
Abstract
Aim:
Ergosterol is a plant sterol with anti-tumor and anti-angiogenic activities, but is poorly soluble. In this study, we attempted to enhance its anti-tumor action and oral bioavailability via poly(lactide-co-glycolide) (PLGA) nanoparticle encapsulation.
Methods:
Ergosterol-loaded PLGA nanoparticles (NPs/Erg) were prepared using the emulsion/solvent evaporation technique. Their physicochemical properties were characterized, and their cytotoxicity against human cancer cell lines was evaluated with MTT assay. The pharmacokinetics and tissue distribution of NPs/Erg were investigated in rats and mice, respectively.
Results:
NPs/Erg were spherical in shape with a particle size of 156.9±4.8 nm and a Zeta potential of -19.27±1.13 mV, and had acceptable encapsulation efficiency and loading capacity. NPs/Erg exerted much stronger cytotoxicity against human cancer cells than the free ergosterol, and showed significantly reduced IC50 values (14.69±0.48 μg/mL in glioma U251 cells; 9.43±0.52 μg/mL in breast cancer MCF-7 cells; 4.70±0.41 μg/mL in hepatoma HepG2 cells). After oral administration of a single dose in rats, NPs/Erg displayed a prolonged plasma circulation with a 4.9-fold increase of oral bioavailability compared with the free ergosterol. After mice received NPs/Erg, the ergosterol in NPs/Erg was rapidly distributed in stomach, kidneys, liver, brain, spleen, and virtually non-existent in heart and lungs. The presence of NPs/Erg in brain was particularly improved compared with the free ergosterol.
Conclusion:
The PLGA nanoparticles serve as a promising carrier for the poorly soluble ergosterol and significantly improve its bioavailability, biodistribution and in vitro anti-tumor activities.
Similar content being viewed by others


Introduction
Recent reports on plant sterols have greatly increased because such sterols could lower blood cholesterol and reduce the risk of cardiovascular and cancer-related diseases1,2,3. Ergosterol (Erg), a prominent phytosterol4, has also shown anti-tumor and anti-angiogenic activities with promising prospects in both in vitro5,6 and in vivo studies7. The ergosterol is now seen as a strong drug candidate with no side effects and has the potential to induce cancer cell death. However, the poor aqueous solubility of the sterol has hindered its wider clinical applications.
Currently, advances in nanotechnology have made it possible to improve the solubility, bioavailability, and absorbency of medicinal herbs as well as couple these advances with using a reduced dose and a sustained steady-state of therapeutic level of drugs over an extended period compared with the traditional methods8,9,10,11. Among the numerous nano-suspensions, the colloidal delivery system of polymeric nanoparticles has attracted much attention as a result of its unique properties, such as high biocompatibility, non-toxic nature, long-term stability, good physiological features, and acceptable control release12,13. The polymeric nanoparticles with smaller particle sizes (approximately 200 nm) could also increase drug accumulation in tumor cells via enhanced permeability and retention (EPR) effects14,15,16. Therefore, nanoparticles could be employed as an effective delivery system to improve phytochemical bioavailability and antitumor activities.
In related studies, formulations such as microemulsions, liposomes and polymeric micelles of ergosterols were developed to increase the solubility and bioavailability of this compound4,6,17. However, the elimination half-life (t1/2) of the sterol could not be significantly improved, which suggested that such formulations could have a less-than-ideal duration of pharmacological action. In the in vivo studies, none of the formulations exhibited the usual sustained release characteristic of drugs (over 24 h). Additionally, there were limited reports on the tissue distribution of these encapsulations, and more importantly, such biodistribution could not significantly broaden the clinical applications of ergosterol. For instance, although significant cancer growth inhibition was observed in U251 glioma cells, the biodistribution studies revealed either no or a small amount of ergosterol deposits in the brain, which indicated the likelihood of a decrease in vivo antitumor activities in the brain cancer cells17. There is therefore a need to actively explore other potential formulations to fully and strategically benefit from the clinical prospects of ergosterol.
Poly (D, L-lactide-co-glycolide) (PLGA), a polymer for developing an array of nanoparticulate drug delivery systems, has presently attracted considerable interest due to its excellent biocompatibility and predictable biodegradability18,19,20. Previous reports have shown that PLGA/Gemcitabine HCl NPs could enhance bioavailability and intestinal uptake of gemcitabine HCl via oral delivery21. Other studies also revealed that small-sized PLGA nanoparticles for the oral delivery of cyclosporine could decrease its nephrotoxicity compared with microemulsion and control the in vivo release over 5 d22. PLGA nanoparticles for the oral delivery of paclitaxel, doxorubicin and docetaxel demonstrated superior in vivo performance, as evidenced by their results of enhanced bioavailability and lower toxicity in addition to a higher drug concentration in the tumor23,24,25. Encapsulating ergosterol in PLGA nanoparticles will therefore benefit from such advantages that are associated with the aforementioned formulations.
In the present study, ergosterol-loaded PLGA nanoparticles (NPs/Erg) were successfully developed for effective oral administration with enhanced bioavailability and selective tissue distribution. The formulated ergosterol significantly increased the susceptibility of cancer cells to the applied sterol. More importantly, the observed biodistribution was highly appreciable in some organs, which could strongly improve the in vivo pharmacological properties, such as the antitumor activities of the ergosterol.
Materials and methods
Materials
Ergosterol (98% purity) was purchased from Acros Organics (Geel, Belguim). PLGA (75:25, with inherent viscosity of 0.4 dL/g) was purchased from Purac Biomaterials (Gorinchem, The Nertherlands). Polyvinyl alcohol 130 (PVA) was purchased from the Research Laboratory (Mumbai, India). Chromatographically pure methanol was obtained from Hanbon Technology Co Ltd (Huaian, China). All other chemicals and reagents used in this study were of analytical grade. The human cancer cell lines (Hepatoma cells, HepG-2; Glioma cells, U251; Breast cancer cells, MCF-7) were obtained from the Cell Bank of the Academy of Science (Shanghai, China). (3-(4, 5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from the Beyotime Institute of Biotechnology (Haimen, China). Fetal bovine serum and Dulbecco's modified Eagle's medium (DMEM) were purchased from the Gibco Company (Grand Island, NY, USA).
