.102). To determine if the impact of enrollment on EFS differed by age group, an interaction term was evaluated in a multivariable model which included enrollment and age. The P-value for interaction was 0.870 suggesting a similar effect across age groups.
When examining the proportion of treatment plans terminated early rather than completed as planned, 249 out of 1408 (17.7%) for those enrolled were terminated early compared to 156 out of 1161 (13.4%) for those not enrolled (P=0.004). When the analysis was restricted to children with ALL treated according to Children’s Oncology Group trials, enrollment was associated with better EFS (HR: 0.61, 95% CI: 0.42–0.88; P=0.008) and OS (HR: 0.46, 95% CI: 0.27–0.76; P=0.003). This benefit was not seen when the analysis was restricted to children treated according to Dana Farber Cancer Institute trials; enrollment was not associated with better EFS (HR: 0.75, 95% CI: 0.35–1.63; P=0.475) or OS (HR: 1.11, 95% CI: 0.39–3.21; P=0.843).
Discussion
We found that enrollment on a therapeutic clinical trial at initial leukaemic diagnosis was independently associated with better EFS for children newly diagnosed with ALL after adjustment for demographic, leukaemic and socioeconomic factors. Improved EFS was also seen when stratified by NCI risk status. This information may be important to families and clinicians when deciding whether to enroll on a clinical trial at diagnosis.
Our results are discordant with (Koschmann et al, 2010) who evaluated the trial effect among paediatric ALL patients treated at Seattle Children’s Hospital from 1997 to 2005. They failed to demonstrate an EFS advantage to participation in studies However, this report was limited since it included only a single centre in the United States. Further, the sample size consisted of only 322 patients (with 48.8% enrolment).
Reasons why enrollment on trials could improve EFS include the following: (1) Treatment effect, in which the interventions being evaluated result in better outcomes compared with standard approaches; (2) Participation effect where enrollment in the trial results in better outcomes due to the effect of the protocol, changes in healthcare professional behavior, changes in patient/family behavior or a placebo effect; and (3) Confounding, if patients enrolled on trials are systematically different than patients not enrolled on trials. The adjusted analyses were important as those enrolled on trials, when compared to those not enrolled, had generally favourable features such as low initial WBC, B precursor immunophenotype and favourable risk cytogenetics. By taking into consideration confounding using several approaches, our study suggests that improved EFS may be the result of the interventions being evaluated in these studies and is consistent with improved outcomes in paediatric ALL with successive clinical trials (Pui et al, 2015). However, we cannot exclude that participation effect may have a role as well. If improved EFS is the result of interventions being evaluated, this finding may not apply to trials examining de-escalation of therapy.
We also found that when patients were treated with COG protocols, patients enrolled had better outcomes compared to those not enrolled, whereas a similar pattern was not seen among those treated according to Dana Farber Cancer Institute protocols. This finding may be related to either the interventions being evaluated in specific trials or may be related to the smaller number of children receiving Dana Farber Cancer Institute studies. We also found that those enrolled on trials were more likely to terminate the treatment plan early compared to those not enrolled on trials. This finding may be an artefact of how the data were collected since if patients enrolled on trials were taken off protocol therapy but continued to follow the same treatment protocol, they were designated as terminating treatment early and starting a new treatment plan as a non-registered patient.
Our results were less definitive regarding whether enrollment on trials improves OS and such an effect is difficult to demonstrate in a disease with high success rates in general. While we found that OS was not improved in the adjusted models, it should be noted that the HR favoured enrollment and that power was limited given excellent survival. Thus, combining the CYP-C data set with other population-based registries may help us understand whether enrollment on trials can reduce mortality.
The strengths of this study are its population-based nature and careful collection of confounders, including leukaemic and socioeconomic factors. Other strengths are the high quality of data and common health care system, which provides universal healthcare. However, these results must be interpreted in light of its limitations. First, potentially important covariates were not available such as minimal residual disease (MRD) status. Second, we did not include adolescent and young adult patients (AYA) in our study. This is important as several studies have identified that AYA patients have lower rates of enrollment on clinical trials (Bleyer, 2002; Downs-Canner and Shaw, 2009; Aristizabal et al, 2015). Third, we lacked immunophenotype information for over 40% of children related to how POGO classifies ALL; this missing data affected the power of analyses, which adjusted for leukaemic-related factors. Finally, we used a census based measure of family income as individually reported data from the family were not available at the population level.
In conclusion, EFS was significantly better in children with ALL enrolled on a clinical trial. Future research should identify barriers to clinical trial enrollment for children with ALL.