Preparation of NPs/Erg
The NPs/Erg were prepared by employing an emulsion/solvent evaporation method as described in previous studies with some modifications26. Briefly, 20 mg of PLGA was added to ergosterol to obtain a mixture of ergosterol/PLGA (10%-50%, w/w). The mixture was dissolved in 1 mL of dichloromethane through ultrasonic agitation. Afterward, the PVA solution (5 mL, 2%, w/v) was added slowly to the product and further sonicated at 200 W for 2 min on ice using a probe sonicator (Xinzhi Biotechnology Co Ltd, China). The resulting O/W emulsion was added drop-wise to another PVA solution (50 mL, 0.5%, w/v) and stirred vigorously for 2 min using a magnetic stirrer to solidify the nanoparticles. The aggregates were removed via filtration using a 0.45-μm cellulose acetate filter membrane. The redundant stabilizer (PVA) was eliminated from the nanoparticles by centrifugation at 15 000 rounds per minute at 4 °C for 30 min. The product was washed (3 times) with double distilled water and re-suspended in Milli-Q water to obtain the PLGA nanoparticles. Unloaded nanoparticles serving as controls were also prepared using the same procedure described above.
In vitro and in vivo HPLC analysis of ergosterol
Ergosterol levels in nanoparticles, plasma and tissue samples were determined using an RP-HPLC method with the Shimadzu Scientific instrument equipped with an LC-20AT pump and an SPD-20A UV-Vis detector (Shimadzu, Kyoto, Japan). The chromatographic conditions were as follows: a Symmetric C18 column (4.6 mm×150 mm, 5 μm, Waters, Milford, MA, USA) and column temperature of 30 °C; a flow rate of 1.0 mL/min; and a detection wavelength of 282 nm. The mobile phase used for the determination of encapsulation efficiency and in vitro release was 98% methanol, while the analysis of bioavailability and tissue biodistribution studies used 93% methanol (brain samples) and 96% methanol (other samples) with the methanol containing 0.05% TFA. The detection sensitivity of ergosterol was approximately 0.02 AUFS.
Morphology, particle size and zeta potential
The morphological properties of the NPs/Erg were examined using a transmission electron microscope, TEM (JEM-2100, JEOL, Tokyo, Japan) at an acceleration voltage of 200 kV. A drop of diluted NPs/Erg solution (0.6 mg/mL) was placed on a copper grid and air-dried at room temperature. The sample was stained with phosphotungstic acid solution (2%, w/v) and subsequently viewed using TEM.
Mean particle size, particle size distribution and the zeta potential of the formulation were measured using the dynamic light scattering (DLS) method via a Zeta Potential/Particle Size analyzer (Brookhaven Instruments Corporation, Holtsville, NY, USA). The freshly prepared NPs/Erg solution was diluted to 0.6 mg/mL to avoid multi-scattering phenomena. The diluted sample was measured using the appropriate instrument. The results were expressed as the means of triplicate samples.
Analysis of process yield
The process yield has been recognized as one of the most basic and common criterion used in the field of pharmacy for industrial production. The process yield was calculated by comparing the total quantity of polymer and drug initially taken for the production process to the quantity of nanoparticles obtained. Thus, process yield=quantity of nanoparticles obtained/total quantity of polymer and drug used×100%.
Drug-loading capacity and encapsulation efficiency
For the estimation of drug loading and encapsulation efficiency, 1 mg of freeze-dried nanoparticles was dissolved in 5 mL of acetonitrile using ultrasound for 15 min. The product was filtered through a 0.45-μm membrane filter. The drug content in the filtrate was determined using HPLC analysis as described in previous sections. The encapsulation efficiency (EE, %) and drug loading capacity (DL, %) were, respectively, calculated using the following equations:
EE (%)=Real drug loading/Theoretical drug loading×100% (A.1)
DL (%)=Weight of drug in nanoparticles/Weight of nanoparticles×100% (A.2)
Diffuse reflectance of infrared by Fourier transform (DRIFT)
Infrared absorption spectra of ergosterol, PLGA nanoparticles, ergosterol/PLGA mixture and ergosterol-loaded PLGA nanoparticles were examined using DRIFT spectroscopy (Spectrum GX Spectrophotometer, Perkin-Elmer, MA, USA) with a diffuse reflectance accessory (Pike Technology model). All samples were initially stored in a desiccator containing silica gel for 48 h at room temperature. Each of the samples (2.4 mg) was mixed with 97.6 mg KBr and dried in an oven for 24 h at 135 °C to keep the samples dry. The instrument was operated at a resolution of 4 cm−1 and 32 scans for each sample. The absorbency scans were analyzed between 400 and 4000 cm−1 for changes in the intensity of the sample peaks.
In vitro release profile of ergosterol-loaded PLGA nanoparticles
In vitro release experiments of ergosterol stock solution (in DMSO), NPs/Erg and free ergosterol suspension [in 0.5% (w/v) CMC–Na solution] were performed using the dialysis method with a water-bathing constant temperature vibrator (SHZ-88, Jintan Medical Instrument Corporation, Jintan, China). An aliquot (1.0 mL, 0.1 mg/mL) of ergosterol stock solution, NPs/Erg and free ergosterol suspension were transferred into dialysis bags (Mw=12 000, Shanghai Green Bird Science and Technology Development, Shanghai, China). The dialysis bags were suspended in 80 mL of 0.9% normal saline containing 50% ethanol (v/v), which was adjusted to different pH values (1.2, 5.0 and 6.8, respectively) and stirred at 37 °C with a constant speed of 70 rounds per minute. At certain time intervals (0, 0.5, 1, 2, 4, 6, 8, 12, 24, 48, 72, 96 and 120 h), 1 mL release media was taken and subsequently replaced with an equal volume of fresh media (preheated to 37 °C). There was also a parallel experiment in which fresh NPs/Erg were directly placed in the release medium without any dialysis bag to rule out the significant contribution of the retardation effect of the membrane barrier. The collected samples at each time point were filtered through a 0.22-μm cellulose nitrate membrane for HPLC analysis. The cumulative release (%) was calculated as the weight ratio of released ergosterol to total ergosterol. The data were presented as the means of triplicate samples.
In vitro cytotoxicity assay
The different cancer cell lines (HepG2, U251, MCF-7 cells) in a logarithmic growth phase were seeded at 4,000 cells/well in DMEM supplemented with 10% (v/v) fetal calf serum, 100 U/mL of penicillin and 100 μg/mL of streptomycin in 96-well plates. The preparation was cultured at 37 °C with 5% CO2 under fully humidified conditions. Following a 24-h incubation, the cells were treated with 100 μL of medium containing each of the respective drug samples (free ergosterol dissolved in DMSO; NPs/Erg at an equivalent dose of ergosterol; and blank PLGA NPs), with each having different concentrations (0.5, 1, 5, 10, 15 and 20 μg/mL). To ensure that the free ergosterol remained soluble in the cell culture, 4 mg/mL of free ergosterol in DMSO was prepared. Then, the free ergosterol solution (0.5 μL) was added to the culture medium (99.5 μL) to give 0.5% DMSO. The DMSO at this concentration was able to maintain the free drug (20 μg/mL and other lower concentrations) in solution. After a 72-h incubation, an MTT solution (20 μL, 5 mg/mL) was added to each well, and the plates were incubated further at 37 °C for 4 h. DMSO (100 μL) was used to solubilize the formazan crystals and the optical density was measured using a microplate reader at 595 nm. The cell viability rate (VR) was calculated according to the following equation:
VR (%)=A/A1×100%, where A refers to the absorbance of treated group and A1 refers to the absorbance of the untreated control group (the vehicle control group). The 50% inhibiting concentration (IC50) of the sample was calculated with the aid of Curve-Expert software (version 1.3; Daniel G HYAMS, Hixson, TN, USA).
Synthesis of suc-ergosterol
Briefly, ergosterol (200 mg) and succinic anhydride (300 mg) were dissolved in 5 mL of anhydrous methylene chloride (CH2Cl2), followed by the addition of triethylamine (50 μL) and 4-dimethyl-aminopyridine (100 mg). The reaction mixture was stirred at room temperature for 24 h under nitrogen purging, which was monitored using TLC with the chloroform-methanol system (20:1, v/v). The spots were detected by charring with 5% phosphomolybdic acid-ethanol solution prior to heating. After concentrating and drying the reaction mixture under a vacuum, the obtained crude suc-ergosterol was purified using silica gel column chromatography and eluted with chloroform-methanol (20:1, v/v). A pale white solid suc-ergosterol (121 mg) was obtained with a yield of 48.41%. The structure of the product was confirmed by NMR, IR, MS and UV, respectively. The IR data were recorded using a FT-IR spectrophotometer, whereas NMR spectra were run at 400 MHz with a NMR spectrometer (Bruker AVANCE II, Switzerland). MS spectra were also measured using a Mass Spectrometer (LCQ, Finnigan, USA), whereas ultraviolet spectral analyses were performed with a spectrophotometer (UV-2201, Shimadzu, Japan) using 1 cm quartz cells.
Sample pretreatment
Blood samples (200 μL each) were taken and added to 50 μL of suc-ergosterol. Absolute methanol (1.0 mL) was added to the mixture to precipitate the proteins. Normal saline (2 mL) was added to the other samples (tissues and excrement; liver- 0.3 g) to prepare the different homogenates. Afterward, suc-ergosterol (50 μL) was added to 0.5 mL of each of the homogenates to form a uniform mixture. Similarly, 4.0 mL of diethyl ether was thoroughly mixed with the product for 5 min. After centrifugation at 3000 rounds per minute for 10 min, the supernatant was dried with nitrogen at 40 °C in a water bath to obtain the residue, which was later reconstituted in 0.4 mL of 96% methanol (containing 0.05% TFA) as mobile phase. Again, the sample was centrifuged at 20 000 rounds per minute for 10 min, and the supernatant was used for HPLC analysis. The interference from endogenous sterols was also examined via HPLC analysis.
Oral pharmacokinetic study of NPs/Erg
Twelve male SD rats (220±20 g) were randomly and equally divided into two groups and then acclimatized to laboratory conditions for 3 d prior to the experiment. The experiments were conducted under acceptable laboratory animal care procedures as approved by the Ethical Committee for Laboratory Animals Care and Use at Jiangsu University. Before oral administration, the two groups of rats were fasted for 12 h and provided with only water. The first group was given a 50 mg/kg dose of free ergosterol suspension [2.5 mg/mL, in 0.5% (w/v) CMC-Na aqueous solution], and the other group was given the same dose of ergosterol in NPs/Erg (2.5 mg/mL). Blood samples (600 μL each) were collected at predetermined time points (0.5, 1, 2, 3,4, 6, 8, 12, 24, 48 and 72 h). The samples were centrifuged at 6000 rounds per minute for 10 min to obtain plasma, which was treated as described under the “Sample pretreatment” section. The sterol content was determined using HPLC analysis. The pharmacokinetic parameters of Cmax, Tmax, AUC0–72 h (the area under the plasma concentration-time curve) and t1/2 (elimination half-life) of ergosterol for the formulation were also determined using BAPP 2.3 pharmacokinetic software (supplied by the Center for Drug Metabolism of the China Pharmaceutical University, China).
Tissue distribution study
Twenty Kunming mice were randomly and equally divided into 2 groups. All mice were fasted for 12 h but were allowed free access to water. The first group was orally administered with NPs/Erg (2 mg/mL of ergosterol content) at a dose of 10 mL/kg, while the other group was treated with the same content of sterol in the free ergosterol suspension [2 mg/mL in 0.5% (w/v) CMC-Na aqueous solution]. At specific times (1 h and 4 h) after the oral administration, the tissues (heart, spleen, lung, liver, kidney, stomach and brain) and excrement samples were removed from the sacrificed mice. All samples were washed with ice-cold saline to remove excess fluid. The samples were also weighed and stored at -20 °C until ready for further analysis (as described under the “Sample pretreatment” section). The drug concentration in the different samples was determined using HPLC analysis.
Statistical analysis
All data are expressed as the mean±SD, where applicable. The differences in means were analyzed using a one-way ANOVA and Dunnett's test or Tukey's test. Statistical analyses were conducted with Prism version 5.0 software. For all statistical tests, P-values <0.05 were considered significant.