Change history
06 March 2018
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Aristizabal P, Singer J, Cooper R, Wells KJ, Nodora J, Milburn M, Gahagan S, Schiff DE, Martinez ME (2015) Participation in paediatric oncology research protocols: Racial/ethnic, language and age-based disparities. Pediatr Blood Cancer 62: 1337–1344.
Bleyer WA (2002) Cancer in older adolescents and young adults: epidemiology, diagnosis, treatment, survival, and importance of clinical trials. Med Pediatr Oncol 38: 1–10.
Borugian MJ, Spinelli JJ, Mezei G, Wilkins R, Abanto Z, Mcbride ML (2005) Childhood leukaemic and socioeconomic status in Canada. Epidemiology 16: 526–531.
Downs-Canner S, Shaw PH (2009) A comparison of clinical trial enrollment between adolescent and young adult (AYA) oncology patients treated at affiliated adult and paediatric oncology centres. J Pediatr Hematol Oncol 31: 927–929.
Koschmann C, Thomson B, Hawkins DS (2010) No evidence of a trial effect in newly diagnosed paediatric acute lymphoblastic leukaemic. Arch Pediatr Adolesc Med 164: 214–217.
Peppercorn JM, Weeks JC, Cook EF, Joffe S (2004) Comparison of outcomes in cancer patients treated within and outside clinical trials: conceptual framework and structured review. Lancet 363: 263–270.
Pole JD, Barber R, Bergeron RE, Carret AS, Dix D, Kulkarni K, Martineau E, Randall A, Stammers D, Strahlendorf C, Strother DR, Truong TH, Sung L (2017) Most children with cancer are not enrolled on a clinical trial in Canada: a population-based study. BMC Cancer 17: 402.
Pui CH, Yang JJ, Hunger SP, Pieters R, Schrappe M, Biondi A, Vora A, Baruchel A, Silverman LB, Schmiegelow K, Escherich G, Horibe K, Benoit YC, Izraeli S, Yeoh AE, Liang DC, Downing JR, Evans WE, Relling MV, Mullighan CG (2015) Childhood acute lymphoblastic leukaemic: progress through collaboration. J Clin Oncol 33: 2938–2948.
Stiller CA, Eatock EM (1994) Survival from acute non-lymphocytic leukaemia, 1971-88: a population based study. Arch Dis Child 70: 219–223.
Vist GE, Bryant D, Somerville L, Birminghem T, Oxman AD (2008) Outcomes of patients who participate in randomized controlled trials compared to similar patients receiving similar interventions who do not participate. Cochrane Database Syst Rev MR000009, Issue 3.
Wagner HP, Dingeldein-Bettler I, Berchthold W, Luthy AR, Hirt A, Pluss HJ, Beck D, Wyss M, Signer E, Imbach P et al (1995) Childhood NHL in Switzerland: incidence and survival of 120 study and 42 non-study patients. Med Pediatr Oncol 24: 281–286.
Acknowledgements
The authors gratefully acknowledge the contributions of study participants, participating paediatric oncology centres, members of the Cancer in Young People in Canada (CYP-C) Management and Steering Committees, the Paediatric Oncology Group of Ontario (POGO) and the five POGO Hospital Partners. The CYP-C is fully funded by the Public Health Agency of Canada. We wish to thank all the data managers at the 17 CYP-C sites for their dedicated work in maintaining the CYP-C data quality and Dr. Mark Bernstein for his leadership in CYP-C development. We also thank Jay Onysko for his management of the CYP-C program and constructive comments on a draft version of this manuscript.
Ethical Approval
The study received Research Ethics Board approval from the coordinating site (The Hospital for Sick Children). For sites outside of Ontario, Research Ethics Board approval was obtained for collection of CYP-C data. For the five Ontario sites, the requirement for Research Ethics Board approval was waived given the Paediatric Oncology Group of Ontario’s status as a prescribed 45 Entity under the Personal Health Information Protection Act, 2004. This status allows the Paediatric Oncology Group of Ontario to be a custodian of personal health information for the province. The requirement for informed consent and assent were waived given the retrospective nature of the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Strahlendorf, C., Pole, J., Barber, R. et al. Enrolling children with acute lymphoblastic leukaemia on a clinical trial improves event-free survival: a population-based study. Br J Cancer 118, 744–749 (2018). https://doi.org/10.1038/bjc.2017.462
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2017.462
Keywords
This article is cited by
-
Creating and adapting an infection management care pathway in pediatric oncology
Supportive Care in Cancer (2022)
-
DKG-Zertifizierung kinderonkologischer Zentren – ein weites Feld …
Bundesgesundheitsblatt - Gesundheitsforschung - Gesundheitsschutz (2020)
-
Haben Studienkinder mit ALL bessere Chancen?
Pflegezeitschrift (2018)