Results
Morphology, particle size and zeta potential
The NPs/Erg morphology using the TEM showed that the NPs were spherical in shape (Figure 1A). The zeta potential of the PLGA nanoparticles was -31.20±4.16 mV, which was significantly higher than that of the NPs/Erg (-19.27±1.13 mV) (Table 1).

Characterization of ergosterol-loaded PLGA nanoparticles. (A) Microphotograph via transmission electron microscopy (×200 k). (B) Particle size and size distribution via DLS.
Drug-loading capacity and encapsulation efficiency of NPs/Erg
The standard curve for the ergosterol loading rate gave a linear equation, Y=27236C−348.9 (n=5, r=0.9998), which was within the range of 0.1–100 μg/mL, where C represents the concentration of ergosterol in NPs, and Y represents the peak area integrated in the chromatogram. The encapsulation efficiency showed a slight reduction with increasing ergosterol concentration for the various formulations, which was contrary to the drug-loading content (Table 2).
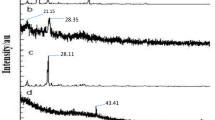
Spectral analysis of ergosterol-load PLGA nanoparticles
The DRIFT spectra of free PLGA nanoparticles showed the characteristic bands of polymer as follows: −CH, −CH2, −CH3 stretching (2850–3000 cm−1), carbonyl –C=O stretching (1700–1800 cm−1), C–O stretching (1050–1250 cm−1), and −OH stretching (3200–3600 cm−1). The ergosterol revealed the characteristic bands corresponding to −OH groups (3700–3000 cm−1), the −CH3 group (2955–2870 cm−1), bands of C=C stretching (1655 cm−1), C–H bending (1459–1369 cm−1) and =C-H bending vibration (800–1000 cm−1) (Figure 2). Concerning the ergosterol/PLGA mixture, the spectra exhibited the characteristic bands of the individual components (Figure 2). The DRIFT spectral of NPs/Erg also showed that the OH stretching band (3200–3600 cm−1) was slightly shifted and increased with regard to the energy absorption (Figure 2). The band corresponding to C=O stretching (1700–1800 cm−1) in the NPs/Erg was broader.

DRIFT spectra of different samples. (A) Free PLGA nanoparticles plus ergosterol; (B) Ergosterol; (C) Free PLGA nanoparticles; and (D) NPs/Erg.
In vitro drug release behavior of NPs/Erg
In the present study, several release media involving 1% SDS, 1% Tween 80 and ethanol solution (10%; 30%) were used, but they all resulted in a low cumulative release (data not shown). The selection of 50% ethanol solution with a solubility of 6.56±0.45 μg/mL gave appropriate cumulative release behaviors at different pH values (1.2, 5.0 and 6.8) (Figure 3). We found that more than 80% of the ergosterol in the stock solution was released into the medium in the first 4 h. However, the highest release rate of the free ergosterol suspension was 19.22% at 24 h in the medium with a pH of 1.2 (similar to artificial gastric fluid) (Figure 3A), followed by the cumulative release of the free ergosterol suspension reaching 14.12% in the medium with a pH of 5.0 (Figure 3B) compared with the lowest release rate (12.14%) in the medium with a pH of 6.8 (similar to artificial intestinal fluid) (Figure 3C). In contrast, the cumulative release rate of the prepared formulation in the medium with a pH of 6.8 (30.32% release rate) (Figure 3C) was far greater than that of the medium with a pH of 1.2 (18.22% release rate) (Figure 3A) within 24 h. After 12 h, the NPs/Erg showed a sustained drug release for 5 d at a constant rate of 6 μg per day. The free drug suspension also appeared to exhibit a sustained release behavior similar to that of the NPs/Erg. Additionally, the parallel NPs/Erg that were directly placed in the release medium gave similar results as that observed for the others using the dialysis method (data not shown).

In vitro release profiles of 0.1 mg/mL of ergosterol stock solution (in DMSO solution), ergosterol-loaded nanoparticles and free ergosterol suspension [in 0.5% (w/v) CMC–Na solution] in 80 mL of the following: (A) 50% ethanol solution at a pH of 1.2; (B) 50% ethanol solution at a pH of 5.0; and (C) 50% ethanol solution at a pH of 6.8, for different time points within 120 h (mean±SD; n=3). The points with significant differences (P<0.05, versus free ergosterol suspension) were labeled as solid.
Cytotoxicity assay
The in vitro cytotoxicity of NPs/Erg in different cancer cell lines (HepG2, U251 and MCF-7 cells) were investigated and compared with the free drug and the blank PLGA nanoparticles. After a 72-h incubation, the cell viability of free drug, NPs/Erg and blank PLGA nanoparticles generally decreased with increasing concentrations of the different samples (Figure 4). At the highest concentration of the blank PLGA NPs in the different cancer cells, the cell viabilities were more than 94%. The NPs/Erg were associated with a lower cell viability in the different human cancer cells compared with the free drug at the same concentration (Table 3). The NPs/Erg exhibited a 8.64%, 19.94% and 26.79% decrease in the IC50 values for U251, MCF-7 and HepG2, respectively, when compared with the free drug.

Cell viability of different samples (blank nanoparticles, free ergosterol and NPs/Erg) with varied concentrations for 72 h using a MTT bioassay against (A) U251 cells, (B) MCF-7 cells and (C) HepG2 cells. *P<0.05 versus blank nanoparticles. #P<0.05 versus free ergosterol.
Synthesis of suc-ergosterol
Suc-ergosterol was synthesized as an internal standard substance for the study. IR, MS, 1H and 13C NMR were some of the commonly used methods in analyzing the chemical composition of suc-ergosterol. The spectral analysis of this compound was as follows: IR (KBr) νmax (cm−1): 3422, 2955, 2870, 1655, 1459, 1369, 1056, 835. ESI-MS, M/Z 991 [2M-H]−; 1H-NMR (CDCl3, 400 MHz) δ: 5.58 (1H, dd, J=5.5, 2.5 Hz, H-6), 5.39 (1H, ddd, J=5.5, 2.8, 2.8 Hz, H-8), 5.26 (1H, dd, J=15.3, 7.0 Hz, H-23), 5.22 (1H, dd, J=15.3, 8.0 Hz, H-22), 4.75 (1H, m, H-3), 2.71 (2H, t, H-2′), 2.64 (2H, t, H-3′), 1.05 (3H, d, J=6.5 Hz, CH3-21), 0.97 (3H, s, CH3-19), 0.95 (3H, d, J=6.8 Hz, CH3-28), 0.87(3H, d, J=6.0 Hz, CH3-26), 0.84 (3H, d, J=6.0 Hz, CH3-27), 0.65 (3H, s, CH3-18); and 13C-NMR (CDCl3, 400 MHz) δ: 177.8(C-4′), 171.55 (C-1′), 141.55 (C-8), 138.40 (C-5), 135.57 (C-22), 131.99 (C-23), 120.28 (C-6), 116.31 (C-7), 73.37 (C-3), 55.71 (C-17), 54.53 (C-14), 46.02 (C-9), 42.82 (C-13), 42.82 (C-24), 39.03 (C-4), 40.45 (C-20), 39.03 (C-12), 36.54 (C-1), 37.08 (C-10), 33.07 (C-25), 29.22 (C-2), 28.29 (C-16), 22.99 (C-15), 21.03 (C-11), 21.03 (C-21), 19.96 (C-26), 19.66 (C-27), 17.61 (C-28), 16.18 (C-19), 12.1 (C-18) (Figure 5).

The structure and 1H NMR data of suc-ergosterol.
In vivo pharmacokinetic study
After oral administration of a single dose of 50 mg/kg, the plasma concentration-time profile of NPs/Erg was greater than that of free ergosterol for the entire study period (Figure 6). The profiles also showed a significant increase in the parameters (AUC0–72 h, Tmax, Cmax) of NPs/Erg. Additionally, the absorption of NPs/Erg showed remarkable characteristics as indicated by the values of Tmax, Cmax and AUC0-72 h (Table 4). Furthermore, the NPs/Erg took a longer time to reach the maximum plasma concentration compared with the free ergosterol. The Cmax of the encapsulated ergosterol was also approximately 1.61-fold higher than the free drug suspension, while the AUC0–72 h also showed an approximate 4.90-fold increase. According to the HPLC data shown in the supporting information (Supplementary Figure S1), there was no interference from endogenous sterols. The comparisons between NPs/Erg and that of other ergosterol-loaded nanoparticles (nanomicelle, liposome or microemulsion) were based on particle size, encapsulation efficiency, loading capacity, Tmax (h), t1/2 (h) and relative bioavailability (Table 5). The NPs/Erg compared with the other formulations had a moderate encapsulation efficiency, exceptional loading capacity, and a longer Tmax (h) and t1/2 (h).

Mean plasma concentration-time profiles of ergosterol after oral administration of a single 50 mg/kg dose each of NPs/Erg and free ergosterol suspension to rats. Mean±SD (n=5). The points with significant differences (P<0.05, versus free ergosterol suspension) were labeled as solid.
Tissue distribution study
The in vivo tissue distribution of ergosterol in the PLGA nanoparticles was rapidly distributed in the stomach, kidney, liver, brain, spleen, and virtually non-existent in the heart and lung (Figure 7). This was contrary to the free ergosterol in which small amount of sterols could be detected in all tissues. Notably, the sterols in the formulation were concentrated in the stomach, brain and liver and to a lesser extent, in the spleen, kidney and lung, or virtually nonexistent in the heart at the time of 4 h. In contrast, the greatest distribution of the free ergosterol was found in the kidney compared with other tissues.

Amount of ergosterol in the heart, liver, spleen, lung, kidney, stomach, brain and excrement following oral administration of a single 50 mg/kg dose of free ergosterol suspension and ergosterol-loaded PLGA nanoparticles to mice at different times. Mean±SD (n=5). *P<0.05 versus free ergosterol.
Discussion
The size of the spherical NPs/Erg droplet under the TEM was smaller than the one observed in DLS. This situation occurred as a result of the associated dehydration and shrinkage in using TEM. The aggregated particles could be caused by natural clustering due to insufficient steric stabilization by a non-ionic surfactant (PVA)27. The hydrophobic moieties of PVA were also interconnected with the PLGA chains to form the matrix, whereas the hydrophilic PVA moieties interacted with the water phase. This arrangement was conducive in the formation of PVA “corona” as observed in TEM and other related studies28. The NPs/Erg did not significantly affect the particle size and distribution, which also indicated an acceptable formulation. The low polydispersity index indicated the uniformity of the nanoparticle size29. Some studies have suggested that lower zeta potential values could augment the uptake of nano-droplets as well as their stability and efficacy30. The negative value of the zeta potential in the NPs/Erg was in agreement with such reports.
As encapsulation efficiency increases, there was a corresponding decrease in drug loading content as stated in other studies31. Fortunately, the process yield could reach the highest value (57.54%) when the drug-polymer ratio was 1:6. At that condition, the drug-loading content and encapsulation efficiency was 10.88%±0.52% and 76.29%±3.68%, respectively, which led to the selection of a 1:6 feeding ratio of drug-polymer as used in previous reports26. The spectral analysis of ergosterol in the nanoparticle was consistent with other similar studies32. The spectral data revealed that the ergosterol had an association with the PLGA polymer via hydrogen bonds. Additionally, the broader band of C=O stretching (1700–1800 cm−1) in the NPs/Erg indicated that the ergosterol had a relationship with the PLGA polymer through interactions between the carbonyl and the carboxyl groups of the sterol and the polymer28. The DRIFT measurements therefore confirmed the successful encapsulation of ergosterol within the PLGA nanoparticles.
Apart from the acceptable cumulative release in the 50% ethanol solution, the medium was also selected because it met the sink condition6. Generally, drug absorption is mostly confined to the intestinal space for a longer time after oral administration21. Therefore, the PLGA nanoparticles had a potential application in enhancing the oral bioavailability of ergosterol because its cumulative release profile at a pH of 6.8 (similar to intestinal fluid) was far better (54.62%) than the free ergosterol (34.23%). The initial drug release of NPs/Erg occurred through diffusion of the drug from the polymer matrix, whereas during the later stages, the release could be mediated through both the diffusion of drug and the degradation of the polymer matrix22. Normally, most of the drug-loaded nanoparticles and microparticulate formulations show a biphasic release pattern wherein there is an initial burst release followed by a sustained release of the drug25. Compared with the free ergosterol suspension, the rapid initial release that was observed in the release profile of ergosterol from the NPs/Erg suspension could indicate that a small amount of compound was closely attached to the surface of the particle.
In the present study, the retardation effect of the membrane barrier did not significantly affect the release profile of the ergosterol, even though the membrane barriers are often related to the dialysis medium. This was clearly evident in the free drug using DMSO that reached equilibrium within a shorter time compared with others. The release profile of NPs/Erg without the membrane barrier also gave similar results as those of others using the dialysis method. Therefore, it is possible that the incomplete drug release could be accounted for by factors such as the drug form in carrier materials and the type of release media. The free ergosterol suspension that seems to show a sustained release behavior could be due to the crystalline solid form of the drug and its barely soluble nature in the suspension. The viscidity of the suspension could also slow down the speed of drug diffusion. Although, the sustained release behavior of the free ergosterol was similar to NPs/Erg, this type of sustained release profile was not pharmaceutically important as witnessed in the formulated drugs. These findings were in agreement with other reports in which the free drug demonstrated an in vitro drug release profile over an extended period of time (beyond 72 h) with a lower cumulative release compared with the encapsulated drugs11.
The cytotoxic studies indicated that the PLGA carrier had no toxic effect on the cells; however, the NPs/Erg were associated with lower cell viability in the different human cancer cell lines (U251, MCF-7 and HepG2) compared with the free drug. The encapsulation of the drug inside the PLGA system was therefore more effective in inhibiting the growth of various cancer cells. The enhanced in vitro cancer growth inhibition of NPs/Erg was possibly due to the different transportation mechanisms between the free ergosterol and PLGA NPs33. The entry of free ergosterol into the cell usually occurs through specific transporters. However, the encapsulated drug enters the cell via other mechanisms such as endocytosis, which could cause the enhanced cytotoxicity of NPs/Erg34. The PLGA NPs could also escape the endo-lysosomal pathway and deliver the formulated drug into the cytoplasm35. These findings suggested that the drug-loaded PLGA nanoparticles were very effective in the in vitro cytotoxicity of cancer cells compared with the free ergosterol.
The spectral analysis of the synthesized suc-ergosterol in this study was consistent with previous reports6. These data supported the successful synthesis of suc-ergosterol as an internal substance. The higher amount of ergosterol from NPs/Erg compared with the free ergosterol in the study suggested a different mode in the absorption and elimination profiles of NPs/Erg. The significant increase in the pharmacokinetic parameters (AUC0–72 h, Tmax, Cmax) of NPs/Erg was in agreement with previous studies regarding the oral administration of PLGA formulations36. We speculate that the improved oral absorption of NPs/Erg resulted in a higher release rate and uptake at the intestinal tract as aided by the inhibition of the P-gp-mediated efflux21,37.
The longer t1/2 (h) of NPs/Erg indicated that the formulation could have a longer circulation time11. Therefore, the mechanism of improving oral bioavailability with NPs/Erg could be due to the enhanced oral absorption and longer circulation time. The delayed Tmax may have occurred because drugs from the free ergosterol suspension were released at the gastric tract. However, the bulk of the formulated drug was probably released at the intestine or directly taken up by the gastrointestinal tract to reach systemic circulation. The entire results showed that the relative bioavailability of NPs/Erg was approximately 490.07% compared with the free drug. Additionally, the working HPLC conditions were appropriate because no interference from endogenous sterols was detected.
The NPs/Erg compared with other similar reported formulations (nanomicelle, liposome or microemulsion) had moderate encapsulation efficiency, extraordinary loading capacity, and a longer Tmax and t1/2, which may be helpful for better absorption and bioavailability (Table 5). Most importantly, the NPs/Erg could be freeze-dried into a solid powder and then mixed into solid dispersions or other quick release dosage forms to produce a highly efficient and long-acting slow-release oral formulation8.
Numerous studies have investigated the enhanced in vivo tumor distribution of drug-loaded PLGA nanoparticles23,24. However, very few reports have focused on the tissue distribution of PLGA nanoparticles, which is of great importance to the evaluation of tissue targeting and toxicity. The biodistribution studies supported the fact that the PLGA nanoparticles could be a potential vehicle in the treatment of high-grade glioma due to the enhanced distribution in the brain and decreased heart, and kidney distribution31. In the present study, the blood brain barrier (BBB) could not restrict the biodistribution of the encapsulated drugs in the brain. These findings were also consistent with other studies in which PLGA NPs could enter the brain34,38,39,40. Although the mechanism that PLGA nanoparticles use to cross the BBB has not yet been fully reported, some studies have indicated that PLGA nanoparticles could improve the permeability and restrain P-gp-mediated efflux21,37. Similarly, certain nanoparticles can facilitate the movement of drugs across the BBB via the blocking of active efflux transporters such as p-glycoprotein41,42,43. These rapid nanoscience developments are likely to enhance the brain endothelial cellular uptake.
In conclusion, ergosterol (an insoluble phytosterol) was successfully encapsulated in polymeric PLGA nanoparticles using an emulsion/solvent evaporation technique. The optimized formulation produced a particle size of approximately 156 nm. The in vitro release profile of ergosterol from the formulation demonstrated a significant improvement in the solubility of ergosterol. The pharmacokinetic study in rats showed that NPs/Erg could achieve a 1.61-fold higher Cmax, a 1.65-fold longer Tmax, a 5.06-fold longer half-life and a 4.90-fold increase in bioavailability compared with the free drug. Additionally, the NPs/Erg were deeply concentrated in the liver, spleen and brain, but they showed less or no accumulation in other related organs such as the kidney, lung and heart. This observation was contrary to the findings for free ergosterol. Similarly, the cytotoxicity of NPs/Erg showed a greater decrease in IC50 among the human cancer cells (U251, MCF-7 and HepG2) compared with the free ergosterol. Therefore, in the present study, the PLGA nanoparticles served as a promising carrier for a poorly soluble plant sterol and significantly improved the bioavailability, biodistribution and in vitro antitumor activities of ergosterol.
Author contribution
Hui-yun ZHANG performed most of the experiments and wrote the manuscript; Caleb Kesse FIREMPONG technically modified the manuscript; Yuan-wen WANG and Miao-miao WANG assisted in the preparation and characterization of NPs/Erg; Wen-qian XU assisted in cell culture experiments; Yuan ZHU and Shan-shan TONG helped with the analysis for the characterization and oral pharmacokinetic study of NPs/Erg; Xia CAO assisted in the analysis of the cell culture results; Jiang-nan YU contributed to the development of the analytical method for in vivo ergosterol; Xi-ming XU designed and directed the study as well as participated in the overall editing and approval of the paper. All authors reviewed the manuscript.
References
Awad AB, Roy R, Fink CS . b-Sitosterol, a plant sterol induces apoptosis and activates key caspases in MDA-MB-231 human breast cancer cells. Oncol Rep 2003; 10: 497–500.
Devaraj S, Jialal I, Vega-López S . Plant sterol-fortified orange juice effectively lowers cholesterol levels in mildly hypercholesterolemic healthy individuals. Arterioscl Throm Vas 2004; 24: e25–8.
Fassbender K, Lütjohann D, Dik MG, Bremmer M, König J, Walter S, et al. Moderately elevated plant sterol levels are associated with reduced cardiovascular risk — the LASA study. Atherosclerosis 2008; 196: 283–8.
Yi C, Zhong H, Tong S, Cao X, Liu H, Fu M, et al. Enhanced oral bioavailability of a sterol-loaded microemulsion formulation of Flammulina velutipes, a potential antitumor drug. Int J Nanomed 2012: 5067–78.
Ma L, Chen H, Dong P, Lu X . Anti-inflammatory and anticancer activities of extracts and compounds from the mushroom Inonotus obliquus. Food Chem 2013; 139: 503–8.
Yi C, Fu M, Cao X, Tong S, Zheng Q, Firempong CK, et al. Enhanced oral bioavailability and tissue distribution of a new potential anticancer agent, Flammulina velutipes Sterols, through liposomal encapsulation. J Agric Food Chem 2013; 61: 5961–71.
Takaku T, Kimura Y, Okuda H . Isolation of an antitumor compound from Agaricus blazei Murill and its mechanism of action. J Nutr 2001; 131: 1409–13.
Cao X, Fu M, Wang L, Liu H, Deng W, Qu R, et al. Oral bioavailability of silymarin formulated as a novel 3-day delivery system based on porous silica nanoparticles. Acta Biomater 2012; 8: 2104–12.
Dasgupta N, Ranjan S, Mundekkad D, Ramalingam C, Shanker R, Kumar A . Nanotechnology in agro-food: From field to plate. Food Res Int.
Shakib K, Tan A, Soskic V, Seifalian AM . Regenerative nanotechnology in oral and maxillofacial surgery. Br J Oral Max Surg 2014; 52: 884–93.
Zhu Y, Peng W, Zhang J, Wang M, Firempong CK, Feng C, et al. Enhanced oral bioavailability of capsaicin in mixed polymeric micelles: Preparation, in vitro and in vivo evaluation. J Funct Foods 2014; 8: 358–66.
Menon JU, Ravikumar P, Pise A, Gyawali D, Hsia CCW, Nguyen KT . Polymeric nanoparticles for pulmonary protein and DNA delivery. Acta Biomater 2014; 10: 2643–52.
Zafar S, Negi LM, Verma AK, Kumar V, Tyagi A, Singh P, et al. Sterically stabilized polymeric nanoparticles with a combinatorial approach for multi drug resistant cancer: In vitro and in vivo investigations. Int J Pharm 2014; 477: 454–68.
Canfarotta F, Whitcombe MJ, Piletsky SA . Polymeric nanoparticles for optical sensing. Biotechnol Adv 2013; 31: 1585–99.
Chen J, Li Y, Zhong W, Hou Q, Wang H, Sun X, et al. Novel fluorescent polymeric nanoparticles for highly selective recognition of copper ion and sulfide anion in water. Sensors Actuators B: Chemical 2015; 206: 230–8.
Dinesh Kumar V, Verma PRP, Singh SK . Development and evaluation of biodegradable polymeric nanoparticles for the effective delivery of quercetin using a quality by design approach. Food Sci Technol 2015; 61: 330–8.
Yi C, Sun C, Tong S, Cao X, Feng Y, Firempong CK, et al. Cytotoxic effect of novel Flammulina velutipes sterols and its oral bioavailability via mixed micellar nanoformulation. Int J Pharm 2013; 448: 44–50.
Acharya S, Sahoo SK . PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliver Rev 2011; 63: 170–83.
Anderson JM, Shive MS . Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliver Rev 2012; 64: 72–82.
Hamdy S, Haddadi A, Hung RW, Lavasanifar A . Targeting dendritic cells with nano-particulate PLGA cancer vaccine formulations. Adv Drug Deliver Rev 2011; 63: 943–55.
Joshi G, Kumar A, Sawant K . Enhanced bioavailability and intestinal uptake of Gemcitabine HCl loaded PLGA nanoparticles after oral delivery. Eur J Pharm Sci 2014; 60: 80–9.
Italia JL, Bhatt DK, Bhardwaj V, Tikoo K, Kumar MN . PLGA nanoparticles for oral delivery of cyclosporine: nephrotoxicity and pharmacokinetic studies in comparison to Sandimmune Neoral. J Control Release 2007; 119: 197–206.
Duong HH, Yung LY . Synergistic co-delivery of doxorubicin and paclitaxel using multi-functional micelles for cancer treatment. Int J Pharm 2013; 454: 486–95.
Fonseca C, Simões S, Gaspar R . Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J Control Release 2002; 83: 273–86.
Musumeci T, Ventura CA, Giannone I, Ruozi B, Montenegro L, Pignatello R, et al. PLA/PLGA nanoparticles for sustained release of docetaxel. Int J Pharm 2006; 325: 172–9.
Jose S, Sowmya S, Cinu TA, Aleykutty NA, Thomas S, Souto EB . Surface modified PLGA nanoparticles for brain targeting of Bacoside-A. Eur J Pharm Sci 2014; 63: 29–35.
Silva LM, Hill LE, Figueiredo E, Gomes CL . Delivery of phytochemicals of tropical fruit by-products using poly (DL-lactide-co-glycolide) (PLGA) nanoparticles: synthesis, characterization, and antimicrobial activity. Food Chem 2014; 165: 362–70.
Gomes C, Moreira RG, Castell-Perez E . Poly (DL-lactide-co-glycolide) (PLGA) nanoparticles with entrapped trans-cinnamaldehyde and eugenol for antimicrobial delivery applications. J Food Sci 2011; 76: N16–24.
Patel RB, Patel MR, Bhatt KK, Patel BG . Formulation consideration and characterization of microemulsion drug delivery system for transnasal administration of carbamazepine. Bull Faculty Pharm, Cairo Univ 2013; 51: 243–53.
Kakumanu S, Tagne JB, Wilson TA, Nicolosi RJ . A nanoemulsion formulation of dacarbazine reduces tumor size in a xenograft mouse epidermoid carcinoma model compared to dacarbazine suspension. Nanomedicine 2011; 7: 277–83.
Jiang X, Xin H, Sha X, Gu J, Jiang Y, Law K, et al. PEGylated poly(trimethylene carbonate) nanoparticles loaded with paclitaxel for the treatment of advanced glioma: in vitro and in vivo evaluation. Int J Pharm 2011; 420: 385–94.
Pool H, Quintanar D, Figueroa JD, Marinho Mano C, Bechara JE, Godínez LA, et al. Antioxidant effects of Quercetin and Catechin encapsulated into PLGA nanoparticles. J Nanomater 2012; 2012: 1–12.
Song XR, Cai Z, Zheng Y, He G, Cui FY, Gong DQ, et al. Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur J Pharm Sci 2009; 37: 300–5.
Paul A, Das S, Das J, Samadder A, Khuda-Bukhsh AR . Cytotoxicity and apoptotic signalling cascade induced by chelidonine-loaded PLGA nanoparticles in HepG2 cells in vitro and bioavailability of nano-chelidonine in mice in vivo. Toxicol Lett 2013; 222: 10–22.
Jin C, Bai L, Wu H, Song W, Guo G, Dou K . Cytotoxicity of paclitaxel incorporated in PLGA nanoparticles on hypoxic human tumor cells. Pharm Res 2009; 26: 1776–84.
Khalil NM, do Nascimento TC, Casa DM, Dalmolin LF, de Mattos AC, Hoss I, et al. Pharmacokinetics of curcumin-loaded PLGA and PLGA-PEG blend nanoparticles after oral administration in rats. Colloid Surface B 2013; 101: 353–60.
Xie X, Tao Q, Zou Y, Zhang F, Guo M, Wang Y, et al. PLGA nanoparticles improve the oral bioavailability of curcumin in rats: characterizations and mechanisms. J Agri Food Chem 2011; 59: 9280–9.
Semete B, Booysen L, Lemmer Y, Kalombo L, Katata L, Verschoor J, et al. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine 2010; 6: 662–71.
Vergoni AV, Tosi G, Tacchi R, Vandelli MA, Bertolini A, Costantino L . Nanoparticles as drug delivery agents specific for CNS: in vivo biodistribution. Nanomedicine 2009; 5: 369–77.
Koziara JM, Lockman PR, Allen DD, Mumper RJ . In situ blood–brain barrier transport of nanoparticles. Pharm Res 2003; 20: 1772–8.
Silva GA . Nanotechnology approaches to crossing the blood-brain barrier and drug delivery to the CNS. BMC Neurosci 2008; 9: S4.
Brigger I, Morizet J, Aubert G, Chacun H, Terrier-Lacombe MJ, Couvreur P, et al. Poly(ethylene glycol)-coated hexadecylcyanoacrylate nanospheres display a combined effect for brain tumor targeting. J Pharm Exp Ther 2002; 303: 928–36.
Krol S, Macrez R, Docagne F, Defer G, Laurent S, Rahman M, et al. Therapeutic benefits from nanoparticles: the potential significance of nanoscience in diseases with compromise to the blood brain barrier. Chem Rev 2012; 113: 1877–903.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant 30973677), the Doctoral Fund of Ministry of Education of China (Grant 20113227110012), and the Industry-University-Research Institution Cooperation (Grants BY2009141 and CY2010023) in the Jiangsu Province and Zhenjiang City. The authors also thank the Jiangsu University Ethics Committee for their kind guidance in the animal experiments.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary information is available at the Acta Pharmacologica Sinica's website.
Supplementary information
Supplementary Information Figure S1
Chromatograms of ergosterol and internal standard in rat plasma, heart, liver, spleen, lung, kidney, stomach, brain and excrement homogenate: (A1) blank rat plasma, (A2)∼(A9): blank heart, liver, spleen, lung, kidney, stomach, brain and excrement homogenate; (B1) blank plasma spiked with ergosterol, and internal standard, (B2)∼(B9): blank heart, liver, spleen, lung, kidney, stomach, brain and excrement homogenate spiked with ergosterol and internal standard; (C1) a rat plasma sample after oral administration, (C2)∼(C9): heart, liver, spleen, lung, kidney, stomach, brain and excrement homogenate samples after oral administration (1, internal standard; 2, ergosterol). (DOC 477 kb)
Rights and permissions
About this article

Cite this article
Zhang, Hy., Firempong, C., Wang, Yw. et al. Ergosterol-loaded poly(lactide-co-glycolide) nanoparticles with enhanced in vitro antitumor activity and oral bioavailability. Acta Pharmacol Sin 37, 834–844 (2016). https://doi.org/10.1038/aps.2016.37
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2016.37
Keywords
This article is cited by
-
A Novel Approach for Delivery of Ergosterol Within Ferritin Cage: Stability, Slow-Release Property, and Cholesterol-Lowering Effect After Simulated Gastrointestinal Digestion
Food Biophysics (2024)
-
Immobilization of psychrophile Psychrobacter sp. ANT206 onto novel reusable magnetic nanoparticles and its application for nitro-aromatic compounds biodegradation under low temperature
Biodegradation (2022)
-
Controlled release and targeted drug delivery with poly(lactic-co-glycolic acid) nanoparticles: reviewing two decades of research
Journal of Pharmaceutical Investigation (2022)
-
Fabrication of Second Generation Smarter PLGA Based Nanocrystal Carriers for Improvement of Drug Delivery and Therapeutic Efficacy of Gliclazide in Type-2 Diabetes Rat Model
Scientific Reports (2019)
